

Abstract
Autotoxic production of proinflammatory mediators during early sepsis induces excessive inflammation, and their later suppression may limit the immune response. We previously reported that sepsis differentially represses transcription and translation of tumor necrosis factor alpha (TNF-α) and interleukin 1β (IL-1β) to reprogram sepsis inflammation. This switch is gene specific and plays a crucial role in the clinically relevant syndrome of endotoxin adaptation/tolerance, multiorgan failure, and poor sepsis outcome. To further define the mechanisms responsible for translation disruption that follows inflammation induction, we used THP-1 human promonocytes as a model of Toll-like receptor 4 (TLR4) responses found in sepsis. We showed that phosphorylation-dependent activation of p38 mitogen-activated protein kinase (MAPK) and translation disruption of TNF-α and IL-6 follow increased MAPK phosphatase 1 (MKP-1) expression and that MKP-1 knockdown rephosphorylates p38 and restores the capacity to translate TNF-α and IL-6 mRNAs. We also observed that the RNA-binding protein motif 4 (RBM4), a p38 MAPK target, accumulates in an unphosphorylated form in the cytosol in endotoxin-adapted cells, suggesting that dephosphorylated RBM4 may function as a translational repressor. Moreover, MKP-1 knockdown promotes RBM4 phosphorylation, blocks its transfer from the nucleus to the cytosol, and reverses translation repression. We also found that microRNA 146a (miR-146a) knockdown prevents and miR-146a transfection induces MKP-1 expression, which lead to increases or decreases in TNF-α and IL-6 translation, respectively. We conclude that a TLR4-, miR-146a-, p38 MAPK-, and MKP-1-dependent autoregulatory pathway regulates the translation of proinflammatory genes during the acute inflammatory response by spatially and temporally modifying the phosphorylation state of RBM4 translational repressor protein.
INTRODUCTION
A normally regulated acute systemic inflammatory response restores immune homeostasis and completes tissue repair (1–3), but its dysregulation during sepsis is often lethal (3–6). Sepsis occurs after overly robust and systemic activation of Toll-like receptors (TLRs) on monocytes/macrophages and neutrophils, which produce proinflammatory cytokines and chemokines required to initiate the disease (7, 8) and promote autotoxic consequences (2, 3, 9, 10). The TLR-mediated signaling path that ignites the acute phase of the sepsis response shifts within hours to a reactive program of reduced inflammation. During this shift, monocytes/macrophages and neutrophils generate heterochromatin to repress transcription of proinflammatory cytokines, like tumor necrosis factor alpha (TNF-α), interleukin 6 (IL-6), and IL-1β, and activate euchromatin of the genes encoding anti-inflammatory cytokines such as IL-10 and IL-RA (11–14). While this gene-specific reprogramming may have evolved to defend the host and then restore homeostasis, it can become lethal by generating too much inflammation early (1, 2, 6) and too much immune suppression and organ failure later (1, 2, 6, 15). This two-phase sepsis response occurs in animals (16) and humans (7), and its repressive component is mirrored by the presence of refractory TLR responses. The sustained repression of proinflammatory genes in blood monocytes (17) and neutrophils (18) during the late phase of acute systemic inflammation is mirrored by endotoxin adaptation or “ tolerance,” a process that can be modeled in cultures of primary monocytes and human monocytic cell lines such as THP-1 cells stimulated by bacterial endotoxin (lipopolysaccharide [LPS]) (13, 19–21). Endotoxin-adapted THP-1 cells, monocytes/macrophages, and neutrophils from septic patients fail to produce proinflammatory TNF-α, IL-6, and IL-1β in response to subsequent stimulation with LPS (13, 18, 22–24). We previously reported that expression of proinflammatory genes in endotoxin-adapted THP-1 cells is differentially repressed at the level of transcription (13, 14) and translation and that disrupted translation is generated by a TLR4-dependent negative feedback path that requires microRNA 146a (miR-146a) induction (25).
The initial TLR4 response of monocytes/macrophages and neutrophils signals gene expression by MyD88, interleukin-1 receptor-associated kinase 1 (IRAK1), IRAK4, tumor necrosis factor receptor-associated factor 6 (TRAF6), NF-κB, and mitogen-activated protein kinases (MAPKs) (26–28). Activated MAPKs, including p38, extracellular signal-regulated kinase, and Jun N-terminal protein kinase (JNK) (29–32), regulate transcription and support proinflammatory cytokine protein synthesis posttranscriptionally by controlling the mRNA stability of genes with AU-rich elements (AREs) at their 3′ untranslated mRNA regions (29, 30). MAPKs inactivate RNA-binding proteins such as tristetraprolin (TTP), which destabilizes and represses translation of target mRNA (30, 33); the inactivation of TTP requires p38-dependent phosphorylation (34). Consistent with this concept, pharmacological inhibition of p38 in LPS-stimulated macrophage cell line RAW264.7 reduces TNF-α mRNA translation (35), and in mice defective in p38, TNF-α protein production is limited and endotoxin shock is prevented (36). Thus, p38 MAPK enhances proinflammatory cytokine protein synthesis in early TLR4 responses by increasing mRNA stability and promoting translation.
An emerging concept supports the hypothesis that TLR4-dependent p38 MAPK inactivation by dephosphorylation disrupts translation of proinflammatory cytokine mRNA (26, 27, 37). MAPK phosphatase 1 (MKP-1) belongs to a group of dual-specificity protein phosphatases (DUSPs) that limit expression of inflammatory cytokines by dephosphorylating and inactivating MAPKs, including p38, on tyrosine and threonine residues (38). Importantly, TLR4 responses induce macrophage MKP-1 expression following p38 activation (38) and increase levels of MKP-1, which correlate with p38 inactivation (35, 38). In line with this, MKP-1 knockout mice (39) and MKP-1-deficient macrophages (31, 35, 37) have sustained p38 activation and overproduce TNF-α and IL-6 in response to LPS stimulation. These data support the hypothesis that MKP-1 promotes endotoxin adaptation by deactivating p38 MAPK, leading to disrupted translation.
We previously discovered that miR-146a induced during endotoxin adaptation of THP-1 human promonocytes represses TNF-α and IL-6 protein synthesis by promoting the assembly of an mRNA-binding translational repressor complex of Ago2 and RNA-binding protein motif 4 (RBM4) proteins (22, 25). MiR-146a induced by TLR4-dependent NF-KB p65 activation directly reduces translation of IRAK1 and TRAF6, which lie upstream of p38 (40). Here, we report a novel TLR4-, miR-146a-, p38 MAPK-, and MKP-1-dependent autoregulatory pathway that represses translation of proinflammatory genes by dephosphorylating the mRNA translational regulatory protein RBM4.
MATERIALS AND METHODS
Cell culture.
The THP-1 human monocytic cell line, obtained from the American Type Culture Collection (Manassas, VA), was maintained in RPMI 1640 medium (Invitrogen, Carlsbad, CA) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine (all from HyClone Laboratories, Logan, UT), and 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) at 37°C and 5% CO2. Cells were made tolerant by overnight incubation with 1 μg/ml of Gram-negative bacterial LPS (Escherichia coli serotype O111:B4; Sigma-Aldrich, St. Louis, MO) as described previously (21). The LPS preparation used in these experiments is TLR4 specific and free of contaminating proteins that activate cells via a non-TLR4-dependent mechanism.
Small interfering RNA and miRNA transfections.
Cells were seeded at 0.5 × 106 cells/ml 1 day prior to transfection. Cells were transfected using HiPerFect transfection reagent per the manufacturer's instructions (Qiagen, Valencia, CA).
For MKP-1 and/or RelB knockdown, endotoxin-adapted THP-1 cells were transfected with pools of MKP-1- and/or RelB-specific small interfering RNAs (siRNAs) or scrambled (control) siRNAs at a 0.5 μM final concentration (Santa Cruz Biotechnology, Santa Cruz, CA).
For miR-146a knockdown, cells were transfected with a negative control or miR-146a-specific antisense 2′-O-methyl oligonucleotides (miR-146a inhibitor; final concentration 100 nM) (Ambion, Austin, TX) for 36 h. For miR-146a overexpression studies, endotoxin-responsive (normal) THP-1 cells were transfected with an miRNA negative control or miR-146a mimics at a 50 nM concentration (Ambion) for 36 h before LPS stimulation.
Preparation of protein extracts.
To prepare whole-cell extract, cells were lysed in 1× radioimmunoprecipitation assay (RIPA) buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholic acid, and 1 mM EDTA (Millipore, Temecula, CA) plus 1× protease inhibitor cocktail. After 30 min on ice, cell lysate was cleared by centrifugation for 5 min at 4°C and 14,000 rpm. Protein concentrations were determined by Bradford assay (Bio-Rad), and aliquots were kept at −20°C.
To isolate cytoplasmic and nuclear proteins, we used the NE-PER nuclear and cytoplasmic extraction kit (Pierce, Rockford, IL) per the manufacturer's instructions. Immediately after harvesting, cells were washed in PBS and resuspended in CER1 lysis buffer with protease inhibitor cocktail and incubated on ice for 1 min. CER2 buffer was added, and the incubation continued for 5 min. Supernatants (cytoplasmic proteins) were recovered by centrifugation for 5 min at 4°C and 14,000 rpm. The nuclear pellets were resuspended in NER lysis buffer with protease inhibitor cocktail and incubated for 40 min on ice with occasional vortexing. The nuclear proteins were recovered by centrifugation for 10 min at 4°C and 14,000 rpm.
Western blot analysis.
Equal amounts of protein extracts were mixed with 5× Laemmli sample buffer, resolved by SDS-10% polyacrylamide gel electrophoresis (Bio-Rad), and transferred to nitrocellulose membranes (Pierce). Membranes were blocked for 1 h at room temperature with 5% milk in Tris-buffered saline/Tween 20 and probed overnight at 4°C with the appropriate primary antibody (p38, phospho p38, MKP-1, RelB [Santa Cruz Biotechnology], Ago2 [MBL International, Woburn, MA], RBM4 [Abcam, Cambridge, MA], or phospho RBM4 antibody) against phospho-serine-309 (custom generated by the Vanderbilt University Antibody and Protein Resources Center). After washing, blots were incubated with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibody (Life Technologies, Grand Island, NY) at room temperature for 2 h. Proteins were visualized with the enhanced chemiluminescence detection system (Pierce) and captured with the Quantity One imaging system (Bio-Rad). Membranes were stripped and reprobed with actin (Sigma-Aldrich) or lamin A (Abcam) antibody as a control.
Measurements of mRNA and miRNA.
The mRNA levels of TNF-α and IL-6, or RelB and RBM4 (for knockdown experiments), were measured by real-time quantitative PCR (qPCR) using TaqMan gene-specific primer/probe sets (Qiagen) as described previously (12). Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA).
MiR-146a expression after miR-146a knockdown in adapted cells or after miR-146a mimic transfection into responsive cells was determined using first-strand synthesis and a miScript II RT kit according to the manufacturer's instructions (Qiagen). The reverse transcription (RT) reaction mixture consisted of 4 μl of polyadenylated miRNA (prepared from total RNA), 3 μl of 25 μM universal RT primer, and 1 μl annealing buffer. The reaction mixture was incubated at 65°C for 5 min followed by the addition of 10 μl of 2× first-strand synthesis reaction mix containing deoxynucleoside triphosphate (dNTP) and 2 μl of SuperScript III RT/RNaseOUT enzyme mix. The reaction mixture (20 μl) was then incubated at 50°C for 50 min, followed by 85°C for 5 min to stop the reaction. The real-time qPCR mixture consisted of 5 μl of a 1:10 dilution of the RT product, 1 μl of 10 μM universal reverse primer, 1 μl of 10 μM miR-146a-specific forward primer (an oligonucleotide identical to the entire mature miRNA sequence, in which U is replaced with T), and 25 μl of SYBR green Fluor qPCR master mix (Qiagen). PCR was run in duplicate at 95°C for 10 min followed by 40 cycles at 95°C for 15 s and 60°C for 1 min using the iCycler iQ5 detection system (Bio-Rad). The relative expression of miR-146a was calculated using the 2−ΔΔCT cycle threshold method after normalization to the endogenous U6 small RNA (as an internal control).
Cytokine measurements.
Levels of TNF-α and IL-6 proteins were determined by enzyme-linked immunosorbent assay using commercially available kits according to the manufacturer's instructions (eBioscience, San Diego, CA). Samples were measured in duplicate.
Statistical analysis.
Data were analyzed with Microsoft Excel. Differences between two groups were analyzed by the unpaired Student t test. One-way analysis of variance (ANOVA) with Tukey's multiple comparison tests was used to analyze data with more than two groups/treatments. Data are presented as mean (± standard deviation [SD]). Differences were considered statistically significant at P values of ≤0.05.
RESULTS
Inactivation of p38 MAPK in endotoxin-adapted cells inhibits proinflammatory cytokine synthesis.
We tested whether TLR4 signaling via p38 activation represses proinflammatory cytokine protein production. To do this, we first assessed p38 expression and phosphorylation in endotoxin-responsive and -adapted THP-1 cells at different times following stimulation with LPS. THP-1 cells were made LPS tolerant/adapted by pretreatment of responsive cells with LPS overnight. As shown in Fig. 1, total p38 was constitutively expressed in responsive and adapted cells during the time course examined. Activation/phosphorylation of p38 was transiently (1 h) induced in responsive cells. We did not detect phospho p38 in endotoxin-adapted cells at any time, indicating that LPS-mediated TLR4 stimulation rapidly downregulates p38; a second LPS stimulation failed to activate p38 in endotoxin-adapted cells.
Fig 1.
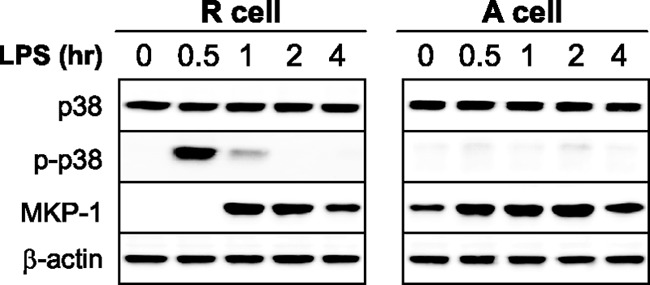
MKP-1 expression is induced by LPS and is sustained in endotoxin-adapted cells concurrent with inactivation of p38 MAPK. Endotoxin adapted/tolerant THP-1 cells were made by pretreatment of responsive (normal) cells overnight with 1 μg/ml of the Gram-negative bacterial LPS. Endotoxin-adapted, along with endotoxin-responsive, cells were then stimulated with 1 μg/ml of LPS for the indicated times and the expression levels of p38, phospho p38, and MKP-1 were measured by Western blot analysis. The results are representative of three experiments. R, endotoxin responsive; A, endotoxin adapted.
To assess whether reduced p38 activation in endotoxin-adapted THP-1 cells is linked to repressed proinflammatory cytokine synthesis, we inhibited p38 activation in endotoxin-responsive cells. Cells were pretreated for 1 h with the p38-specific inhibitor SB203580 and then stimulated with LPS (Fig. 2). Pretreatment with SB203580 (35) completely inhibited p38 phosphorylation and dramatically reduced TNF-α and IL-6 protein levels in response to LPS stimulation (Fig. 2A). Previous studies have shown that proinflammatory cytokine mRNA transcription peaks at 1 h after LPS stimulation in THP-1 cells, whereas the protein production level peaks at a later time (22, 25). Together, these results (Fig. 1 and 2) demonstrate that p38 is inactivated in endotoxin-adapted THP-1 cells and suggest that disrupted p38 activation contributes to repressed proinflammatory cytokine protein synthesis during endotoxin adaptation.
Fig 2.
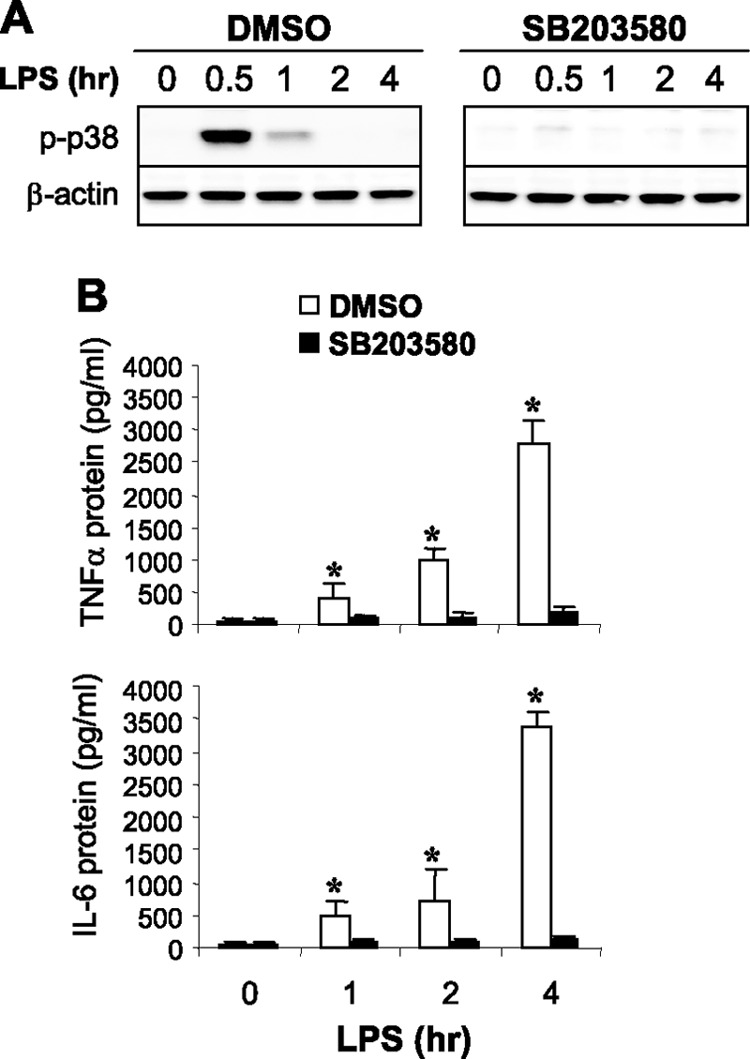
Inhibition of p38 MAPK in endotoxin-responsive cells blocks proinflammatory cytokine synthesis in response to TLR4 stimulation. Endotoxin-responsive THP-1 cells were pretreated for 1 h with 10 μM of the p38 MAPK inhibitor SB203580 and then stimulated with 1 μg/ml of LPS for the indicated times. (A) Expression levels of p38 and phospho p38 MAPK proteins were measured by Western blot analysis. (B) Cells (with the culture supernatants) were harvested and lysed, and then levels of TNF-α and IL-6 proteins were measured by enzyme-linked immunosorbent assay (ELISA). Data are means ± SD of three experiments. *, P < 0.05 compared with treated cells. DMSO, dimethyl sulfoxide.
MKP-1 increases are sustained in endotoxin-adapted cells and its knockdown restores TNF-α protein levels.
To assess the role of MKP-1 in the repression of proinflammatory cytokine synthesis during endotoxin adaptation, we first measured MKP-1 protein in THP-1 cells after LPS stimulation. Western blot analysis did not identify MKP-1 in responsive cells before LPS stimulation, but levels started increasing 1 h after LPS stimulation and slightly decreased by 4 h (Fig. 1) and remained elevated. A second LPS stimulus further increased MKP-1 protein expression. These findings suggested that MKP-1 may directly or indirectly regulate repressed translation during endotoxin adaptation.
To test this hypothesis, we measured TNF-α and IL-6 protein levels after LPS stimulation in endotoxin-adapted cells lacking MKP-1 protein. In addition to translational repression, transcription of proinflammatory cytokines is also repressed in endotoxin-adapted THP-1 cells by the transcriptional repressor RelB-mediated mechanism (12, 25), and RelB knockdown restores mRNA transcription in response to LPS. However, these transcripts are rapidly degraded because of the translational repression (25). As we previously reported (12), knockdown of RelB alone restores TNF-α mRNA but not protein levels. Thus, we knocked down RelB to allow transcription to proceed and concomitantly knocked down MKP-1 to assess its effect on translation (Fig. 3). The low level of TNF-α and IL-6 mRNAs detected at 1 h after LPS stimulation (Fig. 3B) was expected, since the newly transcribed mRNA is rapidly degraded (25). Importantly, knockdown of RelB and MKP-1 in adapted cells restored TNF-α and IL-6 mRNA and protein levels in response to LPS stimulation (Fig. 3A), whereas knockdown of MKP-1 alone did not alter mRNA transcription or translation (Fig. 3C). Together, these results support the hypothesis that by inhibiting p38 activation, MKP-1 promotes translational repression of proinflammatory cytokines during endotoxin adaptation.
Fig 3.

Knockdown of MKP-1 in endotoxin-adapted cells restores proinflammatory cytokine synthesis in response to TLR4 stimulation. Endotoxin-adapted THP-1 cells were made by pretreatment of responsive (normal) cells overnight with 1 μg/ml of LPS. Adapted cells were transfected with a pool of RelB- and/or MKP-1-specific siRNAs or scrambled siRNAs (control knockdown [KD]). After 36 h, cells were stimulated for 0 to 4 h with 1 μg/ml of LPS. After 1 h in LPS, RelB and MKP-1 expression levels were measured by Western blot analysis and TNF-α mRNA levels were measured by real-time PCR. TNF-α and IL-6 mRNA levels were normalized to glyceraldehyde-3-phosphate dehydrogenase expression and are presented relative to control KD (set at 1%). After 4 h in LPS, culture supernatants and cells were harvested and lysed, and then TNF-α and IL-6 protein levels were measured by ELISA. Data are means ± SD of three experiments. *, P < 0.05.
MiR-146a inactivates p38 in endotoxin-adapted cells by inducing MKP-1.
Since it is known that miR-146a contributes to the endotoxin-adapted phenotype in mouse and human monocytes (24, 25, 40), we examined the role of miR-146a in disrupting the p38 phosphorylation axis. We hypothesized that miR-146a expression underlies MKP-1 induction and p38 inactivation during endotoxin adaptation. To test this premise, we knocked down miR-146a in adapted THP-1 cells and measured MKP-1 protein and p38 phosphorylation after LPS stimulation. The knockdown reduced miR-146a levels by ∼80% and concomitantly diminished MKP-1 protein (Fig. 4A) while simultaneously restoring p38 activation after LPS stimulation. Interestingly, we obtained similar results when MKP-1 was knocked down (Fig. 4B), suggesting that MKP-1 is downstream of miR-146a expression. Surprisingly, p38 activation in THP-1 cells after miR-146a or MKP-1 knockdown was markedly increased, even up to 1 h after LPS stimulation compared with responsive cells (Fig. 1). These results support the hypothesis that TLR4-dependent miR-146a induction and sustained MKP-1 protein expression inactivate p38 during endotoxin adaptation.
Fig 4.
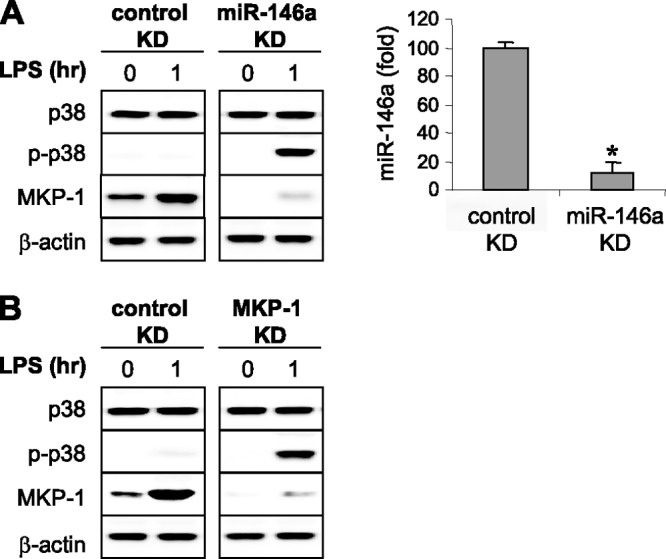
Knockdown of miR-146a or MKP-1 in endotoxin-adapted cells restores p38 MAPK activation. Endotoxin-adapted THP-1 cells were made by pretreatment of responsive cells overnight with 1 μg/ml of LPS. (A) Adapted cells were transfected with anti-miR-146a-specific oligonucleotides (antagomirs) or scrambled anti-miRNA oligonucleotides (control knockdown [KD]). (B) Adapted cells were transfected with a pool of MKP-1-specific or scrambled siRNAs (control KD). Thirty-six hours after transfection of antagomirs or siRNAs, cells were stimulated for 1 h with 1 μg/ml of LPS. Cells were then harvested and expression levels of p38, phospho p38 (p-p38), and MKP-1 were measured by Western blot analysis. The right side of panel A shows miR-146a levels in adapted cells before (control KD) and after its knockdown as determined by real-time PCR. Data are means ± SD of three assays. *, P < 0.05. The results are representative of three experiments.
Knockdown of MKP-1 restores RBM4 phosphorylation and modifies its localization.
We previously reported that translational repression of proinflammatory cytokines in endotoxin-adapted THP-1 cells is induced by the assembly of a translational repressor multicomponent complex, including the RNA-binding proteins Ago2 and RBM4 (25). This repressor complex is gene specific since it does not affect IkBα translation (25; L. Brudecki, D. A. Ferguson, C. E. McCall, and M. El Gazzar, submitted for publication). Whereas we found that Ago2 is constitutively present in the nucleus and cytosol in responsive and adapted THP-1 cells, RBM4 is phosphorylated and shuttles between the nucleus and cytosol in responsive cells but accumulates in the cytosol in adapted cells in an unphosphorylated form, where it interacts with Ago2 and assembles the repressor complex (25). Given that RBM4 localization is regulated by phosphorylation on serine-309 (41, 42), we reasoned that induction of MKP-1 by miR-146a is responsible for RBM4 dephosphorylation and retention in the cytosol in adapted cells. To test this hypothesis, we measured Ago2 and RBM4 proteins in the nucleus and cytosol in adapted THP-1 cells lacking MKP-1. Ago2 was detected in the nucleus and cytosol and its levels did not change after LPS stimulation (Fig. 5). Total RBM4 existed only in the cytosol both before and after LPS stimulation, whereas phospho-serine-309 RBM4 was not detected in the nucleus or cytosol, concurrent with the absence of active p38 (Fig. 1). Importantly, LPS stimulation in cells lacking MKP-1 promoted RBM4 phosphorylation and its re-shuttling between the nucleus and cytosol (Fig. 5). Based on a separate coimmunoprecipitation experiment (data not shown), we observed that anti-total RBM4 antibody did not cross-react with the phosphorylated RBM4, and vice versa. Taken together, these results suggest that MKP-1 induction downstream of miR-146a inactivates p38, thereby leading to accumulation of unphosphorylated RBM4 and assembly of the translation repressor complex.
Fig 5.
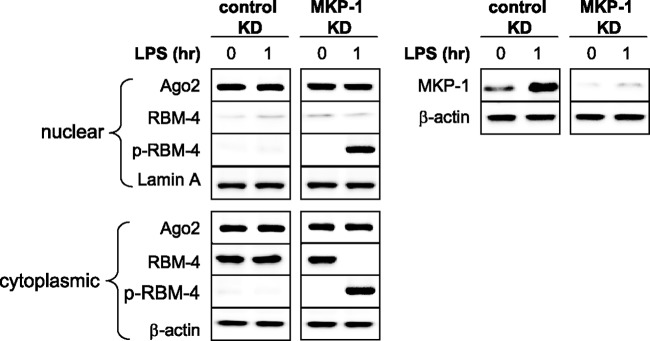
Knockdown of MKP-1 in endotoxin-adapted cells restores RBM4 phosphorylation and modifies its subcellular localization. Endotoxin-adapted THP-1 cells were made by pretreatment of responsive cells overnight with 1 μg/ml of LPS. Adapted cells were transfected with a pool of MKP-1-specific or scrambled siRNAs (control knockdown [KD]). After 36 h, cells were stimulated for 1 h with 1 μg/ml of LPS. Nuclear and cytoplasmic proteins were isolated, and levels of Ago2, RBM4, and p-pRBM4 proteins were measured by Western blot analysis. The right panel shows MKP-1 protein levels before and after the knockdown. The results are representative of three experiments.
Overexpression of miR-146a generates the endotoxin-adapted phenotype and represses proinflammatory cytokine protein synthesis.
To further examine the proximal role of miR-146a in endotoxin adaptation and translation disruption, we overexpressed miR-146a in responsive THP-1 cells using miR-146a mimics. Within 36 h after transfection, miR-146a increased by ∼400-fold (Fig. 6A). Interestingly, MKP-1 protein levels were more rapidly induced (0.5 h) after LPS stimulation than after control transfection. This early MKP-1 induction completely abolished LPS-induced p38 activation (Fig. 6B). In cells without miR-146a overexpression, unphosphorylated RBM4 was detected only in the nucleus, and LPS stimulation induced its phosphorylation and re-shuttling between the nucleus and cytosol (Fig. 6C). This was similar to the pattern observed in endotoxin-adapted cells after MKP-1 knockdown (Fig. 5). In contrast, in cells expressing miR-146a, we did not detect phosphorylated RBM4 in the nucleus or cytosol, but unphosphorylated RBM4 accumulated in the cytosol (Fig. 6C), a result similar to what we observed in adapted cells (Fig. 5). This accumulation paralleled disrupted TNF-α translation (Fig. 6D).
Fig 6.

Overexpression of miR-146a in endotoxin-responsive cells inhibits p38 MAPK activation, prevents RBM4 phosphorylation, and downregulates proinflammatory cytokine synthesis in response to TLR4 stimulation. Endotoxin-responsive THP-1 cells were transfected with an miRNA negative control or miR-146a mimics. After 36 h, cells were stimulated for 0 to 4 h with 1 μg/ml of LPS. (A) Levels of miR-146a were measured after 1 h by real-time PCR. Data are means ± SD of three experiments. *, P < 0.05. (B) Levels of MKP-1, p38, and phospho p38 proteins were measured in whole-cell extract by Western blot analysis. (C) Levels of RBM4 and p-RBM4 (phospho-serine-309) were measured in the nuclear and cytoplasmic fractions by Western blot analysis. (D) After 4 h in LPS, culture supernatants and cells were harvested and lysed, and then TNF-α protein levels were measured by ELISA. Data are means ± SD of three experiments. *, P < 0.05. The results in panels B and C are representative of three experiments.
DISCUSSION
Sustained translational repression of proinflammatory mediators, such as TNF-α, IL-6, and IL-1β, plays a critical role in establishing inflammation adaptation/immunosuppression and perhaps sustaining organ compromise, all of which are prominent features of late severe systemic inflammation in animals and humans (3, 25, 43). In this study, we identified a novel pathway for translation repression following TLR4-mediated responses in a cell model that mimics sepsis. This negative regulatory path requires miR146a-induced expression of the p38 inhibitor MKP-1 and subsequent inactivation of p38, which prevents RBM4 phosphorylation and promotes its cytosolic accumulation and is a rate-limiting contribution to a translational repressor complex. Figure 7 provides a scheme of this autoregulatory path, which is gene specific. Importantly, translation suppression can be reversed by reducing MKP-1 or the upstream miR-146a, providing a strategy for novel therapeutics. Our findings suggest that miR-146a coupled to MKP-1 expression and function is a master checkpoint for switching from TLR4-responsive to TLR4-unresponsive states associated with sepsis in animals (e.g., mice) and humans.
Fig 7.
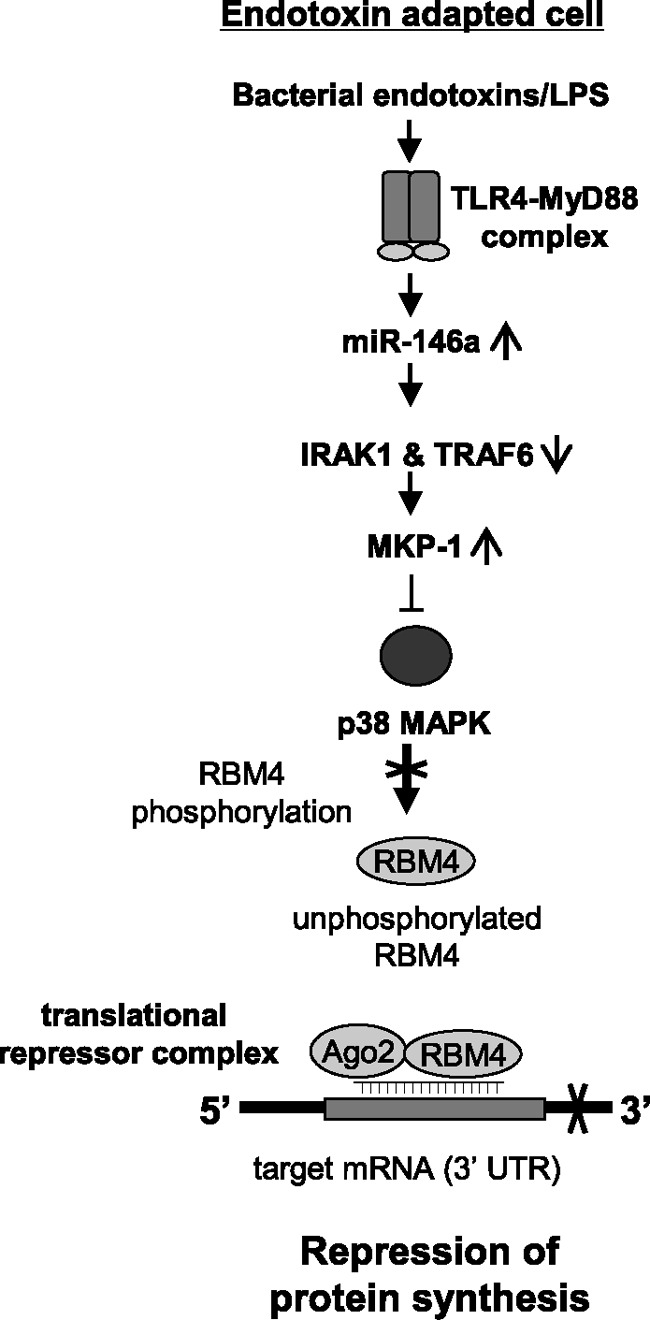
Diagram depicting a TLR4-generated, negative regulatory path leading to repression of proinflammatory protein synthesis during inflammation adaptation. Based on our previous findings and this study, miR-146a expression is induced after the initial activation phase of monocytes/macrophages by bacterial endotoxins and inhibits the signaling proteins IRAK1 and TRAF6. This effect results in the induction of MKP-1, which then dephosphorylates/inactivates p38 MAPK. In the absence of activated p38, phosphorylation of RBM4 is diminished, and thus unphosphorylated RBM4 accumulates in the cytosol in adapted cells, where it binds Ago2 and assembles the translational repressor complex. This complex is recruited to the target mRNA through a target-specific miRNA(s), as part of the complex, and ultimately destabilizes and/or inhibits translation of the target mRNA. In endotoxin-responsive cells, p38 is activated and phosphorylates RBM4, which prevents its binding to Ago2 and assembly of the repressor complex. For simplicity, transcription repression by RelB, which we have described previously (25), is not shown.
RBM4 is a ubiquitously expressed RNA-binding protein that represses mRNA translation by forming repressor complexes with Ago2 (44). Ago2 provides a platform for assembling miRNA-guided translational repressor complexes on target mRNA, and studies suggest that Ago2 (45) or RBM4 (41) is modified to control full assembly of translational repressor complexes. However, we did not observe changes in levels or localization of Ago2 between endotoxin-responsive and -adapted cells, suggesting that endotoxin adaptation may not modify Ago2. In a study similar to ours, Lin et al. (41) found that the Ago2 partner RBM4 is phosphorylated on serine-309 by p38 MAPK in HeLa cells stressed by sodium arsenite exposure, thereby transferring RBM4 to the cytosol and facilitating its binding to Ago2. Previously, we showed that RBM4 is present mainly in the nucleus in responsive/normal THP-1 cells and is phosphorylated and shuttles between the nucleus and cytosol only after the TLR4 response (25). Before TLR4 stimulation, RBM4 does not interact with Ago2, despite the presence of both proteins in the cytosol, during which time TNF-α and IL-1β protein production is normal (25); stimulation of TLR4 then generates the repressor complex. The pattern of RBM4 modification and localization is changed when phospho-serine-309 is lacking, supporting the assembly of the translational repressor complex with Ago2 (25; Brudecki et al., submitted). This study identifies proximal events that spatially and temporally regulate translation.
We found that p38 activation is required to translate proinflammatory TNF-α and IL-6. TLR4 sensing rapidly activates p38 via phosphorylation, but this state is transient due to increasing levels of MKP-1. Consistent with our findings, other studies support the premise that p38 inactivation by an LPS-generated negative regulatory signal inhibits TNF-α and IL-6 protein production in the murine macrophage cell line RAW264.7 (35) and in THP-1 cells (37). Here, we linked this process to the RBM4-Ago2 translational repressor apparatus and identified miR-146a, an NF-κB-dependent miRNA (40), as a proximal coordinator.
MAPKs, including p38, are selectively targeted by MKP-1 phosphatase activity, which potentially dephosphorylates phosphothreonine and phosphotyrosine residues, resulting in inactivation of MAPKs (38, 46). We found that MKP-1 was induced in THP-1 cells at 1 h after LPS stimulation. Although MKP-1 levels decreased slightly over time, MKP-1 accumulated in adapted cells even before a second LPS stimulation, which further increased its levels. MKP-1 induction coincided with dephosphorylation/inactivation of p38, supporting the hypothesis that MKP-1 is primarily responsible for p38 inactivation in our cell model. Importantly, MKP-1 knockdown in endotoxin-adapted THP-1 cells reactivated p38 and restored TNF-α and IL-6 protein levels after LPS stimulation (Fig. 3 and data not shown). Reactivated proinflammatory gene transcription by RelB knockdown did not completely restore TNF-α mRNA levels in endotoxin-adapted cells (Fig. 3), because the newly transcribed mRNA of immediate-response genes is rapidly degraded (27). In the current study, full restoration of TNF-α mRNA and protein translation was observed only when MKP-1 was knocked down along with RelB. Thus, inactivation of p38 by MKP-1 appears to be the primary mechanism for supporting RBM4 compartmentalization and translation suppression of proinflammatory genes during endotoxin adaptation.
Recent studies support the negative regulatory role of MKP-1 during inflammatory responses. Chen et al. (35) reported that overexpression of MKP-1 in the murine macrophages RAW264.7 inactivated p38 and inhibited TNF-α and IL-6 protein production in response to LPS stimulation. Pharmacological inhibition of MKP-1 by triptolide prolongs p38 activation and induces TNF-α and IL-6 production in LPS-stimulated macrophages (35). Based on these and our findings, MKP-1 expression induced by inflammatory stimuli restrains the strength and duration of the acute inflammatory responses (38). MKP-1, unlike the cytosolic dual-specificity phosphatases MKP-3, -4, and -5, is mainly located in the nucleus, where it responds to extracellular stimuli (37, 47). Inactivation of p38 by MKP-1 is likely a direct event, since MKP-1 and p38 can bind to each other (48) and p38 is dephosphorylated and persistently inactivated when MKP-1 levels are sustained (35, 38). Perdiguero et al. (49) reported that inactivating p38 by MKP-1 shifted the early M1 proinflammatory macrophage to an M2 anti-inflammatory/immunosuppressive macrophage during muscle regeneration following injury. This M1-to-M2 shift is similar to the linear phase switching in THP-1 cells and in human and mouse macrophages during sepsis (6, 43).
We and others have established the critical role of miR-146a in the general state of endotoxin adaptation and sepsis by its targeting of IRAK1 and TRAF6 protein expression (24, 25, 40, 50). These proximal signals link to TLR4-dependent NF-κB p65 activation and transactivation of proinflammatory genes. Thus, miR-146a provides a checkpoint to balance inflammatory and immune responses and protect the host against injury. We previously showed that miR-146a knockdown disrupts RBM4-Ago2 interactions and restores TNF-α and IL-1β protein synthesis in response to LPS stimulation (25). Here, we found that the miR-146a knockdown in adapted cells, similar to the MKP-1 knockdown, prolongs reactivation of p38 and that miR-146a overexpression in endotoxin-responsive/normal THP-1 cells induces endotoxin adaptation and represses TNF-α and IL-6 protein synthesis. It is not clear, however, how increases in the miR-146a level support increased expression of MKP-1. Recently, Quinn et al. (51) have shown that miR-146a is also induced by TLR2 activation with bacterial lipoprotein (BLP) and negatively regulates TLR2 by inducing BLP self-tolerance and cross-tolerance to bacteria in THP-1 cells via inhibiting IRAK1 expression. In addition, activation of TLR2 with the bacterial lipopeptide Pam3CSK4 has been shown to inactivate p38 in THP-1 cells and murine bone marrow-derived macrophages (52). In this study, IRAK-M (another IRAK family member, whose activation negatively regulates TLR signaling) was exclusively localized in the cytosol and stabilized MKP-1 protein, resulting in selective inhibition of p38 (52). This effect, however, was independent of the IRAK1 pathway. In this context, IRAK-M-deficient macrophages have been shown to exhibit prolonged MAPK activation and increased inflammatory response and TNF-α production upon stimulation with bacteria or various TLR ligands (53). In addition, a recently reported study demonstrated that IRAK-M expression is upregulated in the human hepatic biliary epithelial cell line HIBEC upon TLR4 stimulation with LPS and contributes to endotoxin tolerance in these cells (54). Thus, it is possible that miR-146a induction by LPS can lead to the inhibition of p38 activation directly by downregulating IRAK1 and/or indirectly by activating IRAK-M to stabilize MAPK-1, which leads to p38 inactivation. Future studies may reveal the full implications of the miR-146a and MKP1 combined checkpoint for inflammation reprogramming and inflammation resolution.
In summary, we have further defined molecular events that inhibit proinflammatory cytokine translation and protein synthesis following the initiation phase of the TLR4-dependent acute inflammatory response. We identified an autoregulatory path prompted by increased miR-146a and MKP1 expression that disrupts cytokine protein synthesis by inactivating p38-dependent control of RBM4 activity. This novel process of translation regulation joins the complex network of events that reprogram chromatin structure to silence transcription of proinflammatory genes and epigenetically activate transcription of genes important in immunosuppression and inflammation resolution. Therapeutic modification at the miR-146a and MKP-1 nexus might rebalance dysregulated inflammation associated with sepsis.
ACKNOWLEDGMENTS
This work was supported in part by National Institutes of Health grant R15 GM100322 (to M.E.) and by funding from the East Tennessee State University College of Medicine.
Footnotes
Published ahead of print 3 July 2013
REFERENCES
- 1.Hotchkiss RS, Karl IE. 2003. The pathophysiology and treatment of sepsis. N. Engl. J. Med. 348:138–150 [DOI] [PubMed] [Google Scholar]
- 2.McCall CE, Yoza BK. 2007. Gene silencing in severe systemic inflammation. Am. J. Respir. Crit. Care Med. 175:763–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Remick DG. 2007. Pathophysiology of sepsis. Am. J. Pathol. 170:1435–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cavaillon JM, Adib-Conquy M. 2006. Bench-to-bedside review: endotoxin tolerance as a model of leukocyte reprogramming in sepsis. Crit. Care 10:233. 10.1186/cc5055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hotchkiss RS, Coopersmith CM, McDunn JE, Ferguson TA. 2009. The sepsis seesaw: tilting toward immunosuppression. Nat. Med. 15:496–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCall CE, Yoza B, Liu T, El Gazzar M. 2010. Gene-specific epigenetic regulation in serious infections with systemic inflammation. J. Innate Immun. 2:395–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cavaillon JM, Adib-Conquy M, Fitting C, Adrie C, Payen D. 2003. Cytokine cascade in sepsis. Scand. J. Infect. Dis. 35:535–544 [DOI] [PubMed] [Google Scholar]
- 8.Sriskandan S, Altmann DM. 2008. The immunology of sepsis. J. Pathol. 214:211–223 [DOI] [PubMed] [Google Scholar]
- 9.Ghisletti S, Huang W, Jepsen K, Benner C, Hardiman G, Rosenfeld MG, Glass CK. 2009. Cooperative NCoR/SMRT interactions establish a corepressor-based strategy for integration of inflammatory and anti-inflammatory signaling pathways. Genes Dev. 23:681–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takeda K, Akira S. 2004. TLR signaling pathways. Semin. Immunol. 16:3–9 [DOI] [PubMed] [Google Scholar]
- 11.Chan C, Li L, McCall CE, Yoza BK. 2005. Endotoxin tolerance disrupts chromatin remodeling and NF-kappaB transactivation at the IL-1beta promoter. J. Immunol. 175:461–468 [DOI] [PubMed] [Google Scholar]
- 12.El Gazzar M, Yoza BK, Hu JY, Cousart SL, McCall CE. 2007. Epigenetic silencing of tumor necrosis factor alpha during endotoxin tolerance. J. Biol. Chem. 282:26857–26864 [DOI] [PubMed] [Google Scholar]
- 13.El Gazzar M, Yoza BK, Chen X, Hu J, Hawkins GA, McCall CE. 2008. G9a and HP1 couple histone and DNA methylation to TNFalpha transcription silencing during endotoxin tolerance. J. Biol. Chem. 283:32198–32208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.El Gazzar M, Yoza BK, Chen X, Garcia BA, Young NL, McCall CE. 2009. Chromatin-specific remodeling by HMGB1 and linker histone H1 silences proinflammatory genes during endotoxin tolerance. Mol. Cell. Biol. 29:1959–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.West MA, Heagy W. 2002. Endotoxin tolerance: a review. Crit. Care Med. 30(1 Suppl):S64–S73 [PubMed] [Google Scholar]
- 16.Brudecki L, Ferguson DA, McCall CE, El Gazzar M. 2012. Myeloid-derived suppressor cells evolve during sepsis and can enhance or attenuate the systemic inflammatory response. Infect. Immun. 80:2026–2034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Munoz C, Misset B, Fitting C, Bleriot JP, Carlet J, Cavaillon JM. 1991. Dissociation between plasma and monocyte-associated cytokines during sepsis. Eur. J. Immunol. 21:2177–2184 [DOI] [PubMed] [Google Scholar]
- 18.McCall CE, Grosso-Wilmoth LM, LaRue K, Guzman RN, Cousart SL. 1993. Tolerance to endotoxin-induced expression of the interleukin-1 beta gene in blood neutrophils of humans with the sepsis syndrome. J. Clin. Invest. 91:853–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biswas SK, Lopez-Collazo E. 2009. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 30:475–487 [DOI] [PubMed] [Google Scholar]
- 20.Fan H, Cook JA. 2004. Molecular mechanisms of endotoxin tolerance. J. Endotoxin Res. 10:71–84 [DOI] [PubMed] [Google Scholar]
- 21.LaRue KE, McCall CE. 1994. A labile transcriptional repressor modulates endotoxin tolerance. J. Exp. Med. 180:2269–2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El Gazzar M, McCall CE. 2010. MicroRNAs distinguish translational from transcriptional silencing during endotoxin tolerance. J. Biol. Chem. 285:20940–20951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mueller LP, Yoza BK, Neuhaus K, Loeser CS, Cousart S, Chang MC, Meredith JW, Li L, McCall CE. 2001. Endotoxin-adapted septic shock leukocytes selectively alter production of sIL-1RA and IL-1beta. Shock 16:430–437 [DOI] [PubMed] [Google Scholar]
- 24.Nahid MA, Pauley KM, Satoh M, Chan EK. 2009. miR-146a is critical for endotoxin-induced tolerance: implication in innate immunity. J. Biol. Chem. 284:34590–34599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.El Gazzar M, Church A, Liu T, McCall CE. 2011. MicroRNA-146a regulates both transcription silencing and translation disruption of TNF-alpha during TLR4-induced gene reprogramming. J. Leukoc. Biol. 90:509–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heagy W, Hansen C, Nieman K, Rodriguez JL, West MA. 2000. Impaired mitogen-activated protein kinase activation and altered cytokine secretion in endotoxin-tolerant human monocytes. J. Trauma 49:806–814 [DOI] [PubMed] [Google Scholar]
- 27.Li L, Cousart S, Hu J, McCall CE. 2000. Characterization of interleukin-1 receptor-associated kinase in normal and endotoxin-tolerant cells. J. Biol. Chem. 275:23340–23345 [DOI] [PubMed] [Google Scholar]
- 28.Su B, Karin M. 1996. Mitogen-activated protein kinase cascades and regulation of gene expression. Curr. Opin. Immunol. 8:402–411 [DOI] [PubMed] [Google Scholar]
- 29.Dong C, Davis RJ, Flavell RA. 2002. MAP kinases in the immune response. Annu. Rev. Immunol. 20:55–72 [DOI] [PubMed] [Google Scholar]
- 30.Lai WS, Parker JS, Grissom SF, Stumpo DJ, Blackshear PJ. 2006. Novel mRNA targets for tristetraprolin (TTP) identified by global analysis of stabilized transcripts in TTP-deficient fibroblasts. Mol. Cell. Biol. 26:9196–9208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salojin KV, Owusu IB, Millerchip KA, Potter M, Platt KA, Oravecz T. 2006. Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. J. Immunol. 176:1899–1907 [DOI] [PubMed] [Google Scholar]
- 32.Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, Strickler JE, McLaughlin MM, Siemens IR, Fisher SM, Livi GP, White JR, Adams JL, Young PR. 1994. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 372:739–746 [DOI] [PubMed] [Google Scholar]
- 33.Carballo E, Lai WS, Blackshear PJ. 1998. Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science 281:1001–1005 [DOI] [PubMed] [Google Scholar]
- 34.Mahtani KR, Brook M, Dean JL, Sully G, Saklatvala J, Clark AR. 2001. Mitogen-activated protein kinase p38 controls the expression and posttranslational modification of tristetraprolin, a regulator of tumor necrosis factor alpha mRNA stability. Mol. Cell. Biol. 21:6461–6469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen P, Li J, Barnes J, Kokkonen GC, Lee JC, Liu Y. 2002. Restraint of proinflammatory cytokine biosynthesis by mitogen-activated protein kinase phosphatase-1 in lipopolysaccharide-stimulated macrophages. J. Immunol. 169:6408–6416 [DOI] [PubMed] [Google Scholar]
- 36.Kotlyarov A, Neininger A, Schubert C, Eckert R, Birchmeier C, Volk HD, Gaestel M. 1999. MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat. Cell Biol. 1:94–97 [DOI] [PubMed] [Google Scholar]
- 37.Nimah M, Zhao B, Denenberg AG, Bueno O, Molkentin J, Wong HR, Shanley TP. 2005. Contribution of MKP-1 regulation of p38 to endotoxin tolerance. Shock 23:80–87 [DOI] [PubMed] [Google Scholar]
- 38.Li L, Chen SF, Liu Y. 2009. MAP kinase phosphatase-1, a critical negative regulator of the innate immune response. Int. J. Clin. Exp. Med. 2:48–67 [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao Q, Wang X, Nelin LD, Yao Y, Matta R, Manson ME, Baliga RS, Meng X, Smith CV, Bauer JA, Chang CH, Liu Y. 2006. MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J. Exp. Med. 203:131–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taganov KD, Boldin MP, Chang KJ, Baltimore D. 2006. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. U. S. A. 103:12481–12486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin JC, Hsu M, Tarn WY. 2007. Cell stress modulates the function of splicing regulatory protein RBM4 in translation control. Proc. Natl. Acad. Sci. U. S. A. 104:2235–2240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin JC, Tarn WY. 2009. RNA-binding motif protein 4 translocates to cytoplasmic granules and suppresses translation via argonaute2 during muscle cell differentiation. J. Biol. Chem. 284:34658–34665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McCall CE, El Gazzar M, Liu T, Vachharajani V, Yoza B. 2011. Epigenetics, bioenergetics, and microRNA coordinate gene-specific reprogramming during acute systemic inflammation. J. Leukoc. Biol. 90:439–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hock J, Weinmann L, Ender C, Rudel S, Kremmer E, Raabe M, Urlaub H, Meister G. 2007. Proteomic and functional analysis of Argonaute-containing mRNA-protein complexes in human cells. EMBO Rep. 8:1052–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rudel S, Meister G. 2008. Phosphorylation of Argonaute proteins: regulating gene regulators. Biochem. J. 413:e7–e9 [DOI] [PubMed] [Google Scholar]
- 46.Keyse SM. 2000. Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Curr. Opin. Cell Biol. 12:186–192 [DOI] [PubMed] [Google Scholar]
- 47.Shanley TP. 2002. Phosphatases: counterregulatory role in inflammatory cell signaling. Crit. Care Med. 30(1 Suppl):S80–S88 [PubMed] [Google Scholar]
- 48.Hutter D, Chen P, Barnes J, Liu Y. 2000. Catalytic activation of mitogen-activated protein (MAP) kinase phosphatase-1 by binding to p38 MAP kinase: critical role of the p38 C-terminal domain in its negative regulation. Biochem. J. 352(Pt 1):155–163 [PMC free article] [PubMed] [Google Scholar]
- 49.Perdiguero E, Sousa-Victor P, Ruiz-Bonilla V, Jardi M, Caelles C, Serrano AL, Munoz-Canoves P. 2011. p38/MKP-1-regulated AKT coordinates macrophage transitions and resolution of inflammation during tissue repair. J. Cell Biol. 195:307–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boldin MP, Taganov KD, Rao DS, Yang L, Zhao JL, Kalwani M, Garcia-Flores Y, Luong M, Devrekanli A, Xu J, Sun G, Tay J, Linsley PS, Baltimore D. 2011. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med. 208:1189–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quinn EM, Wang JH, O'Callaghan G, Redmond HP. 2013. MicroRNA-146a is upregulated by and negatively regulates TLR2 signaling. PLoS One 8:e62232. 10.1371/journal.pone.0062232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Su J, Xie Q, Wilson I, Li L. 2007. Differential regulation and role of interleukin-1 receptor associated kinase-M in innate immunity signaling. Cell. Signal. 19:1596–1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kobayashi K, Hernandez LD, Galan JE, Janeway CA, Jr., Medzhitov R, Flavell RA. 2002. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 110:191–202 [DOI] [PubMed] [Google Scholar]
- 54.Harada K, Isse K, Sato Y, Ozaki S, Nakanuma Y. 2006. Endotoxin tolerance in human intrahepatic biliary epithelial cells is induced by upregulation of IRAK-M. Liver Int. 26:935–942 [DOI] [PubMed] [Google Scholar]