

Abstract
Mutations in the mitochondrial transacylase tafazzin, Taz1p, in Saccharomyces cerevisiae cause Barth syndrome, a disease of defective cardiolipin remodeling. Taz1p is an interfacial membrane protein that localizes to both the outer and inner membranes, lining the intermembrane space. Pathogenic point mutations in Taz1p that alter import and membrane insertion result in accumulation of monolysocardiolipin. In this study, we used yeast as a model to investigate the biogenesis of Taz1p. We show that to achieve this unique topology in mitochondria, Taz1p follows a novel import pathway in which it crosses the outer membrane via the translocase of the outer membrane and then uses the Tim9p-Tim10p complex of the intermembrane space to insert into the mitochondrial outer membrane. Taz1p is then transported to membranes of an intermediate density to reach a location in the inner membrane. Moreover, a pathogenic mutation within the membrane anchor (V224R) alters Taz1p import so that it bypasses the Tim9p-Tim10p complex and interacts with the translocase of the inner membrane, TIM23, to reach the matrix. Critical targeting information for Taz1p resides in the membrane anchor and flanking sequences, which are often mutated in Barth syndrome patients. These studies suggest that altering the mitochondrial import pathway of Taz1p may be important in understanding the molecular basis of Barth syndrome.
INTRODUCTION
Barth syndrome (BTHS) is a recessive X-linked disorder which causes cardiac myopathy, neutropenia, metabolic abnormalities, and atypical mitochondrial function and morphology, with cardiac failure and other potentially fatal complications often, but not always, presenting in infancy and childhood (1–4). The human tafazzin gene (TAZ) was first implicated in the disease by linkage analysis (1, 5). Clinical studies and research in several model organisms, ranging from Saccharomyces cerevisiae to the fly, zebrafish, and mouse, show that mutations in or deletions of the TAZ locus result in abnormal phospholipid species, with the mitochondrial phospholipid cardiolipin (CL) affected the most severely (5–11). Tafazzin functions as a transacylase in the CL maturation pathway by replacing saturated chains with unsaturated chains (7). A hallmark of BTHS is the accumulation of the CL remodeling intermediate monolyso-CL, which lacks a fatty acid (6, 12).
The mitochondrial localization of Taz1p as well as other lipid biogenesis proteins in mitochondria is complex. Cardiolipin synthase, Crd1p, synthesizes CL in the matrix-facing leaflets of the inner membrane (13). Localization studies showed that wild-type Taz1p localizes to the inner and outer mitochondrial membranes, facing the intermembrane space (6, 14, 15). When yeast was used as a system to model Taz1p mutations associated with BTHS, a subset of Taz1p mutants correctly localized to the intermembrane space but assembled in complexes that were not functional (6) or were degraded by the intermembrane space i-AAA protease Yme1 (12). Another subset of mutants localized to the mitochondrial matrix, suggesting that mistargeting of tafazzin resulted in BTHS in a subset of patients (6). Given the unexpected localization for tafazzin within mitochondria and that protein mistargeting could be the cause of BTHS in this subset of patients, a detailed study of Taz1p biogenesis is warranted. Taz1p assembly in the membrane is also unusual in that Taz1p is an interfacial membrane protein, anchored by a hydrophobic membrane anchor that protrudes into but not completely through the lipid bilayer (residues 215 to 232) (6). The membrane anchor of Taz1p is also important for mitochondrial localization because point mutations in the membrane anchor (recapitulating pathogenic point mutations in Taz1p identified in BTHS patients) result in mistargeting to the mitochondrial matrix, where Taz1p either assembles in the leaflet of the inner membrane facing the matrix or forms protein aggregates (6).
The import of mitochondrial proteins is accomplished by a complex translocation system consisting of complexes in the outer membrane, inner membrane, and intermembrane space (16, 17). Whereas many mitochondrial proteins contain a canonical N-terminal targeting sequence, membrane proteins contain internal targeting information that typically resides in the regions near the membrane-spanning domains (18). We have previously shown that Taz1p does not contain a typical N-terminal targeting sequence (6); thus, mitochondrial targeting of Taz1p is directed presumably via an internal targeting sequence.
To date, four general types of outer membrane proteins have been identified in mitochondria (19–22). The targeting and sorting mechanisms that dictate their correct localization often overlap and, in some cases, are not fully characterized. Signal anchored proteins, such as the Tom20p and Tom70p receptors of the TOM complex, have a moderately hydrophobic alpha-helical transmembrane domain in the N terminus, which, in addition to positive charges in the flanking sequences, serves as both an intercellular sorting domain and a membrane anchor. Tail-anchored proteins such as Fis1p, Tom5p, Tom6p, Tom22p, and Gem1p have an alpha-helical C-terminal transmembrane domain and often require a moderately net positive charge located C terminal to the tail-anchor domain (23). Finally, a subset of proteins have multiple transmembrane domains, including those that are alpha-helical (i.e., Fzo1p and Ugo1p) and the β-sheets of β-barrel proteins (i.e., Tom40p and porin). Mim1 mediates the insertion of outer membrane proteins with alpha-helical membrane domains (21, 22). In contrast, the β-barrel proteins have targeting information at the very C terminus and are imported via the TOM complex and then inserted from the intermembrane space side via the sorting and assembly (SAM) complex (24). The small Tim proteins (Tim9p-Tim10p and Tim8p-Tim13p) chaperone the β-barrel proteins through the intermembrane space (25).
Membrane proteins of the inner membrane can use the TIM22 or TIM23 import pathways. The TIM22 pathway (16, 17, 26) mediates the insertion of polytopic carrier proteins such as the ADP/ATP carrier (AAC) and the phosphate carrier. In this pathway, the 70-kDa small Tim chaperone complexes of the intermembrane space, Tim8p-Tim13p and Tim9p-Tim10p, bind to hydrophobic regions of the precursor and mediate translocation to the insertion complex that consists of Tim12p, Tim22p, Tim54p, Tim18p, and a fraction of the small Tim proteins Tim9p and Tim10p. Both matrix targeted and inner membrane proteins that contain an N-terminal targeting sequence are translocated through the TIM23 pathway and may be inserted into the inner membrane by the TIM23 complex or Oxa1p, depending on sorting information (27, 28).
Even though the Tim9-Tim10p complex has been previously implicated in Taz1p biogenesis (15, 29), in this study, we investigated Taz1p import in detail. Taz1p is first imported into the outer membrane using the TOM complex in a Tim9p-Tim10p dependent manner. The membrane anchor and N-terminal flanking sequences are important for targeting. Taz1p then is sorted to the inner membrane via intermediate density membranes to be distributed in both the outer and inner membranes, where it may modify cardiolipin in the membrane leaflets that line the intermembrane space.
MATERIALS AND METHODS
Strains and growth conditions.
Standard conditions were used for the growth, manipulation and transformation of yeast, with the parental strain GA74-1A used except where indicated. Characterization of the following temperature-sensitive or deletion mutant strains with the following relevant genotypes was detailed previously: tim22-4 (30), tim10-1 (31), Δtom20 (YTJB10) and Δtom70 (YVH1) (32), tim23-6 (33), and Δtaz1 (6). All strains were grown in YPEG to mid-log phase and mitochondria were harvested from yeast as previously described (34). Δtaz1 and Δtaz1[pTaz1] strains were described previously (6). The Δtaz1[pTaz1ΔMA215-232] strain was generated by transformation of the Δtaz1 parental strain.
Cloning and in vitro transcription/translation.
The TAZ1 open reading frame was subcloned into an SP6 polymerase in vitro transcription/translation vector with an N-terminal Kozac sequence to enhance transcription (GCC GCC GCC ATG TCT). The V224R mutant was subcloned from pRS425Taz1V224R using NdeI and AvrII restriction enzyme sites. The pRS425Taz1ΔMA215–232 plasmid was made with overlap extension PCR to remove the membrane anchor; briefly, the DNA products from the primers sets A and B (GAAGCCTATCCCAGAAG and ATGCTATTTTGATCATTCTGGTAATACCCC, respectively) and C and D (CAGAATGATCAAAATAGCATCCGAAGCAGTC and GAACCGCTTCCACCATGAGG, respectively) were diluted and used as a template with primer set A and D, and the product of this reaction was cut with Ndel and AvrII and subcloned into the pSP64Taz1 plasmid. The dihydrofolate reductase (DHFR) fusion constructs were subcloned into the pSP65DHFR-Tim23-DHFR vector in which Tim23 was replaced with Taz1 sequences; the inserts were made by amplifying the appropriate Taz1 regions with primers that would add an AvaI restriction enzyme site and a NAAIRS linker region (AATGCTGCTATACGATCG) N terminally and a BamH I site and Gly linker region (GGTGGCGGAGGGGGTGGC) C terminally. To construct the pSP65DHFR-Taz1V224R-DHFR plasmid, the same primers were used to amplify a fragment from the pSP64Taz1V224R template. All 35S-radiolabeled precursors were generated with a Promega TNT kit, but in early experiments (with the tim22-4 and tim10-1 strains), a rabbit reticulocyte lysate was used as previously described (33).
In vitro import assays.
The precursor from the in vitro transcription/translation reaction mixture was incubated at 25°C with isolated mitochondria in import buffer (1 mg/ml of bovine serum albumin, 0.6 M sorbitol, 0.6 M KCl, 10 mM HEPES-KOH [pH 7.1], 10 mM MgCl2, 2.5 mM EDTA, 5 mM l-methionine, 2 mM ATP, and 2 mM NADH). All import reactions were performed at 25°C, and the reactions were quenched at specific time intervals. When an energy-regenerating system was used, the reaction buffer was supplemented with 100 μg/ml of creatine phosphate kinase (CPK), 16 mM creatine Pi, and 10 mM succinate. Where indicated, the potential across the mitochondrial inner membrane was dissipated using 1 μM valinomycin and 25 μM p-trifluoromethoxy carbonyl cyanide phenylhydrazone (FCCP). Nonimported radiolabeled precursor was removed by treatment with 20 μg/ml of trypsin or 5 μg/ml of proteinase K for 30 min on ice. Trypsin was inhibited with 100 μg/ml of soybean trypsin inhibitor and proteinase K with 1 mM phenylmethylsulfonyl (PMSF). For alkali extraction, the samples were first pelleted, resuspended in 100 mM Na2CO3 (pH 10.5), incubated on ice for 30 min, and then centrifuged at high speed (100,000 × g) in an Airfuge. Unless indicated otherwise, all samples were resuspended in SDS sample buffer supplemented with 2-mercaptoethanol (BME), loaded onto 12% polyacrylamide gels, and dried for quantitative autoradiography using a Bio-Rad FX Pro plus fluorimager/phosphorimager. Bio-Rad Quantity 1 software was used to quantitate the gels, and the last time point of import into wild-type mitochondria was set to 100%.
Cross-linking and immunoprecipitation.
The cross-linking and immunoprecipitation protocol was as previously described (30), with the following modifications: the import reaction mixture contained 1 μM valinomycin and 25 μM FCCP to trap the import intermediate. The mitochondria were incubated for 30 min on ice with 1 mM dithiobis[succinimidyl propionate] (DSP), and the reaction was quenched for 15 min with 10 mM Tris-HCl (pH 8.0). For immunoprecipitation, the mitochondria were first solubilized with TNET buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 5 mM EDTA, and 1% Triton X-100) and incubated with the indicated polyclonal rabbit antibodies coupled to protein A-Sepharose beads overnight. Before loading to the 10% polyacrylamide gel, the cross-links of the immunoprecipitation samples were treated with 1 mM 2-mercaptoethanol to reduce the cross-linked products.
Subcellular fractionation, mitoplast manipulation, and digitonin solubilization assays.
Subcellular fractionation was performed as previously described (6). Osmotic shock was performed by incubating the mitochondria after the import assay in 0.03 M sorbitol and 20 mM HEPES-KOH (pH 7.4) for 30 min on ice, with gentle agitation. When necessary, 10 μg/ml of proteinase K with or without 0.1% Triton X-100 was added to the solution. The mitoplasts (mitochondria that have had the outer membrane disrupted) were pelleted in a microcentrifuge and resuspended in sample buffer, and the released soluble proteins were precipitated with 20% trichloroacetic acid (TCA). For the digitonin solubilization assay (35), aliquots were taken from either resuspended mitochondria or the import assay, pelleted, and resuspended in SEMK (250 mM sucrose, 1 mM EDTA, 10 mM morpholinepropanesulfonic acid [MOPS]-KOH [pH 7.2], 100 mM KCl) buffer that contained increasing amounts of digitonin (0% to 0.5% in 0.1% increments). The samples were quickly pelleted in the Airfuge at 100,000 × g at 4°C. The pellet (P) was resuspended in SDS sample buffer supplemented with β-mercaptoethanol, and the supernatants (S) were TCA precipitated.
Blue native gel electrophoresis.
After the import assay, the mitochondria were pelleted and then solubilized in 20 mM HEPES-KOH (pH 7.4), 50 mM NaCl, 10% glycerol, 2.5 mM MgCl2, 1 mM EDTA, 1 mM PMSF, and 1% digitonin for 30 min on ice. Insoluble material was removed by spinning the samples in the microcentrifuge at high speed for 15 min. The solubilized complexes were supplemented with 50 mM 6-aminocaproic acid and 0.5% Coomassie brilliant blue G-250 and then loaded to a 6 to 16% gradient gel at 4°C. The gel was either dried and exposed to film for autoradiography or blotted to polyvinylidene difluoride (PVDF) membranes for Western blot analysis.
RESULTS
Taz1p import is not dependent on a membrane potential.
The atypical localization of Taz1p to both the inner and outer mitochondrial membranes lining the intermembrane space warrants detailed characterization of the biogenesis of Taz1p (6). We first investigated whether a membrane potential (Δψ) was required for Taz1p import. Whereas proteins that are imported across the inner membrane and use the TIM23 or TIM22 translocons require a Δψ for translocation, proteins that are imported directly to the outer membrane do not require a Δψ (16). Radiolabeled Taz1p was imported into isolated mitochondria in which the electrochemical potential was established with NADH or dissipated with valinomycin and p-trifluoromethoxy carbonyl cyanide phenylhydrazone (FCCP). An import time course was performed, nonimported precursor was removed by exogenous treatment with protease, and insertion into the membrane was verified by carbonate extraction (Fig. 1A). Samples were separated by SDS-PAGE and data quantitated following autoradiography. The rates of Taz1p import were similar in the absence or presence of a Δψ, consistent with a previous study (15). Taz1p translocation thus seems to bypass the TIM23 and TIM22 translocons of the inner membrane.
Fig 1.

Taz1p import is not dependent on the Δψ and is first imported through the TOM complex. (A) Radiolabeled Taz1p was incubated with wild-type mitochondria, and aliquots were removed at the indicated time points and incubated with 20 μg/ml of trypsin. Valinomycin at 1 μM and FCCP at 50 μM were added to the reaction buffer to dissipate the Δψ. Samples were separated by SDS-PAGE, and imported Taz1p was detected by autoradiography. Imported Taz1p was quantitated using Bio-Rad Quantity One software, with the last time point in the energized mitochondria set to 100%. (B) Taz1p was imported into isolated yeast mitochondria that were pretreated with 20 μg/μl of trypsin. p, precursor; m, mature form of Su9-DHFR. (C) Import of Taz1p into isolated Δtom20 and Δtom70 mitochondria.
To determine the role of the TOM translocon, we performed “trypsin shaving” in which mitochondria were pretreated with trypsin to remove the TOM receptors prior to import. Pretreatment with protease decreased Taz1p import by 80% compared to import into untreated mitochondria (Fig. 1B), suggesting a requirement for the cytosolic portion of outer membrane receptors in the early steps of Taz1p translocation (36). To dissect transport of Taz1p through the TOM translocon in greater detail, purified mitochondria from both a Δtom20 and a Δtom70 strain were used in the in vitro protein import assay. Tom20p facilitates the import of precursors with a typical N-terminal targeting sequence as well as β-barrel proteins (37), while Tom70p mediates the import of the carrier proteins that have internal targeting sequences (38). As controls, import of the carrier AAC and Su9-DHFR were compromised in Δtom70 and Δtom20 mitochondria, respectively (see Fig. S1 in the supplemental material). In contrast, Taz1p import was deficient in both mutant mitochondria, indicating that both Tom70p and Tom20p facilitate Taz1p targeting to the TOM translocation channel (Fig. 1C).
The TIM23 and TIM22 membrane translocation complexes are not required for Taz1p import.
We took advantage of temperature-sensitive mutants in the TIM22 and TIM23 translocation pathways to assess the requirement of inner membrane translocons. Taz1p was imported into mitochondria isolated from the tim23-6 and tim22-4 mutants (Fig. 2A and B) (30, 33). The impairment of each translocation pathway was demonstrated using AAC and Su9-DHFR precursors for the tim22-4 and tim23-6 mutant strains, respectively (see Fig. S1C and D in the supplemental material). Taz1p import was similar in the mutant and wild-type (WT) mitochondria. Thus, Taz1p import is independent of the TIM22 and TIM23 translocons, suggesting that the inner membrane translocons do not play a direct role in the initial import of Taz1p. In contrast, the import of Taz1p was decreased in the tim10-1 mutant mitochondria (Fig. 2C). This supports our previous result in which Taz1p import was blocked by theTim10p-specific inhibitor MitoBloCK-1 (29). To confirm that Tim10p played a role in Taz1p import, we used mitochondria that contained a suppressor mutation in TIM9 that restored import to the tim10-1 mutant (designated tim10-1tim9S) (29, 31) Indeed, import was restored to levels near that of WT mitochondria (Fig. 2C).
Fig 2.

Taz1p import is independent of the TIM22 and TIM23 translocons. (A to C) Taz1p was imported into tim23-6, tim22-4, tim10-1, and tim10-1tim9S mitochondria as described for Fig. 1A. (D) Taz1p was imported into WT mitochondria followed by cross-linking (XL) with 1 mM DSP and then coimmunoprecipitated (IP) with antibodies directed against Tim10p, Tom40p, cytochrome b2, CCP1, Tim22p, and Tim23p. Cross-links were broken with BME prior to loading the samples to the SDS-PAGE gel to confirm that the conjugates were Taz1p.
To confirm that Tim10p played a direct role in Taz1p import, we used cross-linking and coimmunoprecipitation to characterize import components that were bound to Taz1p. Radiolabeled Taz1p was imported into mitochondria, followed by cross-linking with 1 mM dithiobissuccimidylpropionate (DSP) and quenching with excess Tris buffer. Mitochondria were solubilized and immunoprecipitated with antibodies directed against Tim10p, Tom40p, cytochrome (CYT) b2, cytochrome c peroxidase (CCP1), Tim22p, and Tim23p (Fig. 2D). Taz1p was bound directly to Tim10p and Tom40p but not Tim22p or Tim23p. Taz1p also was not bound to the negative controls CCP1 and CYT b2, verifying a specific interaction of Taz1p with Tom40p and Tim10p. This series of import experiments supports a direct task for Tom40p and Tim10p in the biogenesis of Taz1p.
The Taz1p precursor is initially imported into the outer membrane.
We took advantage of the differing solubilities of the mitochondrial outer and inner membranes to digitonin (35) to substantiate Taz1p import via the outer membrane. Mitochondrial markers from different compartments can be differentially released to the supernatant upon treatment with increasing concentrations of digitonin, because digitonin selectively solubilizes the outer membrane with its high lipid content, followed by the inner membrane with its increased protein concentration. Thus, soluble intermembrane space proteins, such as cytochrome b2, are released to the supernatant following a high-speed spin after solubilization with a low digitonin concentration. Outer membrane proteins that are fully integrated into the membrane, such as porin and OM45, are released at intermediate digitonin concentrations, and finally, inner membrane proteins, such as AAC, are released at a higher detergent concentration (Fig. 3A; see also Fig. S2A in the supplemental material). Endogenous tafazzin had a release profile that is intermediate between the outer and inner membrane markers in both WT and tim23-6 mitochondria (Fig. 3A and C; see also Fig. S2A and C). This profile could be indicative of contact sites or reflect the dual residence of Taz1p on both the outer and inner membranes. However, the Taz1p release profile shifted to that of an outer membrane protein in tim10-1 mitochondria (Fig. 3B; see also Fig. S2B), suggesting that Taz1p distribution is aberrant. We also investigated the distribution of radiolabeled Taz1p following import; similarly, radiolabeled porin and Tim54 were imported as controls for outer and inner membrane markers, respectively (Fig. 3D; see also Fig. S2D). Imported Taz1p was localized to the outer membrane after import into WT mitochondria because Taz1p shared a digitonin solubilization profile with that of imported porin. The Taz1p profile was also similar to another imported outer membrane marker, Tom40p (data not shown). The same analysis was not performed with tim10-1 mitochondria because the import signal was too weak for analysis. We chased the import for up to 2 h, but the imported Taz1p remained in the outer membrane and a distribution profile similar to Taz1p at steady state in WT mitochondria (Fig. 3A; see also Fig. S2A) was not observed. These results suggest that Taz1p is first imported into the outer membrane of the mitochondrion and subsequently traffics into the inner membrane or contact sites.
Fig 3.

Taz1p is first imported into the outer mitochondrial membrane. (A) WT mitochondria (15 mg/ml) were solubilized with the indicated concentration of digitonin, and the soluble proteins were separated from the mitochondrial pellet by centrifugation. Outer (porin and OM45) and inner (Tim45p and AAC) membrane markers were averaged, and cyt b2 marked the intermembrane space. Equal amounts were analyzed by immunoblotting and quantitated with Bio-Rad Quantity 1 software. The percentage solubilized was calculated as [(S)/(S + P) × 100] for each detergent concentration (mean ± SD, n = 3). Immunoblots are provided in Fig. S2A to D in the supplemental material. (B) Same as panel A, with tim10-1 mitochondria. (C) Same as panel A, with tim23-6 mitochondria. (D) Taz1p was imported into WT mitochondria for 15 min, and then fractionation was investigated for the imported Taz1p as in panel A. Markers included imported porin and Tim54p.
Taz1pV224R is imported into the matrix via the Tim23 pathway.
Using yeast as a model, BTHS patients with mutations (V223D, V224R, I226P—equivalent mutations in yeast Taz1p) in the membrane anchor (residues 215 to 232) of Taz1p display an altered protein import pathway in which the mutant protein is imported to the matrix (6). This altered topology suggests that the membrane anchor is important for trafficking in mitochondria. We therefore investigated the role of the membrane anchor in Taz1p assembly by characterizing the import pathway of one representative mutant, Taz1pV224R (Fig. 4). The mutant Taz1pV224R was imported into isolated mitochondria, followed by osmotic shock to disrupt the mitochondrial outer membrane and generate mitoplasts. Centrifugation was used to separate the soluble intermembrane space from the mitoplasts (inner membrane compartment with matrix and intermembrane space proteins that bind to the inner membrane or outer membrane). In WT mitochondria, Taz1p localized to the mitoplast (P) fraction after osmotic shock but was sensitive to protease, confirming a localization facing the intermembrane space (Fig. 4A). In contrast, Taz1pV224R was protected from protease after osmotic shock, indicating a localization facing the matrix (Fig. 4A). As a control, the treatment with Triton X-100 confirmed that Taz1pV224R was sensitive to protease.
Fig 4.

Taz1pV224R is imported into the matrix via the Tim23 pathway. (A) Taz1p and Taz1pV224R were synthesized in vitro and imported into WT mitochondria, which were subjected to osmotic shock, followed by centrifugation to separate the supernatant (S) from the mitoplast pellet (P). Mitochondria were also treated with 20 μg/ml of proteinase K or 20 μg/ml of proteinase K plus 1% Triton X-100. As a control, M was included. (B) Radiolabeled Taz1pV224R was imported into both WT and tim23-6 mitochondria in the presence and absence of a membrane potential, followed by protease treatment to remove nonimported precursor. (C) Taz1pV224R was imported as in panel B in WT, tim10-1, and tim10-1tim9S mitochondria. (D) Taz1p and Taz1pV224R were imported into WT mitochondria, and the samples were solubilized in 1% digitonin and separated on a 6 to 16% blue native gel.
The altered topology of Taz1pV224R suggests that it may be imported independently of Tim10p and rely on the TIM23 import pathway. Indeed, Taz1pV224R import was dependent on a membrane potential and strongly decreased in tim23-6 mutant mitochondria (Fig. 4B). In addition, Taz1pV224R import bypassed the requirement for Tim10p because import was restored in the tim10-1 mitochondria and an additional mutant that contains a suppressing mutation in Tim9p, designated tim10-1tim9S (Fig. 4C) (31). Therefore, the imported mutant is likely found on the inner leaflet of the inner mitochondrial membrane, facing the matrix.
Finally, we investigated assembly of imported Taz1p using blue native gels in WT mitochondria. Taz1p and Taz1pV224R were imported and solubilized in 1% digitonin, followed by centrifugation to remove insoluble material. As reported previously (39), Taz1p assembles in several complexes, including three complexes at approximately 70, 120, and 240 kDa in WT mitochondria (Fig. 4D). However, Taz1pV224R only assembled in the smallest complex (70 kDa) and failed to assemble in larger complexes. Taken together, the import pathway of mutant Taz1pV224R was altered compared to that of wild-type Taz1p, which supports the notion that sequences in the membrane anchor are important for interacting with Tim10p.
The membrane anchor of Taz1p is required for import.
We constructed a version of Taz1p with a deletion in the membrane anchor, designated Taz1pΔMA215–232, and investigated import in vitro and in vivo. When Taz1pΔMA215–232 was expressed from a multicopy plasmid in a strain lacking Taz1p (Δtaz1), the mutant protein was detected at a lower molecular mass faintly in a yeast lysate (Fig. 5A); in contrast, wild-type Taz1p was readily detected when expressed from the same plasmid in the Δtaz1 strain. The yeast lysate was fractionated into a mitochondrial enriched pellet (M) and the postmitochondrial supernatant (PMS) (Fig. 5B). Whereas Taz1p was detected in the mitochondrial fraction, Taz1pΔMA215–232 was not detected after fractionation. However, an RNA transcript for Taz1pΔMA215–232 was detected (see Fig. S3 in the supplemental material). Thus, Taz1pΔMA215–232 is not a stable protein.
Fig 5.

The membrane anchor of Taz1p is required for mitochondrial localization. (A) Taz1pΔMA215–232, which lacks the membrane anchor, was expressed in a Δtaz1 strain and the truncated protein was detected by immunoblot analysis in a total protein lysate. The immunoblot analysis of WT TAZ1 and aconitase (Aco1) serve as controls for blotting and loading, respectively. (B) The lysate (T) of strains from panel A was fractionated into the M and PMS fractions, followed by immunoblotting. Cytosolic Hsp70 served as an additional control for fractionation. (C) Taz1pΔMA215–232 was imported into isolated WT, tim10-1, and tim10-1tim9S mitochondria and subjected to trypsin treatment to remove nonimported precursor. Samples in WT mitochondria were also subject to carbonate extraction (Na2CO3) and separated in supernatant (S) and pellet (P) fractions by centrifugation. Fragments that were initially resistant to protease were labeled “protected.” (D) Taz1pΔMA215–232 was imported as for panel C into WT, Δtom20, and Δtom70 mitochondria.
From the aforementioned evaluation, it was not possible to determine if Taz1pΔMA215–232 was targeted to mitochondria. Consequently, we investigated whether Taz1pΔMA215–232 was imported into isolated WT mitochondria (Fig. 5C). When Taz1pΔMA215–232 was imported and treated with protease to assess translocation across the outer membrane, a reproducible pattern of protected Taz1pΔMA215–232 initially appeared within the first 5 min of import, but it disappeared by the later time point of 30 min (Fig. 5C). The protection pattern resulted from trypsin addition that was added to remove nonimported precursor, suggesting that the Taz1pΔMA215–232 associated with mitochondria but was not imported completely into the intermembrane space. However, Taz1pΔMA215–232 was not inserted into the membrane, because Taz1pΔMA215–232 was not recovered from the pellet after carbonate extraction. Furthermore, this protection pattern was not present in tim10-1 mitochondria, which lack functional Tim9p-Tim10p complex. However, Taz1pΔMA215–232 showed the same protease protection in mitochondria from the suppressor tim10-1tim9S strain (Fig. 5C).
As we previously showed that Tom70p and Tom20p were important for Taz1p translocation, we investigated the import of Taz1pΔMA215–232 in Δtom70 and Δtom20 mitochondria (Fig. 5D). There was a decreased association of Taz1pΔMA215–232 in these mutant mitochondria compared with the WT control. Taken together, the results show that Taz1pΔMA215–232 was targeted to the TOM complex and engaged the Tim9-Tim10p complex but was not translocated across and into the outer membrane. The precursor may become initially “trapped” in the TOM complex, which renders a portion of the substrate protected from protease; however, because of incomplete translocation, the substrate may dissociate from mitochondria at the later time points.
The membrane anchor and flanking sequences are sufficient for import of Taz1p.
To determine if the membrane anchor of Taz1p contained sufficient information for targeting to mitochondria, a series of DHFR fusion proteins were constructed and imported into wild type mitochondria (Fig. 6A). To ensure that there was adequate flexibility of the protein sequence for engaging the mitochondrial translocons, a linker sequence of NAAIRS flanked the N terminus and a glycine linker (GGGGGG) flanked the C terminus of the Taz1p sequence, followed by DHFR fusions on both sides of the Taz1p region. Constructs that contained the membrane anchor (residues 215 to 232) as well as flanking sequences were included. For the constructs that contained only the membrane anchor (DHFR-MA215–232-DHFR) or the membrane anchor with a C-terminal flanking region (DHFR-MA215–265-DHFR), import was not robust. In contrast, constructs that contained the membrane anchor plus the flanking N-terminal region (DHFR-MA204–265-DHFR and DHFR-MA204–232-DHFR) showed strong import. Thus, the membrane anchor and adjacent N-terminal sequence, with several charged amino acid residues, seem to direct Taz1p import.
Fig 6.
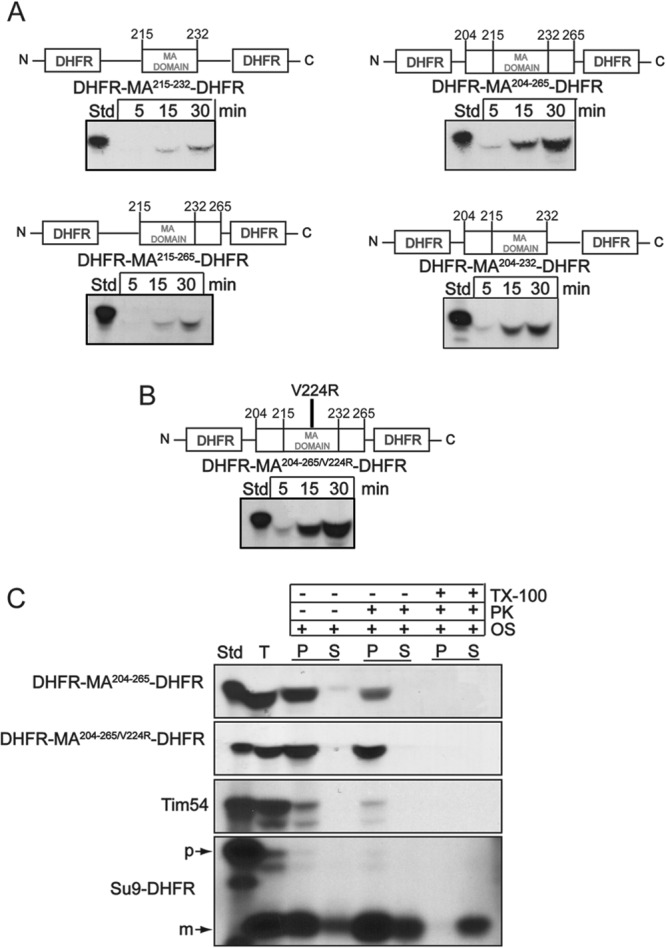
The membrane anchor region and its flanking sequences are sufficient for mitochondrial import. (A) Constructs were made in which the membrane anchor (MA) of Taz1p (amino acids 215 to 232) and flanking sequences (amino acids 215 to 265, 204 to 265, and 204 to 232) were inserted between two DHFR moieties and imported into isolated WT mitochondria. Nonimported precursor was removed by protease treatment. (B) The V224R mutation was introduced, creating DHFR-MA204–265/V224R-DHFR, and imported as for panel A. (C) Imported DHFR-MA204–265-DHFR and DHFR-MA204–265/V224R-DHFR were localized within mitochondria by osmotic shock in the presence and absence of proteinase K and Triton X-100. Controls include Tim54p for the intermembrane space and Su9-DHFR for the matrix.
We also tested the ability of the V224R mutation and flanking sequences to direct import by generating a similar DHFR fusion construct that contained the membrane anchor with the V224R mutation and both N-terminal and C-terminal flanking sequences (DHFR-MA204–265/V224R-DHFR) (Fig. 6B). Again, this region conferred import into mitochondria. Furthermore, the mitochondrial localization was tested with osmotic shock in the presence of protease (Fig. 6C) to assess whether the construct was imported into the matrix or intermembrane space. As predicted, DHFR-MA204–265-DHFR was imported into the intermembrane space, but DHFR-MA204–265/V224R-DHFR was imported into the mitochondrial matrix. As a control, imported Tim54p was detected in the intermembrane space, but Su9-DHFR localized to the matrix. These fusion constructs confirm that the membrane anchor sequence alone is not sufficient for targeting to the mitochondria, but critical targeting information is contained N terminal to the transmembrane domain.
DISCUSSION
The range and variety of Taz1p mutations that result in Barth syndrome hint at both the importance and the complexity of this protein and its mitochondrial function (40). Taz1p has a complex localization pattern in mitochondria such that it localizes to the inner and outer membranes and lines the intermembrane space as an interfacial membrane protein (6). In contrast, cardiolipin synthase has been localized to the inner leaflet of the inner membrane (13). A hot spot for mutations seems to be the membrane anchor domain, and mutations in this region result in mislocalization of Taz1p to the mitochondrial matrix (6). Together, these findings warranted further investigation of the biogenesis of Taz1p, especially to develop strategies to understand BTHS.
This study has shown that Taz1p first binds the TOM complex and that Tom20p and Tom70p are both required. It was important to first implicate the TOM receptors, because some outer membrane proteins, such as the proapoptotic Bcl-2 proteins and various alpha-helical signal- and tail-anchored proteins, may be inserted into the outer membrane without passing through the TOM40 channel (41, 42). Furthermore, this study supports the notion that Taz1p is imported via the TOM translocon because Taz1p was cross-linked to Tom40p during translocation. However, the inner membrane translocons seem to be surprisingly bypassed, because the wild type Taz1p showed no import defects in the tim22-4 or tim23-6 strains, and Taz1p import was independent of a ΔΨ.
The endogenous Taz1p protein resides in both the outer and inner membranes of the mitochondrion, as shown with sucrose gradients, immunoelectron microscopy (6), and, as shown here, a digitonin solubilization assay. The evidence presented in this study indicates that tafazzin first integrates into the outer membrane. Therefore, how Taz1p migrates from the outer membrane to the inner membrane, which is the site of CL synthesis and enrichment and the submitochondrial residence of two binding partners of tafazzin, the ATP synthase and AAC (39), is an open question. Perhaps the enzyme moves through contact sites between the two membranes or is transferred with the lateral migration of phospholipids. Recent studies support the notion that a multisubunit protein complex (termed MitOS/MINOS/MICOS) has a scaffold-like structure to connect outer and inner membranes (43–45), providing a potential conduit for lipids and Taz1p trafficking from the outer to the inner membrane. From the experimental approach in which imported Taz1p was fractionated with digitonin, we attempted to extend the length of the import reaction from 30 min up to 2 h to chase the Taz1p precursor into the inner membrane, but we did not observe a difference in the precursor's distribution profile. It may be reasonable to assume that normal lipid trafficking and diffusion do not occur or cannot be recapitulated in purified mitochondria, even with a ΔΨ.
Taz1p interacts with the Tim9-Tim10p complex after it passes the TOM complex. First, a noticeable import defect was shown with mitochondria from the tim10-1 strain, and Taz1p import was restored in the suppressor strain tim10-1Tim9S, in which the mutation in Tim9p restores function of the Tim9p-Tim10p complex (31). Second, the Taz1p precursor was cross-linked to Tim10p during translocation. Endogenous Taz1p had an altered digitonin solubilization profile in the tim10-1 strain, which we interpreted as an inability to correctly insert into the outer mitochondrial membrane due to the lack of functional Tim10p. This may reveal a role for the Tim9-Tim10p complex post-outer membrane insertion in facilitating the migration of Taz1p to the inner membrane. In the absence of the Tim9-Tim10p complex, Taz1p may be inserted into the outer membrane but not traffic to the inner membrane. The Tim9-Tim10p complex is known to mediate insertion of components of the SAM and TOM40 complex into the outer membrane (17), but tafazzin is one of the few non-β-barrel outer membrane proteins to use the Tim9p-Tim10p complex. Additionally, unlike the β-barrel proteins, tafazzin does not require the SAM complex for membrane integration (15), indicating that Taz1p uses a new mitochondrial import pathway. Whether or not other proteins mediate the insertion of Taz1p into the lipid bilayer is currently unknown and requires additional investigation.
When a single amino acid in the membrane anchor is mutated (V224R), Taz1p alters its normal import course and does not reach the outer mitochondrial membrane. Instead, it interacts with the TIM23 translocon and is pulled into the matrix, where it likely resides on the inner leaflet of the inner membrane. The mislocalization event was seen by subjecting mitochondria to osmotic shock and treating with protease, both in vivo (6) and, in this current study, with an in vitro import assay. We hypothesize that the point mutation, in which a hydrophobic valine is changed to a charged arginine residue, changes the hydrophobicity of this crucial region and interferes with tafazzin's ability to interact with the Tim9p-Tim10p complex, which is known to bind to the hydrophobic regions of membrane proteins and protect them from the aqueous environment of the intermembrane space. In a default pathway, Taz1p is then passed to the TIM23 translocon, which perhaps supports the model that the TIM23 translocon interacts directly with the TOM complex (46). Alternatively, membrane integration may be required for Tim9p-Tim10p complex association with Taz1p. Because the membrane anchor and flanking regions of Taz1p contain several mutations found in BTHS patients (40) and mitochondrial membrane proteins typically have targeting and sorting information in or immediately adjacent to the membrane domains (20), we subsequently focused on the membrane anchor and its role in mitochondrial targeting and sorting. The membrane anchor and the N-terminal region are required for mitochondrial targeting and sorting. When construct Taz1ΔMA215–232 that lacks the membrane anchor was imported into mitochondria, it initially engaged the TOM translocon and was partially protease protected (Fig. 5), but then the construct seemed to disengage from mitochondria because it was subsequently degraded by protease. Moreover, the protection from protease was dependent on Tim10p. We suggest that the Tim9-Tim10p complex binds to the membrane anchor of Taz1p. When Taz1ΔMA215–232 was expressed in a Δtaz1p yeast strain, the protein was quickly degraded by cellular proteases, because the mutant protein was poorly detected in whole-cell extracts and not detected in an enriched mitochondrial fraction. These experiments postulate that the membrane anchor region not only enables the endogenous protein to integrate tightly into the lipid bilayer of the mitochondrial membranes, making it resistant to alkali and high salt extractions, but also is necessary if the protein is to be accurately sorted by the Tim 9/10 import protein machinery. Similar to inner membrane carriers (47), Taz1p likely inserts as a loop across the TOM complex, because appended DHFR at the N and C termini did not inhibit import. Given the abundance of mutations in the membrane anchor and flanking sequences, this study suggests that the molecular basis for BTHS in a subset of patients is caused by defects in mitochondrial targeting and localization. Thus, detailed analysis of Taz1p biogenesis in Saccharomyces cerevisiae provides a framework for further studies to understand the molecular basis of BTHS.
Supplementary Material
ACKNOWLEDGMENTS
We thank Santi Srijumnong for excellent technical assistance and Susan Walsh for technical discussions.
This work was supported by the American Heart Association (0640076N to C.M.K.); California Institute of Regenerative Medicine (RS1-00313 and RB1-01397 to C.M.K.); and National Institutes of Health (R00HL089185 to S.M.C., 1R01GM61721, and 1R01GM073981 to C.M.K., and USPHS NRSA GM07185 to J.D.H.).
Footnotes
Published ahead of print 27 September 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00237-13.
REFERENCES
- 1.Barth PG, Valianpour F, Bowen VM, Lam J, Duran M, Vaz FM, Wanders RJ. 2004. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): an update. Am. J. Med. Genet. A 126A:349–354 [DOI] [PubMed] [Google Scholar]
- 2.Li G, Chen S, Thompson MN, Greenberg ML. 2007. New insights into the regulation of cardiolipin biosynthesis in yeast: implications for Barth syndrome. Biochim. Biophys. Acta 1771:432–441 [DOI] [PubMed] [Google Scholar]
- 3.Claypool SM. 2009. Cardiolipin, a critical determinant of mitochondrial carrier protein assembly and function. Biochim. Biophys. Acta 1788:2059–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schlame M, Ren M. 2009. The role of cardiolipin in the structural organization of mitochondrial membranes. Biochim. Biophys. Acta 1788:2080–2083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barth PG, Wanders RJ, Vreken P, Janssen EA, Lam J, Baas F. 1999. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome). J. Inherit Metab. Dis. 22:555–567 [DOI] [PubMed] [Google Scholar]
- 6.Claypool SM, McCaffery JM, Koehler CM. 2006. Mitochondrial mislocalization and altered assembly of a cluster of Barth syndrome mutant tafazzins. J. Cell Biol. 174:379–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu Y, Condell M, Plesken H, Edelman-Novemsky I, Ma J, Ren M, Schlame M. 2006. A Drosophila model of Barth syndrome. Proc. Natl. Acad. Sci. U. S. A. 103:11584–11588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khuchua Z, Yue Z, Batts L, Strauss AW. 2006. A zebrafish model of human Barth syndrome reveals the essential role of tafazzin in cardiac development and function. Circ. Res. 99:201–208 [DOI] [PubMed] [Google Scholar]
- 9.Gu Z, Valianpour F, Chen S, Vaz FM, Hakkaart GA, Wanders RJ, Greenberg ML. 2004. Aberrant cardiolipin metabolism in the yeast taz1 mutant: a model for Barth syndrome. Mol. Microbiol. 51:149–158 [DOI] [PubMed] [Google Scholar]
- 10.Acehan D, Khuchua Z, Houtkooper RH, Malhotra A, Kaufman J, Vaz FM, Ren M, Rockman HA, Stokes DL, Schlame M. 2009. Distinct effects of tafazzin deletion in differentiated and undifferentiated mitochondria. Mitochondrion 9:86–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Acehan D, Vaz F, Houtkooper RH, James J, Moore V, Tokunaga C, Kulik W, Wansapura J, Toth MJ, Strauss A, Khuchua Z. 2011. Cardiac and skeletal muscle defects in a mouse model of human Barth syndrome. J. Biol. Chem. 286:899–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Claypool SM, Whited K, Srijumnong S, Han X, Koehler CM. 2011. Barth syndrome mutations that cause tafazzin complex lability. J. Cell Biol. 192:447–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schlame M, Haldar D. 1993. Cardiolipin is synthesized on the matrix side of the inner membrane in rat liver mitochondria. J. Biol. Chem. 268:74–79 [PubMed] [Google Scholar]
- 14.Gebert N, Joshi AS, Kutik S, Becker T, McKenzie M, Guan XL, Mooga VP, Stroud DA, Kulkarni G, Wenk MR, Rehling P, Meisinger C, Ryan MT, Wiedemann N, Greenberg ML, Pfanner N. 2009. Mitochondrial cardiolipin involved in outer-membrane protein biogenesis: implications for Barth syndrome. Curr. Biol. 19:2133–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brandner K, Mick DU, Frazier AE, Taylor RD, Meisinger C, Rehling P. 2005. Taz1, an outer mitochondrial membrane protein, affects stability and assembly of inner membrane protein complexes: implications for Barth syndrome. Mol. Biol. Cell 16:5202–5214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. 2009. Importing mitochondrial proteins: machineries and mechanisms. Cell 138:628–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neupert W, Herrmann JM. 2007. Translocation of proteins into mitochondria. Annu. Rev. Biochem. 76:723–749 [DOI] [PubMed] [Google Scholar]
- 18.Hildenbeutel M, Habib SJ, Herrmann JM, Rapaport D. 2008. New insights into the mechanism of precursor protein insertion into the mitochondrial membranes. Int. Rev. Cell Mol. Biol. 268:147–190 [DOI] [PubMed] [Google Scholar]
- 19.Becker T, Gebert M, Pfanner N, van der Laan M. 2009. Biogenesis of mitochondrial membrane proteins. Curr. Opin. Cell Biol. 21:484–493 [DOI] [PubMed] [Google Scholar]
- 20.Walther DM, Rapaport D. 2009. Biogenesis of mitochondrial outer membrane proteins. Biochim. Biophys. Acta 1793:42–51 [DOI] [PubMed] [Google Scholar]
- 21.Papic D, Krumpe K, Dukanovic J, Dimmer KS, Rapaport D. 2011. Multispan mitochondrial outer membrane protein Ugo1 follows a unique Mim1-dependent import pathway. J. Cell Biol. 194:397–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Becker T, Wenz LS, Kruger V, Lehmann W, Muller JM, Goroncy L, Zufall N, Lithgow T, Guiard B, Chacinska A, Wagner R, Meisinger C, Pfanner N. 2011. The mitochondrial import protein Mim1 promotes biogenesis of multispanning outer membrane proteins. J. Cell Biol. 194:387–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horie C, Suzuki H, Sakaguchi M, Mihara K. 2002. Characterization of signal that directs C-tail-anchored proteins to mammalian mitochondrial outer membrane. Mol. Biol. Cell 13:1615–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kutik S, Stojanovski D, Becker L, Becker T, Meinecke M, Kruger V, Prinz C, Meisinger C, Guiard B, Wagner R, Pfanner N, Wiedemann N. 2008. Dissecting membrane insertion of mitochondrial beta-barrel proteins. Cell 132:1011–1024 [DOI] [PubMed] [Google Scholar]
- 25.Hoppins SC, Nargang FE. 2004. The Tim8-Tim13 complex of Neurospora crassa functions in the assembly of proteins into both mitochondrial membranes. J. Biol. Chem. 279:12396–12405 [DOI] [PubMed] [Google Scholar]
- 26.Koehler CM. 2004. New developments in mitochondrial assembly. Annu. Rev. Cell Dev. Biol. 20:309–335 [DOI] [PubMed] [Google Scholar]
- 27.Chacinska A, Lind M, Frazier AE, Dudek J, Meisinger C, Geissler A, Sickmann A, Meyer HE, Truscott KN, Guiard B, Pfanner N, Rehling P. 2005. Mitochondrial presequence translocase: switching between TOM tethering and motor recruitment involves Tim21 and Tim17. Cell 120:817–829 [DOI] [PubMed] [Google Scholar]
- 28.Reif S, Randelj O, Domanska G, Dian EA, Krimmer T, Motz C, Rassow J. 2005. Conserved mechanism of OxaI insertion into the mitochondrial inner membrane. J. Mol. Biol. 354:520–528 [DOI] [PubMed] [Google Scholar]
- 29.Hasson SA, Damoiseaux R, Glavin JD, Dabir DV, Walker SS, Koehler CM. 2010. Substrate specificity of the TIM22 mitochondrial import pathway revealed with small molecule inhibitor of protein translocation. Proc. Natl. Acad. Sci. U. S. A. 107:9578–9583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leuenberger D, Bally NA, Schatz G, Koehler CM. 1999. Different import pathways through the mitochondrial intermembrane space for inner membrane proteins. EMBO J. 17:4816–4822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koehler CM, Merchant S, Oppliger W, Schmid K, Jarosch E, Dolfini L, Junne T, Schatz G, Tokatlidis K. 1998. Tim9p, an essential partner subunit of Tim10p for the import of mitochondrial carrier proteins. EMBO J. 17:6477–6486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lithgow T, Junne T, Wachter C, Schatz G. 1994. Yeast mitochondria lacking the two import receptors Mas20p and Mas70p can efficiently and specifically import precursor proteins. J. Biol. Chem. 269:15325–15330 [PubMed] [Google Scholar]
- 33.Hwang DK, Claypool SM, Leuenberger D, Tienson HL, Koehler CM. 2007. Tim54p connects inner membrane assembly and proteolytic pathways in the mitochondrion. J. Cell Biol. 178:1161–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glick BS, Pon LA. 1995. Isolation of highly purified mitochondria from Saccharomyces cerevisiae. Methods Enzymol. 260:213–223 [DOI] [PubMed] [Google Scholar]
- 35.Glick BS, Brandt A, Cunningham K, Muller S, Hallberg RL, Schatz G. 1992. Cytochromes c1 and b2 are sorted to the intermembrane space of yeast mitochondria by a stop-transfer mechanism. Cell 69:809–822 [DOI] [PubMed] [Google Scholar]
- 36.Pfaller R, Pfanner N, Neupert W. 1989. Mitochondrial protein import. Bypass of proteinaceous surface receptors can occur with low specificity and efficiency. J. Biol. Chem. 264:34–39 [PubMed] [Google Scholar]
- 37.Lithgow T, Schatz G. 1995. Import of the cytochrome oxidase subunit Va precursor into yeast mitochondria is mediated by the outer membrane receptor Mas20p. J. Biol. Chem. 270:14267–14269 [DOI] [PubMed] [Google Scholar]
- 38.Kübrich M, Rassow J, Voos W, Pfanner N, Honlinger A. 1998. The import route of ADP/ATP carrier into mitochondria separates from the general import pathway of cleavable preproteins at the trans side of the outer membrane. J. Biol. Chem. 273:16374–16381 [DOI] [PubMed] [Google Scholar]
- 39.Claypool SM, Boontheung P, McCaffery JM, Loo JA, Koehler CM. 2008. The cardiolipin transacylase, tafazzin, associates with two distinct respiratory components providing insight into Barth syndrome. Mol. Biol. Cell 19:5143–5155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Joshi AS, Zhou J, Gohil VM, Chen S, Greenberg ML. 2009. Cellular functions of cardiolipin in yeast. Biochim. Biophys. Acta 1793:212–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Motz C, Martin H, Krimmer T, Rassow J. 2002. Bcl-2 and porin follow different pathways of TOM-dependent insertion into the mitochondrial outer membrane. J. Mol. Biol. 323:729–738 [DOI] [PubMed] [Google Scholar]
- 42.Otera H, Taira Y, Horie C, Suzuki Y, Suzuki H, Setoguchi K, Kato H, Oka T, Mihara K. 2007. A novel insertion pathway of mitochondrial outer membrane proteins with multiple transmembrane segments. J. Cell Biol. 179:1355–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoppins S, Collins SR, Cassidy-Stone A, Hummel E, Devay RM, Lackner LL, Westermann B, Schuldiner M, Weissman JS, Nunnari J. 2011. A mitochondrial-focused genetic interaction map reveals a scaffold-like complex required for inner membrane organization in mitochondria. J. Cell Biol. 195:323–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.von der Malsburg K, Muller JM, Bohnert M, Oeljeklaus S, Kwiatkowska P, Becker T, Loniewska-Lwowska A, Wiese S, Rao S, Milenkovic D, Hutu DP, Zerbes RM, Schulze-Specking A, Meyer HE, Martinou JC, Rospert S, Rehling P, Meisinger C, Veenhuis M, Warscheid B, van der Klei IJ, Pfanner N, Chacinska A, van der Laan M. 2011. Dual role of mitofilin in mitochondrial membrane organization and protein biogenesis. Dev. Cell 21:694–707 [DOI] [PubMed] [Google Scholar]
- 45.Harner M, Korner C, Walther D, Mokranjac D, Kaesmacher J, Welsch U, Griffith J, Mann M, Reggiori F, Neupert W. 2011. The mitochondrial contact site complex, a determinant of mitochondrial architecture. EMBO J. 30:4356–4370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Albrecht R, Rehling P, Chacinska A, Brix J, Cadamuro SA, Volkmer R, Guiard B, Pfanner N, Zeth K. 2006. The Tim21 binding domain connects the preprotein translocases of both mitochondrial membranes. EMBO Rep. 7:1233–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Curran SP, Leuenberger D, Schmidt E, Koehler CM. 2002. The role of the Tim8p-Tim13p complex in a conserved import pathway for mitochondrial polytopic inner membrane proteins. J. Cell Biol. 158:1017–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.