

Abstract
Microtubules are versatile biopolymers that support numerous vital cellular functions in eukaryotes. The specific properties of microtubules are dependent on distinct microtubule-associated proteins, as the tubulin subunits and microtubule structure are exceptionally conserved. Highly specialized microtubule-containing assemblies are often found in protists, which are rich sources for novel microtubule-associated proteins. A protozoan parasite, Toxoplasma gondii, possesses several distinct tubulin-containing structures, including 22 microtubules closely associated with the cortical membrane. Early ultrastructural studies have shown that the cortical microtubules are heavily decorated with associating proteins. However, little is known about the identities of these proteins. Here, we report the discovery of a novel protein, TrxL1 (for Thioredoxin-Like protein 1), and an associating complex that coats the cortical microtubules. TrxL1 contains a thioredoxin-like fold. To visualize its localization in live parasites by fluorescence, we replaced the endogenous TrxL1 gene with an mEmeraldFP-TrxL1 fusion gene. Structured illumination-based superresolution imaging of this parasite line produced a detailed view of the microtubule cytoskeleton. Despite its stable association with the cortical microtubules in the parasite, TrxL1 does not seem to bind to microtubules directly. Coimmunoprecipitation experiments showed that TrxL1 associates with a protein complex containing SPM1, a previously reported microtubule-associated protein in T. gondii. We also found that SPM1 recruits TrxL1 to the cortical microtubules. Besides SPM1, several other novel proteins are found in the TrxL1-containing complex, including TrxL2, a close homolog of TrxL1. Thus, our results reveal for the first time a microtubule-associated complex in T. gondii.
INTRODUCTION
Toxoplasma gondii is one of the most successful human parasites, infecting ∼20% of the world population. T. gondii infection is asymptomatic in immunocompetent individuals. However, the infection in immunocompromised individuals and unprotected fetuses can be devastating (1, 2). Toxoplasmosis is a leading cause of congenital neurological defects. T. gondii is a member of a large phylum, the Apicomplexa, which contains thousands of species of obligate intracellular parasites. Many of them are serious health threats to humans, including Plasmodium spp., the causative agent for malaria (3). Besides being an important human parasite and a valuable model for understanding the physiology and cell biology of other apicomplexan parasites, T. gondii also has an intriguing cellular architecture, conferring great advantages for studying the structure, function, and biogenesis of eukaryotic cytoskeletons (4–11).
The T. gondii cytoskeleton is formed of several distinct types of biopolymers (tubulin-containing fibers, intermediate filament-like fibers, and actin filaments), which together with their associating proteins drive the dissemination and proliferation of the parasite (7). The actomyosin complexes provide the driving force for parasite movement to invade into and escape from host cells (12–18), and recently they have been shown to be crucial for the proper segregation of the apicoplast, a plastid-like organelle (19). The tubulin- and intermediate filament-containing cortical cytoskeleton provides the framework for constructing new daughter cells and maintains parasite shape (4, 6–11, 20, 21). At least five distinct tubulin-containing structures exist in T. gondii, the spindle pole, centrioles, cortical microtubules, the conoid, and the intraconoid microtubules (Fig. 1A) (5, 6, 9, 22). There are 22 cortical microtubules, and they are closely associated with a set of flattened vesicles (called the inner membrane complex, or IMC) underneath the plasma membrane (Fig. 1A). These microtubules are unusually stable and resistant to harsh detergent extractions (6, 9, 22). Early electron microscopy analyses have shown that the cortical microtubules are heavily decorated with associating proteins (6, 9, 22). However, the identity of these proteins remained unknown for years. Recently, the first two cortical microtubule-associated proteins, SPM1 and SPM2, were discovered (23). SPM1 coats the entire length of the cortical microtubules, while SPM2 only decorates the midsection. Upon the removal of SPM1, the cortical microtubules are no longer resistant to detergent extraction.
Fig 1.

TrxL1 contains a thioredoxin-like fold with a modified catalytic site motif and is well conserved within Apicomplexa. (A) Cartoon drawing of T. gondii showing the apical complex, plasma membrane, inner membrane complex (IMC), and cortical microtubules. For clarity, the apical complex in the cartoon is depicted in the extended state. In most intracellular parasites, the main body of the complex is retracted such that the intraconoid microtubules and the conoid are located posterior to the apical polar ring. (B) Phylogenetic analysis of TrxL domain-containing proteins in Apicomplexa. Classic thioredoxins, hTrx1, TgTRX1, and PfTRX1 are included as an outgroup (green). Sequences for individual TRX or TrxL domains predicted by Pfam were used in the construction of the phylogenetic tree. Bootstrap values above 0.2 are displayed. Maximum bootstrap value, 1. For proteins (e.g., Atrx1) that have multiple TRX domains, each domain is used as a single entry. The TrxL1 clade is shaded. Except for TrxL1 and the proteins from the reported functional studies, all of the other proteins are listed by their gene identifications in EupathDB.org. (C) Multiple alignments of TrxL1 clade from panel B. hTrx1, a classic thioredoxin, is included for comparison purposes. The gray bar below the sequences denotes the TRX domain, and the asterisks mark the position of catalytic residues in hTrx1.
Here, we report another novel cortical microtubule-associated protein, TgTrxL1 (for T. gondii Thioredoxin-Like protein 1). TrxL1 is highly conserved within Apicomplexa. It has a thioredoxin fold, a domain found in members of the thioredoxin (TRX) superfamily. However, TrxL1 lacks the canonical CXXC motif, which is essential for the catalytic function of TRX proteins in controlling the redox state of sulfhydryl (−SH) groups of the target proteins (24, 25). In this study, we characterized the localization and function of TrxL1 and identified multiple novel components of a microtubule-associated complex using TrxL1 as the probe. These results will allow for systematic analysis of the unique structural properties of the parasite cortical microtubules.
MATERIALS AND METHODS
Multiple alignment and phylogenetic analysis.
The list of TrxL domain-containing proteins in apicomplexan parasites used in the phylogenetic analysis shown in Fig. 1B was compiled from the following BLAST searches. (i) BLAST search against the protein database of TgGT1 in ToxoDB, with the full-length amino acid sequence of TgTrxL1 (TGGT1_115220) as the query and an E value cutoff of 0.5. This search generated 5 hits: TGGT1_115220, TGGT1_080930, TGGT1_080000, TGGT1_034060, and TGGT1_088350. (ii) BLAST search against the protein databases of apicomplexan parasites at EupathDB, with the full-length amino acid sequence of TgTrxL1 as the query and an E value cutoff of 0.5. (iii) BLAST search against the protein databases of apicomplexan parasites at EupathDB, with the full-length amino acid sequence of TGGT1_080930, TGGT1_080000, TGGT1_034060, or TGGT1_088350 as the query to retrieve their immediate orthologues in Apicomplexa (i.e., hits with an E value lower than that for any other T. gondii TrxL domain-containing proteins). Redundant sequences were then removed to give the final list. Classic thioredoxins human Trx1 (hTrx1;NP_003320.2) (26), TgTRX1 (TGGT1_074260) (27), and Plasmodium falciparum TRX1 (PfTRX1; PF3D7_1457200) (28) were included in the analysis as an outgroup. Sequences for individual TRX or TrxL domains predicted by Pfam (v27.0) were used in multiple alignments with the Muscle module in MEGA (v5.2.1) using default parameters. The alignment was further edited manually. The phylogenetic tree was constructed from the multiple alignment using the maximum likelihood method. The robustness of the tree was tested by bootstrapping 1,000 times.
Parasite culture and transfection.
T. gondii RHΔhx tachyzoites were used as the parental strain in all experiments. The culture maintenance and parasite transfection were carried out as previously described (29).
Plasmid construction.
The sequences of all PCR primers used in subclonings are listed in Table S1 in the supplemental material.
T. gondii expression plasmids.
For construction of pTKO2-II-mEmeraldFP-TrxL1, ∼2-kb fragments upstream (5′ untranslated region [UTR]) or downstream (3′UTR) of trxl1 were amplified from the RH genomic DNA using primer combinations shown in Table S1 in the supplemental material and subcloned stepwise into the HindIII-ApaI and NotI-EcoRV sites on plasmid pTKO2-II-mCherryFP. (pTKO2-II-mCherryFP has exactly the same structure as pTKO2-II [30], except that an mCherryFP expression cassette replaces a green fluorescent protein [GFP] expression cassette in the vector backbone.) An mEmeraldFP-TrxL1 fusion gene extricated from PUC57-mEmeraldFP-TrxL1 (see below) by PmeI and RsrII digestion was then subcloned into the PmeI-RsrII sites of the resulting vector to generate pTKO2-II-mEmeraldFP-TrxL1. For the generation of PUC57-mEmeraldFP-TrxL1, an mEmeraldFP-TrxL1 fusion gene was synthesized and subcloned into pUC57-simple (Genescript, NJ). There is a linker sequence coding for SGLGS between the coding sequences for mEmeraldFP and TrxL1 and a TrxL1 Kozak sequence at the 5′ end of the mEmeraldFP gene. Silent mutations were introduced in the TrxL1 coding sequence to remove the internal BglII and EcoRI sites.
For construction of pmin-mTagRFP-T-TgCentrin2, an mTagRFP-T fragment (31, 32) with a linker sequence at the 3′ end coding for GHGTGSTGSTSSRS was amplified by PCR, restriction digested, and cloned into the NheI-BglII sites of pmin-mCherryFP-TgCentrin2 to replace mCherryFP.
For construction of pmin-mTagRFP-T-TgMORN1, an mTagRFP-T fragment with a linker sequence at the 3′ end coding for SGLRS was amplified by PCR, restriction digested, and cloned into the NheI-BglII sites of pmin-mCherryFP-TgMORN1 (4) to replace mCherryFP.
For construction of ptub-mCherryFP-mCherryFP, two consecutive coding sequences for mCherryFP with a linker sequence coding for SGLRS between them were cloned into the NheI-AflII sites in a ptub plasmid backbone (30).
For construction of ptub-mCherryFP-TrxL1, the TrxL1 coding region was amplified from the RH cDNA library by PCR, restriction digested, and subcloned into the BglII-AflII sites on plasmid ptub-mCherryFP-TgTUBA1 to replace TgTUBA1. A silent G-to-A mutation at nucleotide position 235 was introduced by PCR in TrxL1 to facilitate the cloning.
For construction of ptub-mEmeraldFP-TrxL1_FL, the mEmeraldFP-TrxL1 fusion gene from pUC57-mEmeraldFP-TrxL1 (described above) was subcloned into the NheI-AflII sites in a ptub plasmid backbone.
For construction of ptub-mEmeraldFP-TrxL1_2-49aa and ptub-mEmeraldFP-TrxL1_2-178aa, the coding region of mEmeraldFP-TrxL1_2-49aa or mEmeraldFP-TrxL1_2-178aa, respectively, was amplified from pUC57-mEmeraldFP-TrxL1 by PCR, digested by NheI and AflII, and cloned into the NheI-AflII sites on a ptub plasmid backbone.
For construction of ptub-mEmeraldFP-TrxL1_50-178aa and ptub-mEmeraldFP-TrxL1_179-220aa, the coding region of TrxL1_50-178aa or TrxL1_179-220aa, respectively, was amplified from pUC57-mEmeraldFP-TrxL1 by PCR, digested by BglII and AflII, and cloned into the BglII-AflII sites on a ptub-mEmeraldFP plasmid backbone.
For construction of ptub-mEmeraldFP-TrxL2 and ptub mEmeraldFP-TLAP1, the coding sequence minus the first methionine for TrxL2 (TGGT1_080930) or TLAP1 (TGGT1_038030), respectively, was amplified from an RH cDNA library by PCR, digested by BglII-AflII or BamHI-AflII, respectively, and cloned into the BglII and AflII sites in ptub-mEmeraldFP-TrxL1_50-178aa.
Bacterial expression plasmids.
For construction of pET22b(+)-FLAG-TrxL1, the coding region of TrxL1 was amplified from ptub-mCherryFP-TrxL1 by PCR, restriction digested, and subcloned into the NheI-NotI sites on plasmid pET22b(+)-FLAG-AKMT (29) to replace AKMT.
Mammalian cell expression plasmids.
For construction of pc22-EGFP-TrxL1 and pc22-mCherryFP-TrxL1, the TrxL1 fragment was released from ptub-mCherryFP-TrxL1 by BglII-AflII digestion and subcloned into the BglII-AflII sites in pc22-EGFP-TgTUBA1 or pc22-mCherryFP-TgTUBA1 (kind gifts from John Murray, University of Pennsylvania), respectively, to replace TgTUBA1.
Generation of loxp_mEmeraldFP-TrxL1_knockin, mEmeraldFP-TrxL1/mTagRFP-T-TgCentrin2, mEmeraldFP-TrxL1/mTagRFP-T-TgMORN1, and Δtrxl1 parasites.
To create the loxp_mEmeraldFP-TrxL1_knockin parasites, ∼1 × 107 RHΔhx parasites were electroporated with 50 μg pTKO2-II-mEmeraldFP-TrxL1 plasmid linearized by NotI, selected with myocophenolic acid (25 μg/ml) and xanthine (50 μg/ml) for two passages, and then sorted by fluorescence-activated cell sorting (FACS). Three mEmerald(+)/mCherry(−) parasites were sorted into each well of a 96-well plate containing confluent human foreskin fibroblasts (HFF) monolayers. The 96-well plate was screened for single plaque-forming clones after 7 to 10 days. Genomic DNA was extracted from the clones using an Extract-N-Amp kit (XNAB-1kt; Sigma, MO) and screened by PCR for clones of parasites in which the endogenous trxl locus had been replaced by the LoxP-mEmeraldFP-TrxL1-HXGPRT-LoxP fragment. One clone was further verified by Southern blotting and used in analysis and subsequent generation of Δtrxl1 parasites.
To create mEmeraldFP-TrxL1/mTagRFP-T-TgCentrin2 parasites and mEmeraldFP-TrxL1/mTagRFP-T-TgMORN1 parasites, loxp_mEmeraldFP-TrxL1_knockin parasites were electroporated with 30 μg either pmin-mTagRFP-T-TgCentrin2 or pmin-mTagRFP-T-TgMORN1 plasmid together with 30 μg pC3 plasmid (conferring pyrimethamine resistance) (33), selected with 1 μM pyrimethamine for two passages, and then sorted by FACS. Three mEmerald(+)/mTagRFP-T(+) parasites were placed into each well of a 96-well plate containing confluent HFF monolayers. A single clone from each parasite line was used in the analysis.
To create the Δtrxl1 parasites, loxp_mEmeraldFP-TrxL1_knockin parasites were electroporated with 30 μg each of pmin-Cre-GFP and ptub-mCherryFP-mCherryFP plasmids together, selected by 6-thioxanthine (80 μg/ml) for 1 passage, and then sorted by FACS 60 h postelectroporation. Three mEmerald(−)/mCherry(+) parasites were sorted into each well of a 96-well plate containing confluent HFF monolayers. Single plaque-forming clones were first screened by fluorescence and then confirmed by genomic PCR for clones of parasites in which the LoxP-mEmeraldFP-TrxL1_HXGPRT_LoxP fragment had been successfully excised. Two clones were further verified by Southern blotting and used in the analysis.
Southern blotting.
Genomic DNA was harvested by a Wizard genomic DNA purification kit (A1120; Promega, CA) and treated with KpnI for 6 h at 37°C. Four μg DNA was loaded on a 0.8% agarose gel and run overnight at 20 V. The gel was transferred to an Immobilon-NY+ charged nylon membrane (INYC00010; Millipore, CA) and probed with TrxL1 probe labeled with biotin using a NEBlot Phototope kit (7550S; New England BioLabs, MA) and developed by a Phototope-Star detection kit (7020S; New England BioLabs, MA) according to the manufacturer's instructions. The blot was stripped at 70°C for 30 min with 0.1% SDS in 0.4N NaOH, reprobed by biotin-labeled 5′UTR probe, and developed as described above.
Mammalian cell transfection.
For transfection of HFF and baby hamster kidney (BHK) cells, 3 μg DNA was mixed with JetPEI transfection reagent (101-05; PolyPlus Transfection, France) in 150 mM NaCl at a JetPEI/DNA ratio of 5:1 and transfected into subconfluent cells grown in glass-bottom dishes (P35G-1.5-20-C; MatTek, MA) per the manufacturer's instructions.
Immunofluorescence labeling.
All immunofluorescence labeling steps were performed at room temperature unless otherwise indicated.
For immunolabeling of intracellular parasites, T. gondii cells growing in an HFF monolayer were fixed in 3.7% (vol/vol) formaldehyde in phosphate-buffered saline (PBS) for 15 min and permeabilized with 0.5% (vol/vol) Triton X-100 (TX-100) in PBS for 15 min. Cells were blocked with 1% (wt/vol) bovine serum albumin (BSA) in PBS for 30 to 45 min and then incubated for ∼60 min first in rabbit anti-IMC1 (a kind gift from Con Beckers, University of North Carolina) diluted 1:1,000 and subsequently in goat anti-rabbit IgG Alexa 568 (A21069; Molecular Probes, OR) diluted 1:1,000. The antibodies were diluted in the blocking buffer.
For immunolabeling of extracellular parasites, ∼1 × 107 parasites were spotted onto clean parafilm and overlaid with poly-l-lysine (P8920; Sigma, MO)-coated coverslips for 30 min. Parasites were fixed in 3.7% formaldehyde in PBS for 15 min and then permeabilized in 0.5% TX-100 for 15 min. For tubulin labeling, coverslips were blocked with 1% BSA in PBS for 30 min. The cells were then incubated for 60 min first in a mouse anti-α-tubulin (T6074; Sigma, MO) and anti-β-tubulin (T5293; Sigma, MO) mixture at 1:300 to 1:1,000 each and subsequently in goat anti-mouse IgG Alexa 568 (A11031; Molecular Probes, OR) diluted 1:500 to 1:1,000. For testing whether TrxL1 stably associates with the cortical microtubules, extracellular parasites on the polylysine-coated coverslip were first extracted in 1% (wt/vol) sodium deoxycholate in PBS for 10 min, fixed in 3.7% formaldehyde in PBS for 15 min, and labeled with tubulin antibodies as described above.
For immunolabeling of HFF and BHK cell cytoskeletons, the cells were first fixed and permeabilized for 2 min at 37°C in PHEM buffer [60 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES), 28 mM HEPES, 10 mM EGTA, 8 mM MgSO4, pH 7.0] containing 3.7% (vol/vol) formaldehyde, 0.075% (vol/vol) gluteraldehyde, and 1% (vol/vol) TX-100, fixed again in PHEM buffer containing 3.7% formaldehyde and 0.075% gluteraldehyde for 15 min at 37°C, and then permeabilized again in PHEM buffer containing 1% TX-100 for 15 min at room temperature. Cells were washed in PBS containing 1 mM MgCl2 (MgPBS) and quenched three times in PBS containing freshly dissolved sodium borohydride at ∼1 mg/ml. Cells were then blocked in MgPBS containing 0.02% (vol/vol) Tween 20 and 2% (wt/vol) BSA for 30 min, incubated in primary antibody for 1 h, washed three times in PBS, and subsequently incubated in secondary antibody for 1 h. Primary antibodies included rat anti-tubulin YL1/2 and YOL-1 hybridoma supernatant mixed at 1:1 (kind gifts from John Murray, University of Pennsylvania). Secondary antibody was goat anti-rat IgG-Alexa 488 (A11006; Molecular Probes, OR) diluted 1:1,000 in PBS with 1% BSA.
Apical complex extension in extracellular parasites.
Extracellular parasites were treated with 5 μM A23187 in culture medium for 15 min and pelleted by centrifugation. The parasite pellet was then resuspended in fresh medium and added to polylysine-coated glass-bottom dishes, incubated for 15 min, fixed in 3.7% formaldehyde in culture medium for 10 min, and finally washed and stored in Dulbecco's PBS until imaging.
Wide-field deconvolution microscopy.
Three-dimensional (3D) image stacks were collected at 35 to 37°C at z increments of 0.3 μm using a Delta Vision imaging station (GE Healthcare-Applied Precision, WA) constructed on an Olympus IX-70 inverted microscope base. A 100× or 60× magnification oil immersion lens and immersion oil at a refractive index of 1.524 or 1.518 were used for all imaging. Deconvolved images were computed using the point-spread functions and software supplied by the manufacturer. The brightness and contrast of images used in the final figures were optimized for color printing.
3D-SIM.
Structured-illumination microscopy (SIM) image stacks were collected at z increments of 0.125 μm using an OMX imaging station (GE Healthcare-Applied Precision, WA). A 100× oil immersion lens (1.4 numerical aperture) and immersion oil at a refractive index of 1.516 were used. Deconvolved images were computed using the point-spread functions and software supplied by the manufacturer.
Recombinant FLAG-TrxL1 expression and purification.
BL21-CodonPlus(DE3)RP strain Escherichia coli (230255; Stratagene, CA) harboring pET22b(+)-FLAG-TrxL1 was grown in LB broth at 37°C until the optical density at 600 nm (OD600) reached 0.6. One mM isopropyl-β-d-thiogalactopyranoside (IPTG) was then added to induce FLAG-TrxL1 expression, and the culture was grown for 16 h at 16°C before harvest. The bacteria were harvested by centrifugation, and the pellet was frozen at −80°C until use. For protein purification, a frozen E. coli pellet was thawed on ice, resuspended, and rotated at 4°C for 10 min in lysis buffer (8 mM Tris-acetate [pH 7.5], 7 mM Tris base [pH unadjusted], 100 mM potassium acetate, 1 mM magnesium acetate) containing 0.5% TX-100 and 25 mg/ml cell lytic express (C1990; Sigma, MO) supplemented with 4 mM dithiothreitol (DTT) and protease inhibitors (0.25 mM phenylmethanesulfonyl fluoride, 10 μg/ml p-toluenesulfonyl-l-arginine methyl ester, 10 μg/ml tosylphenylalanine chloromethyl ketone, and 10 μg/ml n-tosyl-l-lysine chloromethyl ketone). At the end of the incubation, 30 rounds of sonication were applied to ensure complete lysis. The lysate was then clarified by centrifugation and applied to a column loaded with M2-FLAG agarose resin (A2220; Sigma, MO). The column was washed with 3 bed volumes of lysis buffer and eluted with 2 bed volumes of lysis buffer containing 100 μg/ml FLAG peptide (F3290; Sigma, MO). The eluate fractions were analyzed by 4 to 12% gradient Bis-Tris NuPAGE gels (NP0321; Invitrogen, CA), and the fractions containing FLAG-TrxL1 were snap-frozen in liquid nitrogen in aliquots until use.
Microtubule pelleting assays.
Recycled bovine tubulin was polymerized with paclitaxel (T7191; Sigma, MO) as previously reported (34). Recombinant FLAG-TrxL1 was made as described above. One μM FLAG-TrxL1 was precleared and incubated for 20 min at room temperature with microtubules containing 0.5 to 4.0 μM tubulin subunits in BRB80 buffer (80 mM PIPES [pH 6.8], 1 mM MgCl2, 1 mM EGTA) supplemented with 1 mM DTT and 20 μM paclitaxel. The mixtures were spun for 5 min at 45,000 rpm at 22°C in a TLA-100 rotor in a Beckman TLA ultracentrifuge. The pellet and supernatant were separated and solubilized in NuPAGE LDS sample buffer and separated on a 4 to 12% Bis-Tris NuPAGE gel. The gel was fixed and stained with colloidal blue (LC6025; Invitrogen, CA) per the manufacturer's instructions.
Growth competition assay.
loxp_mEmeraldFP-TrxL1_knockin [mEmeraldFP(+)] and Δtrxl1 [mEmerald(−)] parasites were mixed at a 1:1 ratio and inoculated in triplicates into 6-well plates with confluent HFF cells. The cultures were passaged every 46 to 50 h for 20 days. At each passage, a fraction of parasites was removed and analyzed for the proportions of the two parasite lines in the population. To definitively distinguish live parasites from cell debris, the parasites were incubated for 30 min with a cell-permeant nucleic acid stain, SYTO61 (S11343; Molecular Probes, OR), at 750 nM, diluted 6 times with PBS, and then analyzed using an LSRII (BD Biosciences, CA) flow cytometer. The percentage of Δtrxl1 parasites was calculated by percentage of [mEmerald (−)/SYTO61(+)] parasites divided by total parasites [(SYTO61(+))].
Immunoprecipitation of mEmeraldFP-TrxL1 from parasites using anti-GFP antibody.
mEmeraldFP-TrxL1 was immunoprecipitated from loxp_mEmeraldFP-TrxL1_knockin parasites. RHΔhx parasites were used as the negative control. For each immunoprecipitation, ∼5 × 108 parasites were harvested, pelleted, freeze-thawed, resuspended in lysis buffer (150 mM NaCl, 0.5 mM EDTA, 0.5% TX-100, 1 mM NaF, 1 mM Na3VO4, 10% glycerol in 50 mM Tris-HCl [pH 7.5] supplemented with 2.5 mM DTT and the protease inhibitors 0.25 mM phenylmethanesulfonyl fluoride, 10 μg/ml p-toluenesulfonyl-l-arginine methyl ester, 10 μg/ml tosylphenylalanine chloromethyl ketone, and 10 μg/ml n-tosyl-l-lysine chloromethyl ketone), and incubated at 4°C for 10 min. At the end of the incubation, 30 rounds of sonication were applied to ensure complete lysis. The lysate was clarified by centrifugation and then incubated with 60 μl of buffer-exchanged Chromotek-GFP-Trap agarose beads (ACT-CM-GFA0050; Allele Biotechnology, CA) at 4°C for 75 min. The beads were washed five times with lysis buffer and two times with PBS and then eluted with elution buffer (50% H2O, 50% acetonitrile in 0.1%[vol/vol] formic acid) at 70°C for 15 min. The eluates were subjected to NuPAGE or dried in a vacuum at 40°C for MudPIT (for multidimensional protein identification technology) analysis.
MudPIT analysis.
For MudPIT analysis, protein samples prepared as described above were urea denatured, reduced, alkylated, and digested with the endoproteinase Lys-C (11047825001; Roche, IN) followed by modified trypsin (V5111; Promega, CA) as reported previously (35). Peptide mixtures were loaded onto a 250-μm fused silica microcapillary column packed with 5 cm of C18 reverse-phase particles (Aqua, Phenomenex, CA) and strong cation exchange particles (Luna, Phenomenex, CA) connected with a 100-μm fused silica microcapillary column packed with 9 cm of Aqua reverse-phase particles (36). Loaded microcapillary columns were placed in-line with a Quaternary Agilent 1260 series high-performance liquid chromatograph (HPLC) coupled to a Velos Orbitrap Elite mass spectrometer (Thermo, CA) equipped with a nano-liquid chromatography (LC) electrospray ionization source. Fully automated 10-step MudPIT runs were carried out on the electrosprayed peptides as reported previously (35). Full mass spectrometry (MS) spectra were acquired on the peptides over the 400 to 1,600 m/z range in the Orbitrap at 60K resolution, followed by fragmentation in the ion trap on the first to tenth most intense ions selected from the full MS spectrum. Tandem MS (MS/MS) spectra were searched using SEQUEST v.27 (rev.9) (37) with a peptide mass tolerance of 7 ppm and ±0.5 atomic mass units for fragment ions. The sequence database consists of 18,277 T. gondii proteins (combining nonredundant sequences from ToxoDB release 7.2 for the GT1, ME49, and VEG strains and 26 apicoplast proteins), 29,147 Homo sapiens proteins (NCBI; 2011-08-16 release), and 160 usual contaminants (such as human keratins, IgGs, and proteolytic enzymes). To estimate false discovery rates, 47,584 randomized amino acid sequences derived from each nonredundant protein entry were added to the database. Peptide/spectrum matches were sorted, selected, and compared using DTASelect/CONTRAST (38). The false discovery rates of spectral and protein levels were, on average, 0.55% ± 0.22% and 4.3% ± 0.83%, respectively. To estimate relative protein levels, distributed normalized spectral abundance factors (dNSAFs) were calculated for each detected protein as reported previously (39).
RESULTS
TrxL1 is a novel, thioredoxin-like protein.
In a previous proteomic screen, we identified proteins in cytoskeletal fractions of T. gondii either enriched with or depleted of the apical complex (Fig. 1A) (5). In that screen, most cortical microtubule fragments were found in the apical complex-depleted fraction. Therefore, we reasoned that cortical microtubule-associated proteins should be highly represented in this fraction. One of the proteins (ToxoDB entry TGGT1_115220) belongs to the thioredoxin (TRX) superfamily, which we have named TrxL1, for thioredoxin-like protein 1. TrxL1 is predicted to contain 220 amino acids, with residues 69 to 178 forming a thioredoxin-like fold, classified as thioredoxin_8 (Pfam entry PF13905). With TrxL1 as the query and the E value cutoff set at 0.5, a BLAST search in ToxoDB gives five hits, two of which, TrxL1 and TGGT1_034060 (an orthologue of Plrx [40]), have clear orthologues in Plasmodium spp. Only 1 of the 5 hits, ATrx1, an apicoplast protein, has been characterized previously (41). The sequences of this group of proteins are highly diverged from classic thioredoxin proteins (Fig. 1B). Two of them, TrxL1 and TGGT1_089030, do not have a canonical CXXC catalytic site. All of the apicomplexan TrxL1 orthologues have a DPKC motif (residues 82 to 85 in the amino acid sequence of TgTrxL1) (Fig. 1C), suggesting functional divergence of TrxL1 from canonical TRXs during evolution before the speciation of apicomplexan parasites.
TrxL1 is associated with the cortical and intraconoid microtubules.
To characterize cellular distribution of TrxL1, we created a parasite line termed loxp_mEmeraldFP-TrxL1_knockin in which the endogenous trxl1 locus in the RHΔhx parasite was replaced with the coding sequence for mEmeraldFP-tagged TrxL1 plus an expression cassette for the selectable marker HXGPRT flanked by two LoxP sites (Fig. 2A and B; also see Fig. S1 in the supplemental material). Thus, all TrxL1 in this line is expressed under the endogenous promoter and tagged with mEmeraldFP. Further, the flanking LoxP sites allow for subsequent generation of TrxL1 knockout mutants (Δtrxl1) via excision by Cre recombinase (29, 30) (see below).
Fig 2.
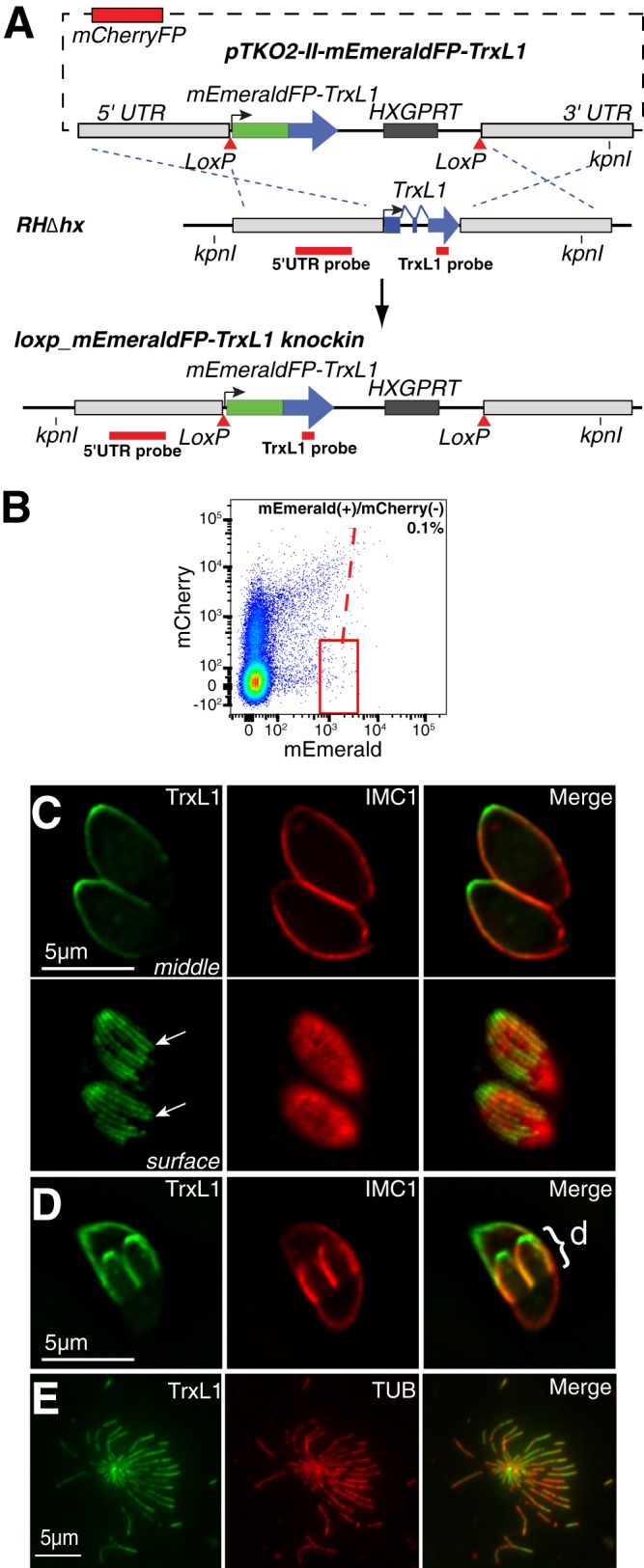
TrxL1 associates with the cortical microtubules in T. gondii. (A) Scheme for generating loxp_mEmeraldFP-TrxL1_knockin parasites using dual-color FACS. The mCherryFP expression cassette on the pTKO2-II-mEmeraldFP-TrxL1 plasmid is used for negative selection, as mCherryFP expression marks the integration of the plasmid into the genome via single crossover or a nonhomologous recombination event instead of the homologous recombination desired. Parasites that express mEmeraldFP but do not express mCherryFP are selected by FACS as shown in panel B. (B) Flow cytometry graph of sorting loxp_mEmeraldFP-TrxL1_knockin clones generated in panel A. [mEmerald(+)/mCherry(−) parasites are within the red frame, which is 0.1% of the total population.] (C) Deconvolved wide-field fluorescence images of the middle (upper row) and surface (lower row) sections of two intracellular interphase loxp_mEmeraldFP-TrxL1_knockin parasites in a parasitophorus vacuole. Arrows indicate two individual cortical microtubules. Green, mEmeraldFP-TrxL1; red, anti-IMC1 labeling. (D) Deconvolved wide-field fluorescence image of the middle section of a dividing loxp_mEmeraldFP-TrxL1_knockin parasite labeled with anti-IMC1 antibody. Green, mEmeraldFP-TrxL1; red, anti-IMC1 labeling; d, daughter parasites. (E) mEmeraldFP-TrxL1 and mouse anti-tubulin (TUB) labeling of a disrupted extracellular loxp_mEmeraldFP-TrxL1_knockin parasite, with the cortical microtubules spreading out in all directions. Parasites were first fixed in 3.7% formaldehyde and then permeabilized with 0.5% TX-100. Green, mEmeraldFP-TrxL1; red, anti-tubulin labeling.
Wide-field fluorescence imaging of intracellular loxp_mEmeraldFP-TrxL1_knockin parasites shows that TrxL1 is localized to multiple fibrous structures radiating from the apex and covering ∼2/3 of the adult parasite body (Fig. 2C) and the full length of the daughter parasite body (Fig. 2D), reminiscent of the distribution pattern of parasite cortical microtubules (4–9, 11). This speculation is confirmed by the colocalization of mEmeraldFP-TrxL1 and anti-tubulin antibody labeling in disrupted extracellular loxp_mEmeraldFP-TrxL1_knockin parasites, where the cortical microtubules are spread out (Fig. 2E). To better resolve TrxL1-containing structures in live parasites, we used three-dimensional structured-illumination microscopy (3D-SIM) to image loxp_mEmeraldFP-TrxL1_knockin parasites and examined the TrxL1 distribution relative to that of other known cytoskeletal structures by expressing mTagRFP-T-tagged TgMORN1, a component of the basal complex and the spindle pole, or TgCentrin2, which marks the centrioles, approximately six peripheral annuli, and the preconoidal ring (Fig. 3) (5). 3D-SIM imaging revealed that besides the 22 cortical microtubules, TrxL1 is localized to a short linear structure at the apical end of the parasite (Fig. 3B to D, insets). We postulated that this structure was composed of the intraconoid microtubules, separated from the roots of the cortical microtubules by less than 150 nm (Fig. 3E). To test this hypothesis, we compared the localization of TrxL1 in the retracted versus extended apical complex (Fig. 3C to F). When the apical complex is retracted, the intraconoid microtubules are positioned posterior to the preconoidal and apical polar rings and are surrounded by the cortical microtubules (22) (Fig. 3E, top). When the apical complex is extended, the intraconoid microtubules move to the anterior of the roots of the cortical microtubules but remain posterior to the preconoidal ring (6, 9, 22) (Fig. 3E, bottom). The positions of the apical linear TrxL1-containing structure in these two states are completely consistent with it being the intraconoid microtubules (Fig. 3C to F). As expected, the two intraconoid microtubules, less than 10 nm apart, cannot be resolved from each other by SIM.
Fig 3.

3D-SIM imaging of mEmeraldFP-TrxL1-expressing parasites. (A) Cartoon drawing showing the main cytoskeletal structures of a parasite at an early stage of daughter construction. The cortical microtubules are present in both the mother and daughters but are not shown to avoid clutter. TgMORN1-containing structures (spindle pole and the basal complex) are labeled in gold. TgCentrin2-containing structures (preconoidal ring, centrioles, peripheral annuli, and a substructure in the basal complex) are labeled in red. AC, apical complex; PM, plasma membrane; pCR, preconoidal ring. (B) Projection of 3D-SIM images of mEmeraldFP fluorescence in loxP_mEmeraldFP-TrxL1_knockin parasites in two adjacent parasitophorous vacuoles, each containing two parasites with retracted apical complex. In the vacuole on the left, the parasites contain well-developed daughters (d). The inset is at 2× magnification. The arrowhead indicates a structure likely to be the intraconoid microtubules viewed from the apical end of a parasite. (C) Projection of 3D-SIM images of two intracellular dividing loxp_mEmeraldFP-TrxL1_knockin parasites stably expressing mTagRFP-T-TgMORN1 with the apical complex in the retracted state. The inset (2× magnification) shows a longitudinal view of the middle section of the parasite apex, where a linear structure (white arrowhead), likely to be the intraconoid microtubules, can be resolved from the surrounding cortical microtubules. Red arrowheads indicate the spindle poles, and red arrows indicate mother and daughter basal complexes. d, daughter parasites; green, mEmeraldFP-TrxL1; red, mTagRFP-T-TgMORN1. (D) Projection of 3D-SIM images of an intracellular dividing loxp_mEmeraldFP-TrxL1_knockin parasite stably expressing mTagRFP-T-TgCentrin2 with the apical complex in the retracted state. Insets (2×) show a longitudinal view of the middle section of the parasite apex, where a linear structure (white arrowhead), likely to be the intraconoid microtubules, is seen located posterior to the mTagRFP-T-TgCentrin2-containing preconoidal ring. Red arrowheads indicate the TgCentrin2-containing peripheral annuli, and red arrows indicate duplicated centrioles. d, daughter parasites; green, mEmeraldFP-TrxL1; red, mTagRFP-T-TgCentrin2. (E) Cartoons depicting the retracted and extended state of the apical complex. An increase in intracellular calcium concentration triggers the switch from the retracted to the extended state. (F) Projection of 3D-SIM images of several extracellular loxp_mEmeraldFP-TrxL1_knockin parasites stably expressing mTagRFP-T-TgCentrin2 treated with the calcium ionophore A23187 to stimulate the extension of the apical complex. In these parasites (oriented at different angles with respect to the coverslip), the apical linear TrxL1-containing structure is seen protruding beyond the roots of the cortical microtubules but remains posterior to the preconoidal ring labeled by mTagRFP-T-TgCentrin2. Insets (2×) show the middle section of the apex indicated by the frames. Green, mEmeraldFP-TrxL1; red, mTagRFP-T-TgCentrin2.
The association between TrxL1 and cortical microtubules is stable, and the TRX-like domain itself is insufficient for mediating this association.
To determine whether the association between TrxL1 and the cortical microtubules is stable, loxp_mEmeraldFP-TrxL1_knockin parasites were extracted with 1% deoxycholate or 1% Triton X-100 detergents. The result shows that TrxL1 remains associated with the microtubules under either condition (Fig. 4A; also see Fig. S2 in the supplemental material). We also treated these parasites with 2.5 μM oryzalin, a dinitroaniline herbicide (5, 8, 10, 42, 43). Although the cortical microtubules of mature parasites are stable and resistant to oryzalin treatment, the drug prevents the formation of new microtubules. Thus, long-term oryzalin treatment results in very distorted parasites, because as the parasites enter cell division, deformed daughters develop and disrupt the shape of the mother parasite when they attempt to bud (5, 8, 10, 42, 43). mEmeraldFP-TrxL1 remains associated with cortical microtubules of the mother parasites upon long-term oryzalin treatment (Fig. 4B). Together, results from the detergent extraction and oryzalin treatment experiments indicate that the interaction between TrxL1 and the cortical microtubules is stable. Truncation analyses show that the TRX-like domain is insufficient for mediating this association. None of the four truncated TrxL1 proteins (TrxL1_2-49 [the N-terminal fragment before the TRX-like domain], TrxL1_2-178 [the N-terminal fragment including the TRX-like domain], TrxL1_50-178 [the TRX-like domain only], and TrxL1_179-220 [the C-terminal fragment after the TRX-like domain]) showed cortical microtubule localization (see Fig. S3). This indicates that either both N- and C-terminal fragments or full-length TrxL is required for the microtubule association.
Fig 4.
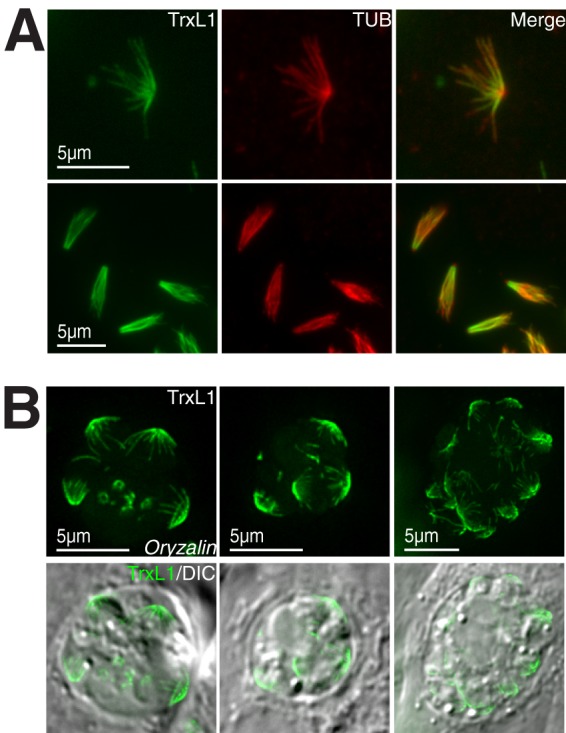
TrxL1 stably associates with the cortical microtubules. (A) mEmeraldFP-TrxL1 (green) and mouse anti-tubulin (red) labeling of extracellular loxp_mEmeraldFP-TrxL1_knockin parasites extracted with 1% (wt/vol) deoxycholate. (Upper row) a parasite with cortical microtubules well spread. (Lower row) Parasites with cortical microtubules collapsed, which is more typical after deoxycholate extraction. (B) Image of mEmeraldFP-TrxL1 (green) in loxp_mEmeraldFP-TrxL1_knockin parasites treated with 2.5 μM oryzalin for 18 h. Three different parasitophorous vacuoles are shown. In adult parasites, mEmeraldFP-TrxL1 remains associating with cortical microtubule after treatment. DIC, differential interference contrast.
No direct association between TrxL1 and microtubules is observed.
To test if TrxL1 can bind to microtubules directly, we first expressed mCherryFP (Fig. 5A)- or enhanced GFP (data not shown)-tagged TrxL1 in HFF and BHK cells. Surprisingly, we found that TrxL1 fusion proteins localized diffusely throughout the cell with no distinct microtubule labeling (Fig. 5A) regardless of the expression level. One explanation for this result is that TrxL1 does not bind to microtubules directly. Alternatively, TrxL1 can bind microtubules directly but is outcompeted by endogenous microtubule binding proteins in mammalian cells. If the second possibility is true, then TrxL1 should be able to bind to microtubules in vitro. To address this possibility, we expressed and purified recombinant FLAG-TrxL1 from E. coli and assessed the binding of TrxL1 to in vitro-polymerized microtubules using a sedimentation assay (Fig. 5B). Recombinant FLAG-TrxL1 did not pellet with microtubules together even when the tubulin subunits were at a 4-fold molar excess; thus, FLAG-TrxL1 does not bind to microtubules in vitro, supporting the hypothesis that TrxL1 associates with the cortical microtubules in an indirect manner in the parasite.
Fig 5.

No direct interaction between TrxL1 and microtubules is detected. (A) Ectopic expression of mCherryFP-TrxL1 in HFF (upper row) and BHK (lower row) cells labeled with anti-tubulin antibodies. Red, mCherryFP-TrxL1; green, anti-tubulin labeling. (B) Microtubule pelleting assay of FLAG-TrxL1. One μM recombinant FLAG-TrxL1 purified from E. coli was incubated with in vitro-polymerized microtubules at the indicated concentrations followed by centrifugation. M, molecular size marker. P, pellet; S, supernatant.
TrxL1 is dispensable for T. gondii growth in culture and is not required for maintaining cortical microtubule stability.
The apparent high-affinity binding of TrxL1 to the cortical microtubules in T. gondii suggests that it is important for maintaining the structure and function of the cortical microtubules, yet the likely indirect nature of this association argues otherwise. Therefore, we decided to investigate the effect of TrxL1 removal on parasite growth and microtubule stability. To generate the Δtrxl1 parasite, Cre recombinase was transiently expressed in loxp_mEmeraldFP-TrxL1_knockin parasites to excise the DNA fragment flanked by the two loxP sites (i.e., the mEmeraldFP-TrxL1 coding sequence and the HXGPRT expression cassette) (29, 30) (Fig. 6A). Although in principle the knockout parasite can be distinguished from the loxp_mEmeraldFP-TrxL1_knockin parasite based on the presence [mEmerald(+)] or absence [mEmerald(−)] of green fluorescence, we found that this criterion alone cannot distinguish nonfluorescent cell debris from mEmerald(−) parasites. To address this problem, a plasmid expressing cytoplasmic mCherryFP was cotransfected with the Cre plasmid. As only live parasites can express mCherryFP, this strategy allows us to reliably distinguish the knockout parasite [mEmerald(−)/mCherry(+)] from nonfluorescent particles [mEmerald(−)/mCherry(−)] based on mCherryFP fluorescence and from the loxp_mEmeraldFP-TrxL1_knockin parasite [mEmerald(+)] based on mEmeraldFP fluorescence (Fig. 6B). The deletion of the TrxL1 gene was confirmed by Southern blotting (Fig. 6C) and genomic PCR (see Fig. S1 in the supplemental material). Two independent clones obtained from two different electroporations were used in subsequent analysis.
Fig 6.

Generation and characterization of Δtrxl1 parasites. (A) Scheme for generating Δtrxl1 parasites. Parental loxp_mEmeraldFP-TrxL1_knockin parasites were cotransfected with a plasmid transiently expressing Cre recombinase (to excise the genome fragment between the two loxP sites) and ptub-mCherryFP-mCherryFP (for transiently expressing mCherryFP and distinguishing knockout parasites from nonfluorescent cell debris). Clonal Δtrxl1 parasites were selected by FACS as shown in panel B. (B) Flow cytometry graph of sorting Δtrxl1 parasites generated in panel A. [mEmerald(−)/mCherry(+) parasites are within the red frame, which is 0.6% of the total population.] (C) Southern blotting analyses of the trxl1 locus in parental RHΔhx (P), loxp_mEmeraldFP-TrxL1_knockin (KI), and Δtrxl1 (KO) parasites. Genomic DNA of the parasites was digested with KpnI. The blot was first hybridized with a TrxL1 probe (left; see the diagram in panel A) and then stripped and rehybridized with a 5′UTR probe (right; see the diagram in panel A). The predicted KpnI fragment size is 5.5 kb in RHΔhx, 7.7 kb in loxp_mEmeraldFP-TrxL1_knockin, and 3.8 kb in Δtrxl1 parasites. As expected, no signal is seen in Δtrxl1 parasites when hybridized with the TrxL1 probe. (D) Competition growth assay between Δtrxl1 and loxp_mEmeraldFP-TrxL1_knockin parasites. x axis, number of days in culture after the mixing of Δtrxl1 and loxp_mEmeraldFP-TrxL1_knockin parasites in equal proportion on day 0. y axis, average percentage of Δtrxl1 in the population in 3 independent experiments. Error bars indicate standard deviations. (E) Deconvolved wide-field fluorescent image of a middle (left) and surface (right) section of two intracellular Δtrxl1 parasites expressing mCherryFP-TgTubA1. (F) Extracellular Δtrxl1 parasites extracted with deoxycholate and labeled with mouse anti-tubulin antibodies. DOC, deoxycholate treatment.
To assess the effect of Trxl1 on parasite growth, we mixed Δtrxl1 and loxp_mEmeraldFP-TrxL1_knockin parasites and measured the proportion of each parasite line in the population over the course of 20 days. We observed only very subtle disadvantages for the Δtrxl1 parasites in the competition (Fig. 6D). Because the canonical function of thioredoxin family members is to maintain the reduced state of the target proteins, we also examined, by plaque assay, the growth rate of Δtrxl1 mutants and two parental strains (RHΔhx and loxP_mEmeraldFP-TrxL1_knockin) in the presence of t-butanol peroxihydrogen or phenazine methosulfate, which are oxidative stressors. We found that plaque-generating capabilities of these different parasite lines are indistinguishable from each other under all conditions (data not shown), indicating that TrxL1 is not a major factor for neutralizing oxidative stress.
To determine whether TrxL1 is involved in maintaining the stability of the cortical microtubules, we examined tubulin localization by expressing mCherryFP-TUBA1 in Δtrxl1 parasites. We found that the morphology of mCherryFP-TUBA1-containing structures in the knockout mutant is indistinguishable from that of wild-type parasites (Fig. 6E). Furthermore, we extracted the Δtrxl1 mutant with deoxycholate and found that the cortical microtubules of the Δtrxl1 mutant were stable upon detergent extraction (Fig. 6F), similar to those of parental parasites (Fig. 4A).
TrxL1 is a part of a protein complex coating the cortical microtubules in T. gondii.
Due to the stable yet likely indirect nature of the association of TrxL1 with the cortical microtubules, we reasoned that there should be adaptor proteins recruiting TrxL to the microtubules and that TrxL1 can be a useful probe for uncovering new microtubule binding proteins. In order to identify TrxL1-interacting proteins, we immunoprecipitated mEmeraldFP-TrxL1 from loxP_mEmeraldFP-TrxL1_knockin parasites using anti-GFP antibody conjugated to agarose beads (Fig. 7). SDS-PAGE analysis showed that several proteins specifically coprecipitated with mEmeraldFP-TrxL1 (Fig. 7A). MudPIT (44, 45) was used to identify these proteins (Fig. 7B). Apart from mEmeraldFP-TrxL1 itself, the most abundant protein in the precipitate was SPM1, a microtubule binding protein recently identified by the Morrissette laboratory (23). Not surprisingly, α- and β-tubulins are also found in the complex. When expressed in the SPM1 knockout mutant (Δspm1) (23), mEmeraldFP-TrxL1 is almost completely cytosolic, revealing that SPM1 is the adaptor for recruiting TrxL1 to the cortical microtubules (Fig. 7C).
Fig 7.

Identification and characterization of TrxL1 binding partners. (A) Whole-cell lysates from loxp_mEmeraldFP-TrxL1_knockin parasites (mE-TrxL1) were immunoprecipitated with anti-GFP antibody. The lysates from RHΔhx parasites were used as the negative control (Ctl). Immunoprecipitated materials were analyzed with SDS-PAGE (A) or MudPIT (B). (B) Proteins from mEmeraldFP-TrxL1 immunoprecipitation with more than two peptides identified in each replicate by MudPIT. (C) The proper localization of TrxL1 to the microtubules requires SPM1. (Upper) Middle section and surface view of RHΔhx parasites expressing mEmerald-TrxL1. (Lower) Middle section and surface view of Δspm1 parasites expressing mEmerald-TrxL1. (D) Projections of 3D-SIM images of T. gondii expressing mEmeraldFP-tagged TrxL2 (left) and TLAP1 (right) showing localization to the cortical microtubules.
Besides SPM1 and α- and β-tubulins, there were five other proteins consistently present in the pulldown, including a close homolog of TrxL1, which we named TrxL2 (TGGT1_080930) (Fig. 1B). Interestingly, similar to TrxL1, TrxL2 does not have a CXXC motif in the thioredoxin-like fold. Instead, it contains HSKC in the corresponding region. A BLAST search shows that besides its orthologue in Neospora canium (a coccidian closely related to T. gondii), TrxL2 shares the highest sequence similarity with TgTrxL1, suggesting that these two genes were derived from a gene duplication event. This event might have occurred before the speciation of N. canium and T. gondii but after the separation of coccidians from other subclasses of apicomplexans, such as Plasmodium spp. Alternatively, the duplication might have occurred in an ancestral apicomplexan before the differentiation of the subclasses, but the gene coding for TrxL2 was lost in noncoccidian parasites during evolution. Similar to TrxL1, mEmeraldFP-tagged TrxL2 is localized to the cortical microtubules in a highly specific fashion (Fig. 7D, left), suggesting that these two homologs are functionally redundant. The four additional proteins present in the TrxL1-containing complex are hypothetical proteins with no recognizable domains. We named them TrxL1-associating proteins (TLAPs) (TLAP1, TGGT1_038030; TLAP2, TGGT1_115520; TLAP3, TGGT1_070610; and TLAP4, TGGT1_019570). Among them, TLAP1 and TLAP2 are well conserved between T. gondii and Plasmodium spp. TgTLAP3 and its putative Plasmodium orthologues share only weak homology. TLAP4 seems to be specific to the coccidian subclass. mEmeraldFP-tagged TLAP1 shows clear localization to the cortical microtubules (Fig. 7D, right), strongly supporting that TrxL1 and its associating proteins form a microtubule-coating complex.
DISCUSSION
In this study, we identified and characterized a novel microtubule-associated protein, TrxL1, in Toxoplasma gondii. Using structured illumination-based superresolution imaging in live parasites, we determined that TrxL1 is associated with the cortical microtubules and the intraconoid microtubules. This demonstrates tremendous potential in revealing and studying fine structures in these small organisms using superresolution techniques, as the intraconoid microtubules are separated by only ∼150 nm from the roots of the cortical microtubules (6, 9, 22), too close to be resolved by conventional wide-field microscopy. We found that although TrxL1 is stably associated with the cortical microtubules, this interaction is likely to be indirect, consistent with the lack of a recognizable microtubule binding domain in its primary sequence. In the process of characterizing TrxL1, we further improved the Cre-LoxP-based gene knockout method that we previously implemented in T. gondii (29, 30) by tagging the target gene with fluorescent protein in the LoxP knock-in line (Fig. 2A). The fluorescent tag driven by the endogenous promoter of the target gene in itself is already very useful for imaging, counting molecules, and immunoprecipitation. Combined with the flanking LoxP sites, it also becomes a powerful intermediate for isolating knockout mutants, because after Cre expression, the loss of fluorescence indicates successful excision of the target gene (Fig. 6). The knockout parasites can be isolated from the rest of the population by FACS. If the target gene is essential and the knockout mutant fails to grow as a clone, one can still use the mixed population of the Cre-transfected LoxP knock-in line to identify the mutants under the microscope based on fluorescence and analyze the knockout phenotype in live cells. This strategy can be used in any Cre-LoxP-based technique regardless of how Cre expression is controlled (i.e., by transient transfection as described previously [29, 30] and in this work or by conditional expression, as in the case of the DiCre system first developed for mouse genetics [46] and recently implemented in T. gondii and P. falciparum [47, 48]).
TRX proteins constitute a large, conserved protein family that maintains the balance of redox potential in the cell. Canonical TRX proteins have a CXXC active-site motif. The −SH group in the first cysteine initially “attacks” a disulfide bond in a substrate and forms an intermolecular disulfide bond, which is then reduced by the −SH group in the second cysteine that forms an intramolecular disulfide bond with the first cysteine, leaving the target cysteine in the substrate in the reduced state (24, 25). This activity allows TRXs to regulate the redox and functional states of proteins in the cytoplasm as well as in various organelles, such as mitochondria, choloroplast, and endoplasmic reticulum (41, 49). It has been shown that the function of certain cytoskeletal proteins is affected by their redox states, which in turn can be controlled by the activity of members of the TRX family (50–53). For instance, upon oxidation by ONOO− and H2O2 in cysteines, tau and microtubule-associated protein-2 (MAP2) lose their ability to promote microtubule polymerization in vitro, an effect that can be reversed by treatment with human TRX and TRX reductase in the presence of NADH (53). TRX-related proteins were also found to associate with dynein heavy chains in the outer dynein arms in the flagellum. It was postulated that these proteins control the redox state of the components of the outer dynein arms and are important for regulating the beating of the flagellum (50–52). The only TRX domain-containing protein known to interact with microtubules is the human thioredoxin-like 2 protein, a bifunctional protein that contains a canonical thioredoxin domain and a nucleoside-diphosphate kinase domain (54). Although the function of this protein is still unknown, another thioredoxin-nucleoside diphosphate kinase (TXNDC3) appears to be involved in the assembly of the outer dynein arm of the flagellum. A nonsense mutation in TXNDC3 has been correlated with primary ciliary dyskinesia, a disease caused by defects in cilia/flagella in respiratory epithelium, embryonic nodes, and sperm (55). The microtubule localization of TrxL1 and TrxL2 reveals that the cytoskeletal association of this class of proteins might be more common than previously appreciated. Interestingly, both TrxL1 and TrxL2 lack the CXXC motif. In TrxL1, a well-conserved DPKC motif is in place of CXXC. The functional importance of this replacement has yet to be determined, but it suggests that these two proteins are not functional thioredoxin enzymes, consistent with our finding that Δtrxl1 parasites do not sensitize the parasite to oxidative stressors. However, it is also possible that they reduce substrate in an unconventional fashion (56), as certain monocysteine glutaredoxins can reduce substrates by forming an intermolecular disulfide bond with a cofactor (57).
Although TrxL1 associates with microtubules in T. gondii, when expressed in mammalian cells it is completely cytosolic. As the major tubulins in T. gondii are ∼97% identical to human tubulin, it is unlikely that TrxL1 binds directly to the microtubule lattice. Indeed, we discovered that the recruitment of TrxL1 to the cortical microtubules is dependent on SPM1, a previously identified cortical microtubule-associated protein (23). Unlike SPM1, however, the removal of TrxL1 does not affect microtubule stability. This might reflect a functional redundancy between TrxL1 and its close homolog, TrxL2, present in the same microtubule-associated complex, which also provides a plausible explanation for the lack of a significant growth phenotype of the TrxL knockout mutant despite the high degree of conservation among TrxL1 orthologues within Apicomplexa. Another intriguing possibility is that the association of TrxL1 and TrxL2 with microtubules does not reflect their role in regulating microtubule function per se; rather, it permits localized enzyme activity, analogous to the cytoskeletal association of glycolytic enzymes (58–62).
The identification of TrxL1 and its associating proteins revealed an intriguing protein complex that includes SPM1, TrxL2, and several novel proteins. Interestingly, although SPM1 is required for recruiting TrxL1 to the cortical microtubules, previous work suggests that its association with the microtubule is likely to be indirect (23), which means that an additional member(s) of the TrxL1-containing complex is responsible for the direct microtubule binding. Further study of TrxL1 and its interacting proteins will provide important insights into the construction and maintenance of the cortical microtubules in T. gondii.
Supplementary Material
ACKNOWLEDGMENTS
We thank Naomi Morrissette (University of California, Irvine) for the SPM1 knockout parasite, Con Beckers (University of North Carolina) for the rabbit anti-IMC1 antibody, Richard Day (Indiana University School of Medicine) and Mike Davidson (Florida State University, National High Magnetic Field Laboratory) for an mTagRFP-T-containing plasmid, John Murray (University of Pennsylvania) for YL1/2 and YOL-1 tubulin antibodies and ptub-mTagRFP-T-TgCentrin2, pc22-EGFP-TubA1, and pc22-mCherryFP-TubA1 plasmids, and Jim Power and the LMIC at Indiana University for imaging support.
This study was supported by the March of Dimes (6-FY12-258) and NIH-NIAID (R01-AI098686).
Footnotes
Published ahead of print 19 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00082-13.
REFERENCES
- 1.Luft BJ, Hafner R, Korzun AH, Leport C, Antoniskis D, Bosler EM, Bourland DD, III, Uttamchandani R, Fuhrer J, Jacobson J, Morlat P, Vilde JL, Remington JS, ACTG 077p/ANRS 009 Study Team 1993. Toxoplasmic encephalitis in patients with the acquired immunodeficiency syndrome. Members of the ACTG 077p/ANRS 009 Study Team. N. Engl. J. Med. 329:995–1000 [DOI] [PubMed] [Google Scholar]
- 2.Olariu TR, Remington JS, McLeod R, Alam A, Montoya JG. 2011. Severe congenital toxoplasmosis in the United States: clinical and serologic findings in untreated infants. Pediatr. Infect. Dis. J. 30:1056–1061 [DOI] [PubMed] [Google Scholar]
- 3.Levine ND. 1988. Progress in taxonomy of the Apicomplexan protozoa. J. Protozool. 35:518–520 [DOI] [PubMed] [Google Scholar]
- 4.Hu K. 2008. Organizational changes of the daughter basal complex during the parasite replication of Toxoplasma gondii. PLoS Pathog. 4:e10. 10.1371/journal.ppat.0040010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu K, Johnson J, Florens L, Fraunholz M, Suravajjala S, DiLullo C, Yates J, Roos DS, Murray JM. 2006. Cytoskeletal components of an invasion machine–the apical complex of Toxoplasma gondii. PLoS Pathog. 2:e13. 10.1371/journal.ppat.0020013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morrissette NS, Murray JM, Roos DS. 1997. Subpellicular microtubules associate with an intramembranous particle lattice in the protozoan parasite Toxoplasma gondii. J. Cell Sci. 110(Part 1):35–42 [DOI] [PubMed] [Google Scholar]
- 7.Morrissette NS, Sibley LD. 2002. Cytoskeleton of apicomplexan parasites. Microbiol. Mol. Biol. Rev. 66:21–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morrissette NS, Sibley LD. 2002. Disruption of microtubules uncouples budding and nuclear division in Toxoplasma gondii. J. Cell Sci. 115:1017–1025 [DOI] [PubMed] [Google Scholar]
- 9.Nichols BA, Chiappino ML. 1987. Cytoskeleton of Toxoplasma gondii. J. Protozool. 34:217–226 [DOI] [PubMed] [Google Scholar]
- 10.Nishi M, Hu K, Murray JM, Roos DS. 2008. Organellar dynamics during the cell cycle of Toxoplasma gondii. J. Cell Sci. 121:1559–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shaw MK, Compton HL, Roos DS, Tilney LG. 2000. Microtubules, but not actin filaments, drive daughter cell budding and cell division in Toxoplasma gondii. J. Cell Sci. 113:1241–1254 [DOI] [PubMed] [Google Scholar]
- 12.Meissner M, Schluter D, Soldati D. 2002. Role of Toxoplasma gondii myosin A in powering parasite gliding and host cell invasion. Science 298:837–840 [DOI] [PubMed] [Google Scholar]
- 13.Dobrowolski JM, Sibley LD. 1996. Toxoplasma invasion of mammalian cells is powered by the actin cytoskeleton of the parasite. Cell 84:933–939 [DOI] [PubMed] [Google Scholar]
- 14.Herm-Gotz A, Weiss S, Stratmann R, Fujita-Becker S, Ruff C, Meyhofer E, Soldati T, Manstein DJ, Geeves MA, Soldati D. 2002. Toxoplasma gondii myosin A and its light chain: a fast, single-headed, plus-end-directed motor. EMBO J. 21:2149–2158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wetzel DM, Hakansson S, Hu K, Roos D, Sibley LD. 2003. Actin filament polymerization regulates gliding motility by apicomplexan parasites. Mol. Biol. Cell 14:396–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gaskins E, Gilk S, DeVore N, Mann T, Ward G, Beckers C. 2004. Identification of the membrane receptor of a class XIV myosin in Toxoplasma gondii. J. Cell Biol. 165:383–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frenal K, Polonais V, Marq JB, Stratmann R, Limenitakis J, Soldati-Favre D. 2010. Functional dissection of the apicomplexan glideosome molecular architecture. Cell Host Microbe 8:343–357 [DOI] [PubMed] [Google Scholar]
- 18.Heaslip AT, Leung JM, Carey KL, Catti F, Warshaw DM, Westwood NJ, Ballif BA, Ward GE. 2010. A small-molecule inhibitor of T. gondii motility induces the posttranslational modification of myosin light chain-1 and inhibits myosin motor activity. PLoS Pathog. 6:e1000720. 10.1371/journal.ppat.1000720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacot D, Daher W, Soldati-Favre D. 2013. Toxoplasma gondii myosin F, an essential motor for centrosomes positioning and apicoplast inheritance. EMBO J. 32:1702–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mann T, Gaskins E, Beckers C. 2002. Proteolytic processing of TgIMC1 during maturation of the membrane skeleton of Toxoplasma gondii. J. Biol. Chem. 277:41240–41246 [DOI] [PubMed] [Google Scholar]
- 21.Mann T, Beckers C. 2001. Characterization of the subpellicular network, a filamentous membrane skeletal component in the parasite Toxoplasma gondii. Mol. Biochem. Parasitol. 115:257–268 [DOI] [PubMed] [Google Scholar]
- 22.Hu K, Roos DS, Murray JM. 2002. A novel polymer of tubulin forms the conoid of Toxoplasma gondii. J. Cell Biol. 156:1039–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tran JQ, Li C, Chyan A, Chung L, Morrissette NS. 2012. SPM1 stabilizes subpellicular microtubules in Toxoplasma gondii. Eukaryot. Cell 11:206–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holmgren A. 1989. Thioredoxin and glutaredoxin systems. J. Biol. Chem. 264:13963–13966 [PubMed] [Google Scholar]
- 25.Carvalho AP, Fernandes PA, Ramos MJ. 2006. Similarities and differences in the thioredoxin superfamily. Prog. Biophys. Mol. Biol. 91:229–248 [DOI] [PubMed] [Google Scholar]
- 26.Hashemy SI, Holmgren A. 2008. Regulation of the catalytic activity and structure of human thioredoxin 1 via oxidation and S-nitrosylation of cysteine residues. J. Biol. Chem. 283:21890–21898 [DOI] [PubMed] [Google Scholar]
- 27.Akerman SE, Muller S. 2005. Peroxiredoxin-linked detoxification of hydroperoxides in Toxoplasma gondii. J. Biol. Chem. 280:564–570 [DOI] [PubMed] [Google Scholar]
- 28.Kawazu SI, Takemae H, Komaki-Yasuda K, Kano S. 2010. Target proteins of the cytosolic thioredoxin in Plasmodium falciparum. Parasitol. Int. 59:298–302 [DOI] [PubMed] [Google Scholar]
- 29.Heaslip AT, Nishi M, Stein B, Hu K. 2011. The motility of a human parasite, Toxoplasma gondii, is regulated by a novel lysine methyltransferase. PLoS Pathog. 7:e1002201. 10.1371/journal.ppat.1002201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heaslip AT, Dzierszinski F, Stein B, Hu K. 2010. TgMORN1 is a key organizer for the basal complex of Toxoplasma gondii. PLoS Pathog. 6:e1000754. 10.1371/journal.ppat.1000754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kremers GJ, Hazelwood KL, Murphy CS, Davidson MW, Piston DW. 2009. Photoconversion in orange and red fluorescent proteins. Nat. Methods 6:355–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shaner NC, Lin MZ, McKeown MR, Steinbach PA, Hazelwood KL, Davidson MW, Tsien RY. 2008. Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat. Methods 5:545–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Donald RG, Roos DS. 1993. Stable molecular transformation of Toxoplasma gondii: a selectable dihydrofolate reductase-thymidylate synthase marker based on drug-resistance mutations in malaria. Proc. Natl. Acad. Sci. U. S. A. 90:11703–11707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heaslip AT, Ems-McClung SC, Hu K. 2009. TgICMAP1 is a novel microtubule binding protein in Toxoplasma gondii. PLoS One 4:e7406. 10.1371/journal.pone.0007406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Florens L, Washburn MP. 2006. Proteomic analysis by multidimensional protein identification technology. Methods Mol. Biol. 328:159–175 [DOI] [PubMed] [Google Scholar]
- 36.McDonald WH, Ohi R, Miyamoto DT, Mitchison TJ, Yates JR., III 2002. Comparison of three directly coupled HPLC MS/MS strategies for identification of proteins from complex mixtures: single-dimension LC-MS/MS, 2-phase MudPIT, and 3-phase MudPIT. Int. J. Mass Spectrom. 219:245–251 [Google Scholar]
- 37.Eng JK, McCormack AL, Yates JR., III 1994. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5:976–989 [DOI] [PubMed] [Google Scholar]
- 38.Tabb DL, McDonald WH, Yates JR., III 2002. DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 1:21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y, Wen Z, Washburn MP, Florens L. 2010. Refinements to label free proteome quantitation: how to deal with peptides shared by multiple proteins. Anal. Chem. 82:2272–2281 [DOI] [PubMed] [Google Scholar]
- 40.Becker K, Kanzok SM, Iozef R, Fischer M, Schirmer RH, Rahlfs S. 2003. Plasmoredoxin, a novel redox-active protein unique for malarial parasites. Eur. J. Biochem. 270:1057–1064 [DOI] [PubMed] [Google Scholar]
- 41.DeRocher AE, Coppens I, Karnataki A, Gilbert LA, Rome ME, Feagin JE, Bradley PJ, Parsons M. 2008. A thioredoxin family protein of the apicoplast periphery identifies abundant candidate transport vesicles in Toxoplasma gondii. Eukaryot. Cell 7:1518–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stokkermans TJ, Schwartzman JD, Keenan K, Morrissette NS, Tilney LG, Roos DS. 1996. Inhibition of Toxoplasma gondii replication by dinitroaniline herbicides. Exp. Parasitol. 84:355–370 [DOI] [PubMed] [Google Scholar]
- 43.Morrissette NS, Mitra A, Sept D, Sibley LD. 2004. Dinitroanilines bind alpha-tubulin to disrupt microtubules. Mol. Biol. Cell 15:1960–1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wolters DA, Washburn MP, Yates JR., III 2001. An automated multidimensional protein identification technology for shotgun proteomics. Anal. Chem. 73:5683–5690 [DOI] [PubMed] [Google Scholar]
- 45.Washburn MP, Wolters D, Yates JR., III 2001. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19:242–247 [DOI] [PubMed] [Google Scholar]
- 46.Jullien N, Goddard I, Selmi-Ruby S, Fina J-L, Cremer H, Herman J-P. 2007. Conditional transgenesis using dimerizable Cre (DiCre). PLoS One 2:e1355. 10.1371/journal.pone.0001355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Andenmatten N, Egarter S, Jackson AJ, Jullien N, Herman J-P, Meissner M. 2013. Conditional genome engineering in Toxoplasma gondii uncovers alternative invasion mechanisms. Nat. Methods 10:125–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Collins CR, Das S, Wong EH, Andenmatten N, Stallmach R, Hackett F, Herman J-P, Muller S, Meissner M, Blackman MJ. 2013. Robust inducible Cre recombinase activity in the human malaria parasite Plasmodium falciparum enables efficient gene deletion within a single asexual erythrocytic growth cycle. Mol. Microbiol. 88:687–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Powis G, Montfort WR. 2001. Properties and biological activities of thioredoxins. Annu. Rev. Biophys. Biomol. Struct. 30:421–455 [DOI] [PubMed] [Google Scholar]
- 50.Patel-King RS, Benashki SE, Harrison A, King SM. 1996. Two functional thioredoxins containing redox-sensitive vicinal dithiols from the Chlamydomonas outer dynein arm. J. Biol. Chem. 271:6283–6291 [DOI] [PubMed] [Google Scholar]
- 51.King SM. 2010. Sensing the mechanical state of the axoneme and integration of Ca2+ signaling by outer arm dynein. Cytoskeleton 67:207–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wakabayashi KI, King SM. 2006. Modulation of Chlamydomonas reinhardtii flagellar motility by redox poise. J. Cell Biol. 173:743–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Landino LM, Skreslet TE, Alston JA. 2004. Cysteine oxidation of tau and microtubule-associated protein-2 by peroxynitrite: modulation of microtubule assembly kinetics by the thioredoxin reductase system. J. Biol. Chem. 279:35101–35105 [DOI] [PubMed] [Google Scholar]
- 54.Sadek CM, Jimenez A, Damdimopoulos AE, Kieselbach T, Nord M, Gustafsson JA, Spyrou G, Davis EC, Oko R, van der Hoorn FA, Miranda-Vizuete A. 2003. Characterization of human thioredoxin-like 2. A novel microtubule-binding thioredoxin expressed predominantly in the cilia of lung airway epithelium and spermatid manchette and axoneme. J. Biol. Chem. 278:13133–13142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duriez B, Duquesnoy P, Escudier E, Bridoux A-M, Escalier D, Rayet I, Marcos E, Vojtek A-M, Bercher J-F, Amselem S. 2007. A common variant in combination with a nonsense mutation in a member of the thioredoxin family causes primary ciliary dyskinesia. Proc. Natl. Acad. Sci. U. S. A. 104:3336–3341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Collet JF, Messens J. 2010. Structure, function, and mechanism of thioredoxin proteins. Antioxid. Redox Signal. 13:1205–1216 [DOI] [PubMed] [Google Scholar]
- 57.Mesecke N, Mittler S, Eckers E, Herrmann JM, Deponte M. 2008. Two novel monothiol glutaredoxins from Saccharomyces cerevisiae provide further insight into iron-sulfur cluster binding, oligomerization, and enzymatic activity of glutaredoxins. Biochemistry 47:1452–1463 [DOI] [PubMed] [Google Scholar]
- 58.Knull HR, Walsh JL. 1992. Association of glycolytic enzymes with the cytoskeleton. Curr. Topics Cell. Regul. 33:15–30 [DOI] [PubMed] [Google Scholar]
- 59.Kusakabe T, Motoki K, Hori K. 1997. Mode of interactions of human aldolase isozymes with cytoskeletons. Arch. Biochem. Biophys. 344:184–193 [DOI] [PubMed] [Google Scholar]
- 60.Waingeh VF, Gustafson CD, Kozliak EI, Lowe SL, Knull HR, Thomasson KA. 2006. Glycolytic enzyme interactions with yeast and skeletal muscle F-actin. Biophys. J. 90:1371–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pomel S, Luk FCY, Beckers CJM. 2008. Host cell egress and invasion induce marked relocations of glycolytic enzymes in Toxoplasma gondii tachyzoites. PLoS Pathog. 4: e1000188. 10.1371/journal.ppat.1000188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Starnes GL, Coincon M, Sygusch J, Sibley LD. 2009. Aldolase is essential for energy production and bridging adhesin-actin cytoskeletal interactions during parasite invasion of host cells. Cell Host Microbe 5:353–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.