

Abstract
ExsA activates type III secretion system (T3SS) gene expression in Pseudomonas aeruginosa and is a member of the AraC family of transcriptional regulators. AraC proteins contain two helix-turn-helix (HTH) DNA binding motifs. One helix from each HTH motif inserts into the major groove of the DNA to make base-specific contacts with the promoter region. The amino acids that comprise the HTH motifs of ExsA are nearly identical to those in LcrF/VirF, the activators of T3SS gene expression in the pathogenic yersiniae. In this study, we tested the hypothesis that ExsA/LcrF/VirF recognize a common nucleotide sequence. We report that Yersinia pestis LcrF binds to and activates transcription of ExsA-dependent promoters in P. aeruginosa and that plasmid-expressed ExsA complements a Y. pestis lcrF mutant for T3SS gene expression. Mutations that disrupt the ExsA consensus binding sites in both P. aeruginosa and Y. pestis T3SS promoters prevent activation by ExsA and LcrF. Our combined data demonstrate that ExsA and LcrF recognize a common nucleotide sequence. Nevertheless, the DNA binding properties of ExsA and LcrF are distinct. Whereas two ExsA monomers are sequentially recruited to the promoter region, LcrF binds to promoter DNA as a preformed dimer and has a higher capacity to bend DNA. An LcrF mutant defective for dimerization bound promoter DNA with properties similar to ExsA. Finally, we demonstrate that the activators of T3SS gene expression from Photorhabdus luminescens, Aeromonas hydrophila, and Vibrio parahaemolyticus are also sensitive to mutations that disrupt the ExsA consensus binding site.
INTRODUCTION
The pathogenic lifestyles of Pseudomonas aeruginosa, Yersinia enterocolitica, Yersinia pestis, and Yersinia pseudotuberculosis are each dependent upon a type III secretion system (T3SS) (1). The T3SS is thought to function like a molecular syringe to inject effector proteins into host cells (2). Although the T3SS structural components are highly conserved between P. aeruginosa and the yersiniae, the repertoires of translocated effectors are distinct. A primary role for the effector proteins from each of these organisms is to inhibit phagocytosis, thereby allowing the bacteria to replicate to high numbers and overwhelm the host (1, 3, 4). T3SSs have also been described as contact-dependent secretion systems because physical contact between the bacterium and the host cell serves as a signal to initiate translocation of the effectors and to induce high levels of T3SS gene expression (5). The host contact signal can be mimicked by growing P. aeruginosa or the yersiniae under calcium-limiting conditions (6). Host contact and calcium limitation also trigger the secretion/translocation of regulatory proteins that inhibit T3SS gene expression (7). This feature serves as an efficient mechanism to couple gene expression to secretory activity. Despite the many similarities between the P. aeruginosa and yersiniae T3SSs, however, the mechanisms involved in regulating T3SS gene expression are distinct.
The primary transcriptional activator of T3SS gene expression in P. aeruginosa is ExsA (8). ExsA-dependent transcription is activated in response to inducing conditions (i.e., low Ca2+ or host cell contact) by a partner-switching mechanism involving three additional proteins: ExsC, ExsD, and ExsE (7). ExsD functions as an antiactivator by directly binding to the amino-terminal domain (NTD) of ExsA and inhibiting ExsA-dependent transcription (9, 10). ExsC is an anti-antiactivator that binds to and antagonizes the inhibitory activity of ExsD (11–13). The final component of the cascade, ExsE, is a secreted substrate of the type III export machinery. ExsE also binds to ExsC and prevents formation of the ExsC-ExsD complex (14, 15). The current model proposes that nonpermissive conditions (i.e., high Ca2+) prevent T3SS gene expression through formation of the inhibitory ExsD-ExsA and ExsC-ExsE complexes (16). Conversely, inducing conditions trigger ExsE secretion and the partner-switching mechanism whereupon formation of the ExsD-ExsC complex is favored and free ExsA is available to activate transcription.
The yersiniae have an ExsA homolog (LcrF/VirF) that also activates T3SS gene expression but lack homologs of ExsE, ExsC, and ExsD and rely upon alternative mechanisms to regulate LcrF-dependent transcription. Expression of the yersiniae T3SS is induced in response to elevated temperature (i.e., 37°C). This induction is due, at least in part, to thermoregulation of LcrF expression and occurs at both the transcriptional and posttranscriptional level. LcrF is encoded as the last gene of the yscW-lcrF operon, with expression of the operon being directly repressed by YmoA, a small nucleoid-associated protein (17–19). YmoA is degraded by the Clp and Lon proteases at 37°C, thereby resulting in increased yscW-lcrF transcription (20). Posttranscriptional control of LcrF expression involves an inhibitory secondary structure that forms in the 5′-untranslated leader region of the lcrF mRNA and prevents LcrF translation at moderate temperatures (19, 21). At 37°C, the mRNA undergoes a structural alteration that provides ribosomal access to the Shine-Dalgarno sequence, resulting in elevated LcrF translation. Neither transcriptional nor posttranscriptional regulation of LcrF expression, however, is directly linked to the activity of the secretion machinery. Instead, T3SS gene expression in the yersiniae is coupled to secretory activity via the combined action of two substrate chaperone complexes (YopD-SycD and LcrQ-SycH) that function, in part, to suppress T3SS gene expression prior to activation of the secretion process (22, 23). Although the mechanism by which these regulatory complexes suppress T3SS gene expression is not fully understood, recent studies suggest that these complexes directly interact with the 5′-untranslated regions of target mRNAs to block translation and enhance mRNA degradation (24). Upon contact with a eukaryotic cell, secretion of YopD and LcrQ is triggered, resulting in the disassembly of the YopD-SycD and LcrQ-SycH negative regulatory complexes and facilitating high-level T3SS gene expression. Secretion is further regulated by the YopN-SycN-YscB-TyeA complex that acts as a plug by blocking the Ysc secretion channel and preventing YopN transport in the presence of calcium and prior to contact with eukaryotic cells (25). Upon induction (i.e., low calcium/host cell contact), free YopN is subsequently secreted through an open channel, allowing Yop effectors to freely travel through the injectisome and into the host cell.
ExsA and LcrF are both members of the AraC/XylS family of transcriptional regulators. Prototypical AraC/XylS proteins consist of two distinct domains (amino- and carboxy-terminal domains [CTD]) separated by a flexible linker (26). The amino-terminal domain of AraC proteins is usually involved in self-association. Previous studies have established that ExsA is monomeric in solution but self-associates as a dimer when bound to DNA (27). In contrast, purified VirF/LcrF is thought to be dimeric in solution (28). In some cases, the NTD of AraC proteins can also serve as in input for regulatory signals. For instance, binding of arabinose to the NTD of AraC results in activation of the PBAD promoter (29). Conversely, binding of ExsD to the NTD of ExsA inhibits DNA binding activity (9, 30). A regulatory role for the NTD of LcrF has not been described.
The carboxy-terminal domain of ExsA contains two helix-turn-helix (HTH) DNA binding motifs (26). Each HTH has a recognition helix that makes base-specific contacts with the target DNA (i.e., promoter region). Recent studies have characterized the interaction of ExsA with several P. aeruginosa T3SS promoters. Each ExsA-dependent promoter consists of two adjacent binding sites for monomeric ExsA (27). Binding site 1 is centered ∼41 bp upstream of the transcription start, and binding site 2 is centered at the −65 position. An alignment of all 10 ExsA-dependent promoter sequences identified a consensus ExsA binding site, AaAAAnwmMygrCynnnmTGayAk, with the nucleotide positions indicated in bold typeface being required for maximal ExsA binding (27) and the uppercase letters representing more highly conserved positions than the lowercase letters. The conserved GnC and TGnnA sequences constitute binding site 1 and are recognized by the two HTH motifs of a single ExsA monomer (31). The adenine-rich sequence is positioned within binding site 2. The precise nature of the interaction of ExsA with binding site 2, however, is unclear and appears to differ between promoters (32). Occupation of the promoter region by ExsA occurs in an ordered manner whereby one ExsA monomer binds to site 1 and then recruits a second monomer to site 2. Efficient occupation of site 2 requires self-association of the ExsA monomers that is mediated by the NTD (33). The ExsA monomers are positioned in a head-to-tail orientation when bound to sites 1 and 2 (31). Once bound to the promoter region, ExsA activates transcription by recruiting the RNA polymerase-σ70 complex to the promoter (34, 35).
We hypothesized that the detailed information available regarding ExsA binding activity might be applicable to the interaction of LcrF/VirF with promoter DNA. Using promoter fusions, mutagenesis, and in vitro DNA binding assays, we find that LcrF binds to and activates transcription of ExsA-dependent promoters in P. aeruginosa and that plasmid-expressed ExsA complements a Y. pestis lcrF mutant for T3SS gene expression. Promoter mutations that disrupt ExsA-dependent activation also inhibit activation by LcrF, suggesting that ExsA and LcrF recognize a common DNA sequence. In support of this conclusion, each of the core T3SS promoter regions from the yersiniae possesses well-defined ExsA consensus binding sites. Our findings with LcrF are also applicable to the activators of T3SS gene expression from Photorhabdus luminescens, Aeromonas hydrophila, and Vibrio parahaemolyticus.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The bacterial strains used in this study are provided in Table S1 in the supplemental material. Escherichia coli strains were cultured in LB-Miller (LB) broth or agar supplemented with ampicillin (100 μg/ml) or gentamicin (15 μg/ml) as required. P. aeruginosa strains were cultured on Vogel-Bonner minimal medium agar containing gentamicin (100 μg/ml) and/or carbenicillin (300 μg/ml) as required (36). To measure T3SS gene expression, P. aeruginosa strains were cultured at 30°C in Trypticase soy broth (TSB) supplemented with 100 mM monosodium glutamate, 1% glycerol, and 2 mM EGTA to an optical density at 600 nm (OD600) of approximately 1.0. β-Galactosidase activity was measured using the 2-nitrophenyl-β-d-galactopyranoside substrate as previously described (12). All values reported within this study represent the average from at least three independent experiments.
The Y. pestis strains used in this study are Pgm− and avirulent by peripheral routes of infection (37). Y. pestis KIM5-3001 and derivatives of these strains were routinely grown in heart infusion broth (HIB) liquid medium or on tryptose blood agar (TBA) plates at a temperature of 28°C. For secretion experiments, Y. pestis strains were grown in the presence or absence of 2.5 mM calcium chloride in thoroughly modified Higachi's (TMH) defined medium as previously described (25).
SDS-PAGE and immunoblotting.
Whole-cell lysates of P. aeruginosa were prepared by harvesting 1.25 ml of cell culture (OD600 of 1.0) by centrifugation (16,000 × g, 5 min, 23°C), suspending in 0.25 ml 2× SDS-PAGE sample buffer, and sonicating for 5 s. Secreted protein samples were prepared from 1 ml of cell-free culture supernatant fluid by adding 350 μl of 50% trichloroacetic acid and incubating overnight at 4°C. Precipitated protein was collected by centrifugation (16,000 × g, 15 min, 23°C), washed with acetone, dried, and suspended in 15 μl 2× SDS-PAGE sample buffer. Samples were analyzed by 15% SDS-PAGE and subjected to immunoblotting or silver staining.
Construction of lcrF deletion mutants.
Deletion of the lcrF coding sequence and insertion of a kan (KIM5-3233-F2) or dhfr (KIM5-3001-F1) gene was accomplished using lambda red-mediated recombination essentially as described by Datsenko and Wanner (38). PCR products used to construct gene replacements were generated from template plasmid pKD4 (kan) or by using the EZ::TN<DHFR> transposon (Epicentre, Madison, WI) as a template for PCR. Primers P1 and P2 (see Table S2 in the supplemental material) were used to amplify the kan and dhfr PCR products. Y. pestis KIM5-3001 and KIM5-3233 were electroporated with pKD46, encoding the red recombinase. Y. pestis strains carrying pKD46 were grown in HIB at 28°C to an OD620 of 0.5 and then for 2 h with 0.2% l-arabinose. Electrocompetent cells were prepared as previously described and electroporated with purified PCR products (39). Gene replacements were confirmed by PCR using oligonucleotides LcrF-F1 and LcrF-R1. The FLP recombination target (FRT)-flanked kan cassette was removed via FRT-mediated recombination using plasmid pCP20 as previously described (40). Plasmids pKD46 and pCP20 were cured from the Y. pestis deletion mutants by overnight growth at 39°C.
Plasmid construction and site-directed mutagenesis.
All of the reporter and plasmid constructs used in this study are provided in Tables S3 and S4, respectively, in the supplemental material. The pBAD30-LcrF expression vector was constructed by amplifying a 0.9-kb LcrF-encoding DNA fragment amplified from plasmid pCD1 (41) using PCR primers LcrF-KpnI (TTTGGTACCTTTAGATTTTTAGGACAGTAT) and LcrF-HindIII (TTTAAGCTTACTTTATAGTCCAAAAGTGTC) (see Table S2 in the supplemental material). The PCR product was cloned into the KpnI/HindIII restriction sites of pBAD30 (42), generating plasmid pBAD30-LcrF. The LcrF expression vector (pJK32) was constructed by first performing site-directed mutagenesis on pBAD30-LcrF (QuikChange system) using primer pair 81126787-81126788 (see Table S2) to destroy the NdeI restriction site (through introduction of a silent mutation) within lcrF. The resulting plasmid (pJK29) was used in a subsequent PCR with primer pair 81559809-81126789 (see Table S4) to amplify lcrF. The PCR product was cloned into the NdeI/SacI restriction sites of pEB131, which replaces the exsA coding sequence with lcrF, resulting in pJK32. To generate the pET16blcrF expression plasmid, primer pair 81559809-82001905 (see Table S4) was used to amplify lcrF, and the resulting PCR product was cloned into NdeI/BamHI restriction sites in pET16b. To generate the pVxsA plasmid, vxsA was PCR amplified from V. parahaemolyticus genomic DNA using primer pair 85928866-86519886 (see Table S4), and the PCR product was cloned into pEB131 vector so that translation was controlled by the native ExsA ribosomal binding site.
The Y. pestis transcriptional reporters were made by PCR amplifying the PyopN, PlcrG, and PyscN promoter regions using the primer sets indicated in Table S3 in the supplemental material. PCR products were cloned into mini-CTX-lacZ as EcoRI/HindIII restriction fragments and integrated onto the PA103 chromosome as previously described (43). PyscN promoter mutations were introduced using a two-step PCR method. The PyscN 5′ Hind primer (88203767) and the specific primers indicated in Table S2 in the supplemental material (minictxJK503-511) were used to generate megaprimers. The megaprimers were used in a second PCR with the PyscN 3′ Eco primer. The resulting PCR products were cloned as HindIII/EcoRI restriction fragments into mini-CTX-lacZ and integrated onto the PA103 chromosome as described above.
Protein expression and purification.
E. coli Tuner(DE3) carrying either the pET16bexsA or pET16blcrF expression vector was grown at 30°C in LB supplemented with ampicillin (200 μg/ml). When the culture OD600 reached 0.5, isopropyl-β-d-thiogalactopyranoside (IPTG) (1 mM final concentration) was added to induce ExsA or LcrF expression. Cultures were incubated an additional 2 to 4 h at 30°C. Cells were harvested by centrifugation (6,000 × g, 10 min, 4°C) and suspended in 30 ml ExsA buffer (20 mM Tris-HCl [pH 8.0], 500 mM NaCl, 20 mM imidazole, 0.5% Tween 20) supplemented with two protease inhibitor cocktail tablets (Roche Diagnostics, Indianapolis, IN). Cells were lysed using a Microfluidizer (Microfluidics, Newton, MA), and the suspension was centrifuged (20,000 × g, 20 min, 4°C) to remove cell debris. ExsA and LcrF were purified from the cleared lysates using Ni2+-affinity chromatography and dialyzed overnight at 4°C in ExsA buffer lacking imidazole and supplemented with 1 mM dithiothreitol (DTT) as previously described (27). Protein concentration was determined using a BCA protein assay (Thermo Scientific, Rockford, IL).
Circular permutation and EMSAs.
Probes for the circular permutation assays and the methodology were performed as previously described (27). DNA probes for electrophoretic mobility shift assays (EMSAs) were generated by standard PCR using the primer pairs listed in Table S5 in the supplemental material. Specific promoter probes containing the ExsA or LcrF binding sites were ∼200 bp, and the nonspecific probe (160 bp) was derived from the algD promoter region. The nonspecific portions of the probes described in Fig. 5B were also derived from the algD promoter region. PCR products were gel purified using the QIAquick gel extraction method (Qiagen, Valencia, CA). Short promoter probes (∼50 bp) were generated by annealing complementary oligonucleotides (25 pmol each) diluted in duplex buffer (30 mM HEPES [pH 7.5], 100 mM potassium acetate; 50 μl final volume). The primer mixture was heated to 95°C for 5 min and gradually cooled to 25°C at a rate of 1°C/min. Reactions were purified using the Qiagen nucleotide removal kit. Promoter probes were end labeled with 10 μCi of [γ-32P]ATP as previously described (27). EMSA reactions (20 μl) contained 0.06 nM of the nonspecific and/or specific probes, 10 μl 2× DNA binding buffer (20 mM Tris [pH 7.5], 100 mM KCl, 2 mM EDTA, 2 mM DTT, and 10% glycerol), 25 ng/μl poly(2′-deoxyinosinic 2′-deoxycytidylic acid), and 100 μg/ml bovine serum albumin. Reaction mixtures were incubated at room temperature for 5 min, and then ExsA or LcrF was added at the specified concentrations. Reaction mixtures were incubated at room temperature for 15 min and analyzed on 5% polyacrylamide glycine gels. Gels were analyzed with an FLA-7000 phosphorimager (Fujifilm) and Multigage version 3.0 software (Fujifilm).
Fig 5.

LcrF binds to the P. aeruginosa PexoT promoter probe as a dimer. (A) EMSA using 50-bp radiolabeled probes derived from the ExsA-dependent PexoT promoter. Probes (0.05 nM each) were incubated in the presence of 12, 36, 108, 324 nM LcrF (lanes 2 to 5) or LcrFm (lanes 6 to 10) for 15 min at 25°C. Samples were analyzed by native polyacrylamide gel electrophoresis and phosphorimaging. (B) Diagram depicting PexoT promoter probes with truncations that destroy binding sites 1 and/or 2. Solid and dotted lines represent native and nonnative PexoT sequences, respectively. The adenine-rich region and conserved GnC and TGnnA sequences are indicated in bold. (C) EMSA using 60-bp radiolabeled probes derived from the ExsA-dependent PexoT promoter. Probes (0.05 nM each) were incubated in the presence of 90 nM ExsA(A), LcrF(F), or LcrFm for 15 min at 25°C. Samples were analyzed and imaged as described above.
Cross-linking experiments.
Purified LcrF was exchanged into cross-linking buffer (20 mM HEPES [pH 7.9], 500 mM NaCl, 0.5% Tween 20, 1 mM DTT) using Micro Bio-Spin 6 columns (Bio-Rad). Cross-linking reactions were performed in cross-linking buffer by incubating LcrF (800 nM) with the indicated concentration of Sulfo-EGS [ethylene glycol bis(sulfosuccinimidylsuccinate)], DSS (disuccinimidyl suberate), or DMP (dimethyl pimelimidate) for 60 min. Samples were immediately loaded onto a 12% SDS-polyacrylamide gel and subjected to immunoblot analyses using LcrF polyclonal antiserum.
RESULTS
LcrF activates T3SS gene expression in P. aeruginosa.
The AraC family of transcriptional regulators is characterized by the presence of a DNA binding domain consisting of two helix-turn-helix (HTH) motifs (26). The first helices of each HTH motif (termed the recognition helix) serve as the primary determinants of binding specificity and function by inserting into adjacent major grooves of the DNA to make base-specific contacts with the promoter region (44). We observed that the amino acid sequences comprising the recognition helices of ExsA are virtually identical to the activators of T3SS gene expression (LcrF/VirF) in the pathogenic yersiniae (see Fig. S1 in the supplemental material). Based on this observation, we hypothesized that LcrF/VirF recognizes a nucleotide sequence similar to the ExsA consensus binding site. To test this idea, we performed a complementation experiment by introducing an LcrF expression plasmid (pLcrF) into a PA103 exsA::Ω mutant carrying the ExsA-dependent PexsC-lacZ, PexsD-lacZ, or PexoT-lacZ transcriptional reporter. The resulting strains were cultured under noninducing (high Ca2+/−EGTA) and inducing (low Ca2+/+EGTA) conditions for T3SS gene expression and assayed for β-galactosidase activity. As shown in Fig. 1A to C, plasmid-expressed ExsA and LcrF both complemented the exsA::Ω mutant for expression of the PexsC-lacZ, PexsD-lacZ, and PexoT-lacZ reporters with two notable differences. First, LcrF-dependent reporter activity was significantly elevated compared to ExsA-dependent activity. Second, whereas activation of the reporters by ExsA was further enhanced when cells were grown under low Ca2+ conditions (3-, 7-, and 7-fold for PexsC-lacZ, PexsD-lacZ, and PexoT-lacZ, respectively), activation of the same reporters by LcrF was only modestly elevated in the absence of Ca2+ (1.3-, 1.4-, and 1.4-fold, respectively). A trivial explanation for the LcrF-dependent increase in reporter activity is that the steady-state expression levels of LcrF are elevated relative to ExsA. Immunoblotting of whole-cell extracts using purified LcrF and ExsA as standards, however, indicated that the amounts of LcrF and ExsA are similar in P. aeruginosa (see Fig. S2 in the supplemental material).
Fig 1.
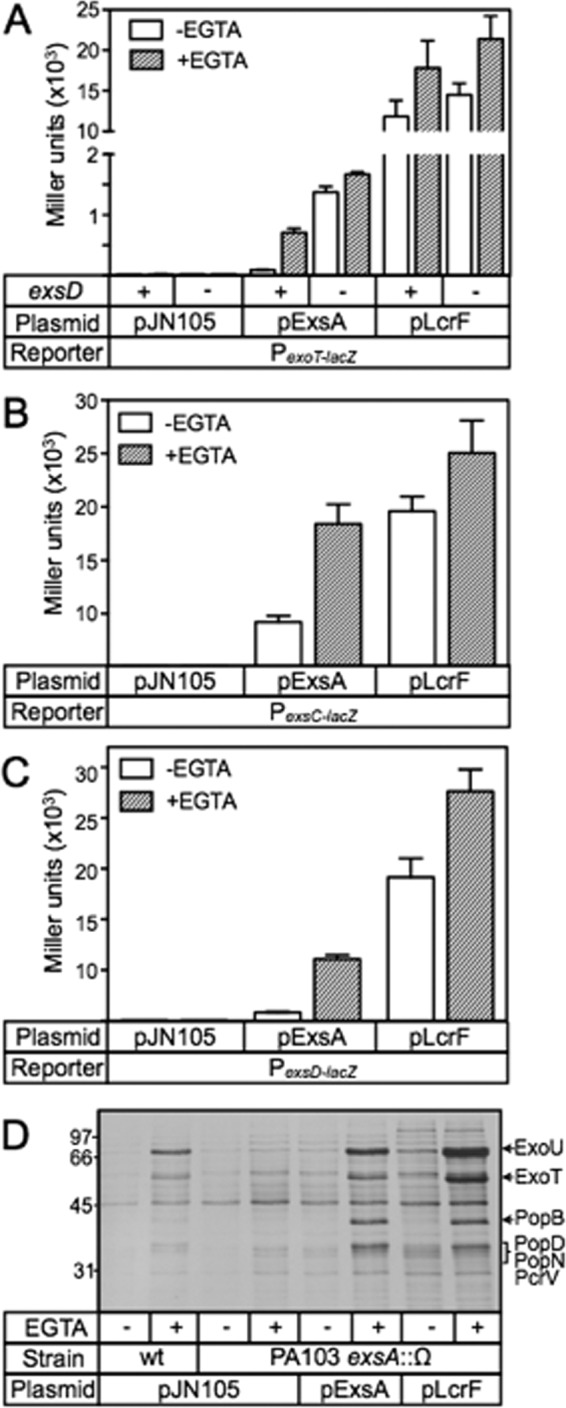
LcrF complements an exsA mutant for T3SS gene expression. (A to C) The PA103 exsA::Ω strain carrying either the PexoT-lacZ (A), PexsC-lacZ (B), or PexsD-lacZ (C) transcriptional reporter was transformed with a vector control (pJN105), an ExsA expression vector (pExsA), or an LcrF expression vector (pLcrF). The resulting strains were cultured under noninducing (−EGTA, open bars) or inducing (+EGTA, hatched bars) conditions for T3SS gene expression and assayed for β-galactosidase activity as reported in Miller units. *, P < 0.001; ** P < 0.01. (D) Silver-stained gel of concentrated culture supernatant fluid prepared from wild-type PA103 or the PA103 exsA::Ω strain carrying the indicated plasmids following growth under noninducing (−EGTA) or inducing (+EGTA) conditions for T3SS gene expression. The positions of molecular mass standards are indicated on the left side of the gel, and the type III secreted proteins ExoU, ExoT, PopB, PopD, PopN, and PcrV are labeled on the right side of the gel (49).
Since the increase in LcrF-dependent reporter activity could not be linked to elevated LcrF levels, we next addressed the possibility that LcrF is insensitive to the ExsD antiactivator. ExsD inhibits ExsA-dependent activation under high Ca2+ conditions but is largely inactive under low Ca2+ conditions, owing to the action of the ExsC/ExsE regulatory cascade (16). In the absence of exsD, therefore, ExsA-dependent activation of the PexoT-lacZ reporter is derepressed and largely insensitive to Ca2+ levels (Fig. 1A). LcrF-dependent activation of the PexoT-lacZ reporter, however, was similar in both the presence and absence of exsD (Fig. 1A). Based on this finding, we conclude that LcrF is not regulated by ExsD, thereby accounting for the insensitivity of LcrF to Ca2+ levels.
The P. aeruginosa T3SS regulon consists of 10 ExsA-dependent promoters that control the expression of the secretion machinery, effectors, chaperones, and translocator proteins (45). To determine whether plasmid-expressed LcrF activates the entire T3SS regulon, culture supernatant samples were collected from cells grown under noninducing and inducing conditions for T3SS gene expression and subjected to SDS-PAGE and silver staining. As shown in Fig. 1D, expression of either ExsA or LcrF in the exsA mutant resulted in elevated secretion of the ExoU/ExoT effectors, the PopB/PopD/PcrV translocators, and the PopN regulator relative to the vector control (pJN105). The finding that LcrF supported a higher level of secretion than ExsA is consistent with the reporter data presented in Fig. 1A to C and indicates that LcrF activates all 10 of the ExsA-dependent promoters.
LcrF DNA binding properties.
We next compared the DNA binding properties of ExsA and LcrF using electrophoretic mobility shift assays (EMSAs). ExsA and LcrF were expressed as amino-terminal histidine-tagged fusion proteins in E. coli and purified by Ni2+-affinity chromatography (see Fig. S3A in the supplemental material). To independently confirm a previous report that purified LcrF is dimeric (28), we performed cross-linking studies using a panel of amine-reactive cross-linkers of various spacer arm lengths (DSS, DMP, and Sulfo-EGS). Purified LcrF (with a molecular mass of 30 kDa) was incubated with the cross-linkers for 60 min, electrophoresed on denaturing polyacrylamide gels, and immunoblotted for LcrF. Whereas incubation of LcrF with either Sulfo-EGS (see Fig. S3B) or DMP (data not shown) resulted in a large amount of an ∼60-kDa cross-linked species, the cross-linked species was absent from the sample containing the vehicle (dimethyl sulfoxide [DMSO]) alone. In contrast, cross-linking experiments with LcrFm, a monomeric variant described below, resulted in only small amounts of the ∼60-kDa cross-linked species, thereby confirming that purified LcrF is dimeric in solution, while LcrFm is primarily monomeric.
EMSAs were performed by incubating ExsA or LcrF with a nonspecific (160-bp) control probe derived from the algD promoter region and a specific (∼200-bp) probe derived from the ExsA-dependent PexsC, PexoT, and PexsD promoters. Samples were subjected to electrophoresis on nondenaturing polyacrylamide gels and phosphorimaging. Previous studies with ExsA found that the PexsC, PexoT, and PexsD promoter regions each contain two binding sites for monomeric ExsA (27, 31). Binding to the PexoT and PexsD promoters occurs through a monomer assembly pathway whereby one ExsA monomer binds to site 1 (resulting in shift product 1 in Fig. 2C and E) and then recruits another ExsA monomer to a second binding site (site 2) that is positioned just upstream of site 1 (represented as shift product 2). ExsA likely binds to the PexsC promoter via monomer assembly as well but occurs in a highly cooperative manner such that the binding kinetics are very rapid, thereby resulting in the formation of an abundance of shift product 2 and only fleeting amounts of shift product 1 (Fig. 2A).
Fig 2.

DNA binding properties of purified ExsA and LcrF. EMSAs were performed using radiolabeled probes derived from the ExsA-dependent PexsC (A and B), PexoT (C and D), and PexsD (E and F) promoters. A nonspecific probe (Non-Sp) derived from the algD promoter region was included in all binding reactions as a negative control. Probes (0.05 nM each) were incubated in the absence or presence of 11, 23, 45, 90, 180, or 360 nM ExsA (A, C, and E) or LcrF (B, D, and F) for 15 min at 25°C. Samples were analyzed by native polyacrylamide gel electrophoresis and phosphorimaging. The positions of shift products 1 and 2 are indicated. The asterisk indicates background shifting of the nonspecific probe by LcrF.
Whereas ExsA binding resulted in the formation of shift products 1 and 2, LcrF binding resulted in a single predominant shift product when bound to the PexoT, PexsD, and PexsC promoter probes (Fig. 2B, D, and F). Lower mobility complexes were also evident with LcrF, but the precise nature of those complexes was not examined. For reasons described below, the predominant shift products formed by LcrF were designated shift product 2. The mobilities of shift product 2 formed by ExsA and LcrF were indistinguishable from one another when using the PexsC promoter probe (Fig. 2A and B, lanes 8 and 9). Since both the isoelectric points (8.6 and 9.1) and molecular masses (31.6 and 30.8 kDa) of ExsA and LcrF are similar, this finding suggested that shift product 2 represents two molecules bound to the PexsC promoter probe. The manner in which shift product 2 is formed, however, differs in that formation of shift product 2 by ExsA results from the sequential binding of two ExsA monomers, whereas occupation by LcrF results from the binding of a single LcrF dimer.
In contrast to our findings for the PexsC promoter probe, the primary LcrF promoter probe complexes observed for PexoT and PexsD (shift product 2) had reduced mobility compared to that of shift product 2 generated by ExsA (Fig. 2C to F, lanes 8 and 9). In a previous study, we found that the PexsC, PexsD, and PexoT promoter probes, while not inherently bent, do bend when bound by ExsA (27). While ExsA and LcrF appear to bend the PexsC promoter probe to a similar level (Fig. 2A and B), we hypothesized that LcrF bends the PexoT and PexsD promoter probes to a higher degree than ExsA and that this increase in bending accounted for the difference in the mobility of shift product 2. To test this idea, we performed EMSAs using smaller 50-bp promoter probes that were previously shown to negate the effect that ExsA-dependent bending has on mobility owing to their smaller size (3). When using the smaller PexsC, PexsD, and PexoT promoter probes, the mobilities of shift product 2 formed by ExsA and LcrF were similar in each case (Fig. 3A to C, lanes 3 and 7). This finding suggested that the reduced mobility of the LcrF promoter probe complexes observed in Fig. 2 results from increased DNA bending by LcrF relative to ExsA.
Fig 3.
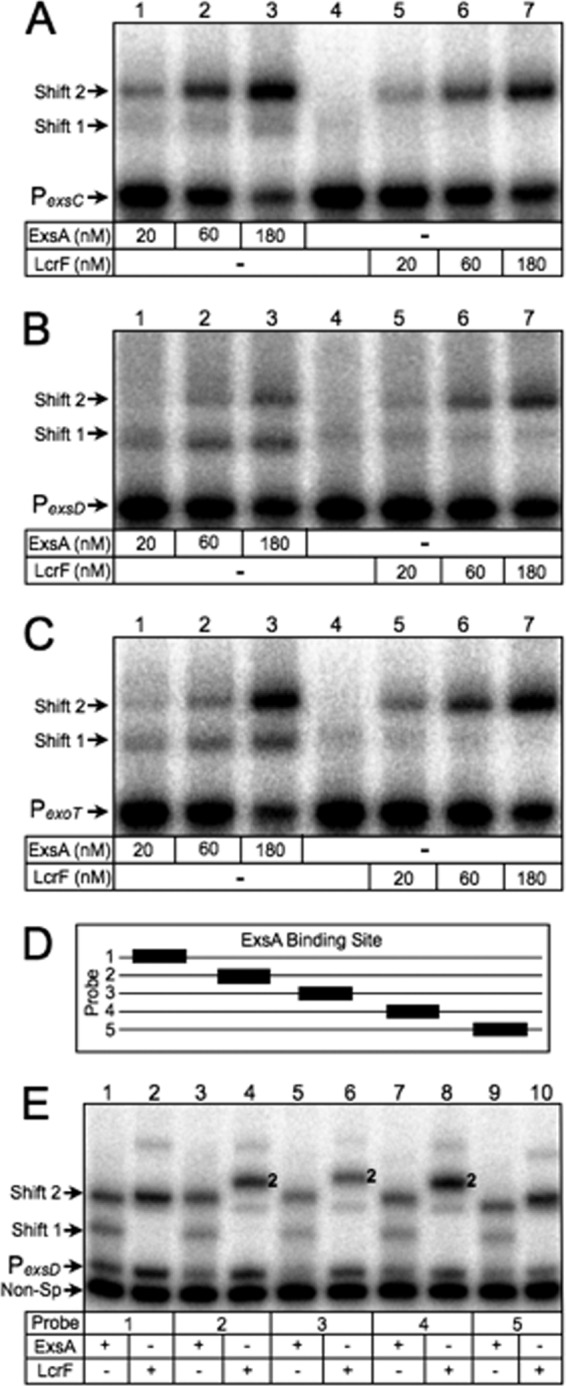
DNA bending properties of ExsA and LcrF. (A to C) EMSAs using 50-bp radiolabeled probes derived from the ExsA-dependent PexsC (A), PexsD (B), and PexoT (C) promoters. Probes (0.05 nM each) were incubated in the presence of 20, 60, or 180 nM ExsA (lanes 1 to 3 in each panel) or LcrF (lanes 5 to 7 in each panel) for 15 min at 25°C. Samples were analyzed by native polyacrylamide gel electrophoresis and phosphorimaging. (D) Diagram depicting the position of the ExsA binding site (black box) derived from the PexsD promoter within probes 1 to 5 (solid line). (E) Circular permutation experiment performed using probes 1 to 5 (0.05 nM each) incubated in the presence of 180 nM ExsA (odd-numbered lanes) or LcrF (even-numbered lanes) for 15 min at 25°C. Samples were analyzed by native polyacrylamide gel electrophoresis and phosphorimaging. The positions of shift products 1 and 2 are indicated.
To further confirm the differential effect that ExsA and LcrF have on bending, we performed circular permutation assays using the PexsD promoter probe. Circular permutation is based on the observation that the electrophoretic mobility of bent DNA is more severely retarded when the bend is located in the center of a DNA fragment (46). To test for differential bending, we used a panel of five PexsD promoter probes in which the ExsA binding site was positioned at evenly spaced intervals across an ∼200-bp DNA fragment (Fig. 3D). As expected, the mobility of shift product 2 formed by LcrF was most highly retarded when the ExsA binding site was positioned toward the center of the EMSA probes (Fig. 3E, probes 2 to 4) and retarded to a higher extent than seen with ExsA. These combined data demonstrate that ExsA and LcrF have differential effects on DNA bending and show that these effects are promoter dependent.
The EMSA data generated using the shorter, 50-bp probes (Fig. 3A to C) and circular permutation probes (Fig. 3E) further support the conclusion that ExsA and LcrF bind as monomers and dimers, respectively. The primary basis for this conclusion is the absence of shift product 1 when using LcrF for the EMSA reactions presented in Fig. 3A to C and E. While some faint bands in the area of shift product 1 can be seen for LcrF in Fig. 3B and C, the same bands are also present in the lane that contains the promoter probes alone (lane 4 compared to lane 5).
Genetic determinants for LcrF binding.
ExsA-dependent activation of the PexoT-lacZ reporter requires two highly conserved sequences (GnC and TGnnA) that constitute binding site 1 (Fig. 4A). To determine whether LcrF is sensitive to the same mutations, a panel of mutant PexoT-lacZ reporter strains was transformed with the pLcrF expression plasmid, cultured under inducing (+EGTA) conditions for T3SS gene expression, and assayed for β-galactosidase activity. Similar to our previous findings with ExsA (27), activation of the PexoT-lacZ reporter by LcrF was highly sensitive to mutations that disrupted the core GnC and TGnnA sequences (Fig. 4B). The C-39A substitution, which represents a weakly conserved position in the ExsA consensus binding sequence, had an intermediate effect on activation by LcrF. Finally, the C-50G substitution, which was included as a negative control, had no effect on activation by ExsA or LcrF.
Fig 4.

ExsA- and LcrF-dependent activation is sensitive to nucleotide substitutions in the ExsA consensus site. (A) Sequence of the PexoT promoter showing the conserved GnC and TGnnA sequences (highlighted in bold) and the nucleotide substitutions indicated with an arrow. (B) The PA103 exsA::Ω strain carrying the indicated PexoT-lacZ reporters was transformed with pExsA or pLcrF. The resulting strains were cultured in the presence of EGTA and assayed for β-galactosidase activity. Activation by ExsA (open bars) and LcrF (hatched bars) is reported as the percent activity normalized to the activity of the wild-type PexoT-lacZ reporter. (C and D) EMSAs using radiolabeled probes derived from the mutant PexoT promoters. The nonspecific PalgD probe (Non-Sp) was included as a negative control. Probes (0.05 nM each) were incubated in the presence of 45 nM ExsA (C) or LcrF (D) for 15 min at 25°C. Samples were analyzed by native polyacrylamide gel electrophoresis and phosphorimaging. The positions of shift products 1 and 2 are indicated.
To confirm that the activation defects observed in Fig. 4B correlated with reduced DNA binding, we used promoter probes derived from the mutant PexoT-lacZ reporters in EMSAs. Because occupation of binding site 2 by ExsA is dependent upon prior occupation of site 1, mutations in site 1 result in a significant decrease in the formation of shift products 1 and 2 (Fig. 4C) (27). Binding of LcrF to the same promoter probes, however, was largely unaffected by substitutions that disrupt the ExsA consensus site (Fig. 4D). We speculated that LcrF, being dimeric in solution, might be less sensitive to substitutions in site 1, because additional interactions at site 2 compensate for binding defects at site 1. To test this hypothesis, we generated a monomeric variant of LcrF based upon prior knowledge of AraC dimerization. AraC dimerizes through an antiparallel, coiled-coil region that is stabilized by leucine triads located at each end of the interface (47). Two of the predicted leucine triad residues in LcrF (L136 and L144) were changed to alanine, and the resulting protein was designated LcrFm (monomeric). LcrFm was significantly impaired for activation of the PexoT-lacZ reporter in an exsA mutant but was stably expressed (see Fig. S3C in the supplemental material). LcrFm was purified by Ni2+-affinity chromatography and found to be largely monomeric in cross-linking studies (see Fig. S3A and B). In EMSAs, the binding properties of monomeric ExsA and LcrFm were similar, resulting in the formation of shift products 1 and 2 at the PexoT promoter probe (Fig. 2C and Fig. 5A, lanes 6 to 10). In contrast, LcrF formed only shift product 2 (Fig. 5A, lanes 1 to 5).
Although LcrFm preferentially formed shift product 1 at the PexoT promoter, it was unclear whether occupation was occurring at binding site 1 or 2. To examine this further, we used a panel of PexoT promoter probes with truncations that disrupt binding sites 1 and/or 2 (Fig. 5C). The probe lengths were maintained at 60 bp by replacing the deleted regions with nonspecific DNA. The minimal promoter probe consisted of the adenine-rich, GnC, and TGnnA sequences and supported formation of shift products 1 and 2 by ExsA and LcrFm (Fig. 5C, lanes 1 and 3). The same probes were previously used to show that occupation of site 2 by ExsA is dependent upon the presence of binding site 1 (27), and we demonstrate similar findings here for LcrFm. Probe 2, which lacks a functional site 2, resulted in a complete lack of shift product 2 formation by ExsA and LcrFm but still supported formation of shift product 1. In contrast, ExsA and LcrFm binding was significantly impaired using probes lacking either site 1 (probe 3) or sites 1 and 2 (probe 4). In contrast to our findings for ExsA and LcrFm, removal of either binding site 1 or 2 (probe 2 and 3) had only modest effects on formation of shift product 2 by wild-type LcrF (Fig. 5C, lanes 2, 5, 8), and it was not until both sites were removed (probe 4) that a significant reduction in LcrF binding was observed (Fig. 5C, lane 11). These findings demonstrate that dimeric LcrF is more tolerant of substitutions that disrupt the ExsA consensus binding site and that LcrFm preferentially binds to site 1. Having established the latter point, we next asked whether LcrFm was sensitive to nucleotide substitutions that disrupt the ExsA consensus binding site using the panel of mutant PexoT promoter probes described in Fig. 4A. Whereas native LcrF binding was largely unaffected by substitutions in the ExsA consensus region (Fig. 4D), binding by LcrFm was significantly reduced (Fig. 5D), demonstrating that the ExsA consensus binding site is required for maximal occupation of site 1.
The ExsA consensus binding site is present in Y. pestis LcrF-dependent promoters.
The finding that LcrF is sensitive to mutations that disrupt the ExsA consensus binding site suggested that the same recognition sequence exists in the yersiniae. To examine this further, we searched the PyopN, PlcrG, PyscN, and PyscB promoter regions from Y. pestis and identified a nearly perfect match to the ExsA consensus binding sequence in each promoter (Fig. 6). As previously observed for ExsA-dependent promoters (27), the consensus TGnnA sequence in each Y. pestis promoter was separated from the putative Pribnow (TATAAT) boxes by ∼21 to 22 bp. This prompted us to test whether PyopN-lacZ, PlcrG-lacZ, and PyscN-lacZ transcriptional reporters were responsive to ExsA. For these experiments, the ExsA and LcrF expression plasmids were introduced into an exsA::Ω ΔexsD double-mutant background to avoid feedback regulation on ExsA activity by ExsD. Although both ExsA and LcrF resulted in significant activation of the PyopN-lacZ, PlcrG-lacZ, and PyscN-lacZ reporters, LcrF-dependent activity at each of the promoters was significantly elevated compared to ExsA-dependent activity (Fig. 7A to C).
Fig 6.

Sequence alignment of the ExsA consensus binding sequence in ExsA (Pseudomonas aeruginosa)-, LcrF (Yersinia pestis)-, PxsA (Photorhabdus luminescens)-, AxsA (Aeromonas hydrophila)-, and VxsA (Vibrio parahaemolyticus)-dependent promoters. ExsA binding sites 1 and 2 are indicated with arrows. The consensus sequence is indicated in red, and −10 regions are underlined. The boxed regions in the P. aeruginosa PpcrG and Y. pestis PlcrG promoters correspond to regions protected by ExsA and LcrF/VirF from DNase I cleavage, respectively (see Fig. S5 in the supplemental material) (31, 48).
Fig 7.

ExsA activates transcription of Y. pestis transcriptional reporters. (A to C) The PA103 exsA::Ω strain carrying either the PyopN-lacZ (A), PyscN-lacZ (B), or PlcrG-lacZ (C) reporters was transformed with pJN105, pExsA, or pLcrF. The resulting strains were cultured under noninducing (−EGTA, open bars) or inducing (+EGTA, hatched bars) conditions for T3SS gene expression and assayed for β-galactosidase activity. Values were reported in Miller units. (D to F) EMSAs using radiolabeled probes derived from the LcrF-dependent PyopN (D), PyscN (E), and PlcrG (F) promoters. The PalgD probe was included in all binding reactions as a nonspecific (Non-Sp) control. Probes (0.05 nM each) were incubated in the absence (lane 2) or presence of 11, 23, 45, or 90 nM ExsA (lanes 3 to 6 in each panel) or LcrF (lanes 7 to 10 in each panel) for 15 min at 25°C. Samples were analyzed by native polyacrylamide gel electrophoresis and phosphorimaging. The positions of shift products 1 and 2 are indicated.
To further examine the interaction of ExsA and LcrF with the Y. pestis PyopN, PlcrG, and PyscN promoter regions, we performed EMSAs. Binding of ExsA to each promoter probe resulted in the generation of shift products 1 and 2 (Fig. 7D to F, lanes 3 to 6). Lower mobility promoter probe complexes were also apparent with the PyopN and the PyscN promoter probes at the highest concentrations of ExsA (Fig. 7D to F, lane 6). These products likely represent nonspecific interactions with ExsA. Binding of LcrF to the Y. pestis promoter probes resulted in only a single predominant species (shift product 2) that exhibited reduced mobility relative to the ExsA-promoter probe complexes (Fig. 7D to F, lanes 5 and 8). These findings further support the conclusion that ExsA binds as a monomer, whereas LcrF binds as a dimer, and that LcrF has increased DNA bending activity relative to that of ExsA.
Since ExsA activates transcription of LcrF-dependent promoters, we next tested whether the nucleotides that comprise the ExsA binding consensus are required for activation of the Y. pestis PyscN promoter. Similar to our findings with the mutant PexoT-lacZ reporters in Fig. 4B, mutations that disrupted the core GnC and TGnnA sites in the PyscN-lacZ transcriptional reporter also resulted in a significant decrease in ExsA- and LcrF-dependent activity (Fig. 8A and B). Conversely, mutations that altered the nonconserved −50 and poorly conserved −39 positions had less severe effects on ExsA- and LcrF-dependent activity.
Fig 8.
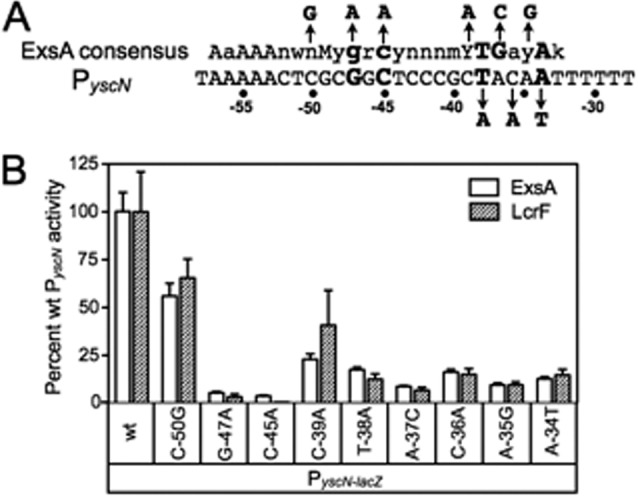
Y. pestis PyscN reporter activity is sensitive to substitutions that disrupt the ExsA binding site. (A) The PyscN promoter sequence showing the ExsA consensus sequence in binding site 1. The GnC and TGnnA sequences are highlighted in bold, and each promoter nucleotide substitution is indicated with an arrow. (B) The PA103 exsA::Ω strain carrying the indicated mutant PyscN-lacZ transcriptional reporters was transformed with either pExsA or pLcrF. The resulting strains were cultured in the presence of EGTA and assayed for β-galactosidase activity. Activation by pExsA (open bars) and pLcrF (hatched bars) is reported as the percent activity of the mutant promoters normalized to the activity at the wild-type PyscN promoter.
ExsA complements a Y. pestis lcrF mutant for T3SS gene expression.
The finding that LcrF complements a P. aeruginosa exsA mutant for T3SS gene expression led us to examine whether ExsA complements a Y. pestis lcrF mutant. An lcrF deletion mutant was generated by gene replacement in a strain bearing a yopM::lacZYA transcriptional reporter. The wild-type and ΔlcrF yopM::lacZYA reporter strains were transformed with the pExsA and pLcrF expression plasmids and assayed for reporter activity following growth in the absence of Ca2+. Similar to our finding that LcrF complements an exsA mutant, ExsA was able to restore yopM::lacZYA reporter activity in the Y. pestis ΔlcrF mutant (Fig. 9A), albeit to a lesser extent than LcrF. To determine if ExsA could fully activate the Y. pestis T3SS, strains were cultured in the presence or absence of Ca2+, separated into cell lysate and supernatant fractions, and analyzed by immunoblotting for YopM and YopN. As shown in Fig. 9B, LcrF and ExsA both complemented the ΔlcrF mutant for expression and low Ca2+-dependent secretion of YopM and YopN. Based on these data, we conclude that ExsA complements a Y. pestis ΔlcrF mutant for T3SS gene expression albeit at reduced levels relative to LcrF, likely owing to poor ExsA expression in Y. pestis (Fig. 9B).
Fig 9.
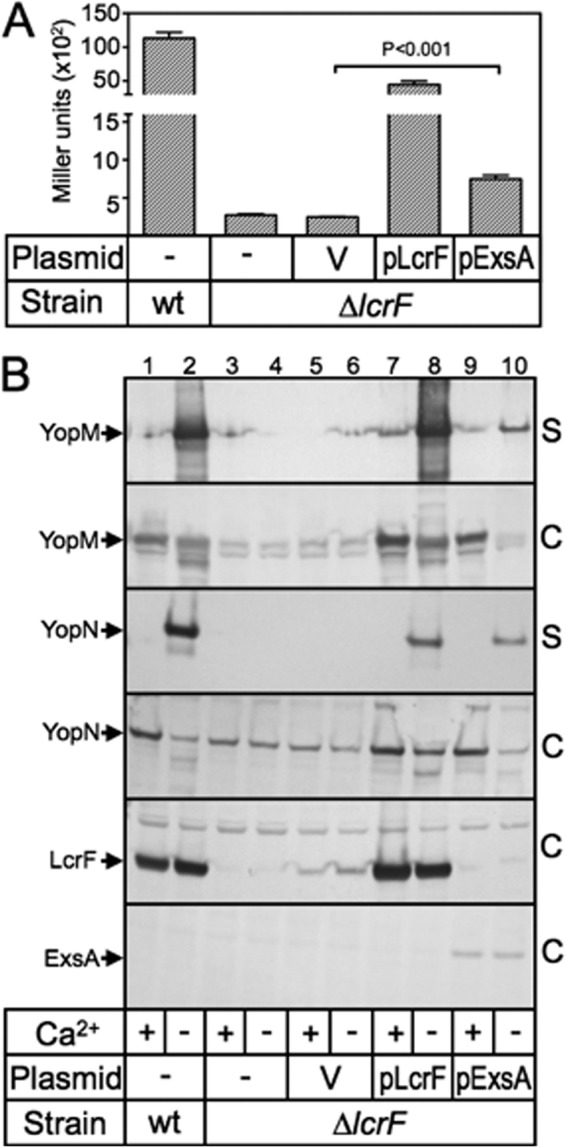
ExsA activates expression of the Y. pestis T3SS. (A) Y. pestis KIM5-3001 (yopM::lacZYA) and KIM5-3233-F2 (ΔlcrF yopM::lacZYA) carrying a vector control (V), pLcrF, or pExsA were cultured under inducing conditions for T3SS gene expression and then assayed for β-galactosidase activity as reported in Miller units. (B) Wild-type Y. pestis KIM5-3001 (lanes 1 and 2) or KIM5-3001-F1 (ΔlcrF) (lanes 3 to 10) lacking a vector (lanes 1 to 4) or carrying a vector control (lanes 5 and 6), pLcrF (lanes 7 and 8), or pExsA (lanes 9 and10) were grown under noninducing conditions (+calcium) or inducing (−calcium; odd-numbered lanes) conditions for T3SS gene expression. Concentrated supernatant fluid (S) and cell-associated fractions (C) were prepared and subjected to SDS-PAGE and immunoblot analysis. Arrows indicate protein products.
ExsA homologs from other pathogens are also sensitive to substitutions in the PexoT promoter.
The core structural components of the P. aeruginosa, yersiniae, A. hydrophila, P. luminescens, and V. parahaemolyticus T3SSs are encoded within 4 highly conserved operons. Based on the P. aeruginosa designations, those operons consist of popN-pcrR, pcrG-popD, pscN-pscU, and exsD-pscM (see Fig. S4 in the supplemental material). The only apparent variance in genetic organization among the five organisms is the yersiniae yscB-yscM operon, which lacks exsD. An alignment of the promoter regions located upstream of each operon revealed nearly perfect ExsA consensus binding sites in each organism (Fig. 6). Bolstering our confidence in the alignment is that the spacing seen between the TGnnA sequence and the putative −10 Pribnow boxes is 20 to 22 bp. As mentioned earlier, a similar spacing arrangement is seen in all 10 ExsA-dependent promoters. Finally, the derived consensus from an alignment of all 20 promoter regions shown in Fig. 6 is essentially identical to the ExsA consensus derived using only the P. aeruginosa promoters. Based on these observations, we tested whether the activators from A. hydrophila, P. luminescens, and V. parahaemolyticus are sensitive to the same PexoT-lacZ reporter mutations that disrupt ExsA- and LcrF-dependent activation. Like ExsA and LcrF, AxsA (A. hydrophila), PxsA (P. luminescens), and VxsA (V. parahaemolyticus) were sensitive to substitutions in the GnC and TGnnA sequences of the PexoT-lacZ reporter, while the poorly conserved −39 and nonconserved −50 positions had intermediate and no effects on activity, respectively (Fig. 10).
Fig 10.
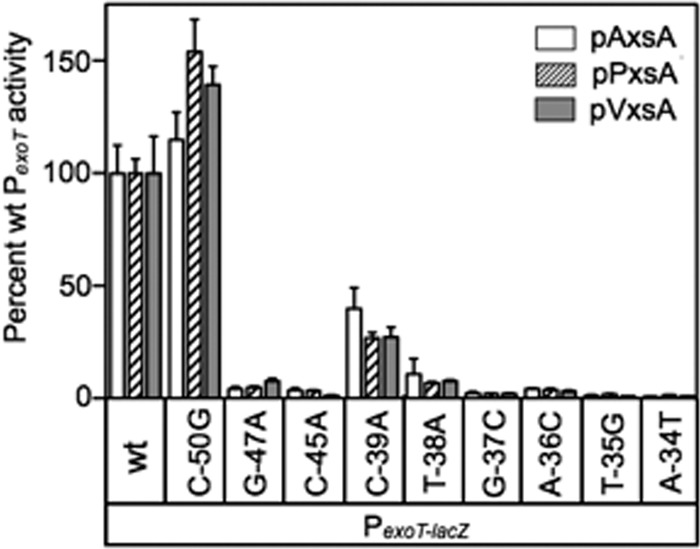
The PA103 exsA::Ω strain carrying the indicated PexoT-lacZ transcriptional reporters was transformed with AxsA (pAxsA), PxsA (pPxsA), or VxsA (pVxsA) expression vectors. The resulting strains were cultured in the presence of EGTA and assayed for β-galactosidase activity. Activation by pAxsA (open bars), pPxsA (hatched bars), and pVxsA (gray bars) is reported as the percent activity of the mutant promoters normalized to the activity of the wild-type PexoT promoter.
DISCUSSION
Several regions in the yopE, lcrG, virC, and yopH promoter regions were previously shown to be protected from DNase I cleavage by Y. enterocolitica VirF (28, 48). The protected regions contained a putative consensus binding sequence (TTTTaGYcTgTat, where uppercase letters indicate more highly conserved positions than lowercase letters) that was present in at least three copies per promoter and organized as either single sites or inverted repeats. The authors proposed that each monomer of the VirF dimer bound to a consensus site (half-site) and that high-affinity binding occurred at inverted repeats bearing the strongest match to consensus (48). That study also noted, however, that the consensus binding sequences were highly degenerate and variably spaced from the transcription starts sites and acknowledged that the consensus site assignment remained questionable. The high degree of similarity between the HTH motifs of LcrF/VirF and ExsA prompted us to test the hypothesis that each protein recognizes a similar DNA sequence. Herein, we demonstrate that the ExsA consensus binding site (AaAAAnwmMyGrCynnnmTGayAk) is present in Y. pestis promoter regions and is required for LcrF-dependent activation of transcription.
Purified ExsA generates two promoter probe complexes in EMSAs, representing monomeric ExsA bound to site 1 (product 1) and both sites 1 and 2 (product 2) (27). In contrast, purified LcrF resulted in the appearance of one primary promoter probe complex which we designated shift product 2 (Fig. 2 and 7). A previous study suggested that purified LcrF is dimeric in solution (28). That finding is consistent with our own cross-linking studies with purified LcrF (see Fig. S3B in the supplemental material). The simplest interpretation of our binding data, therefore, is that shift product 2 results from the binding of one LcrF dimer. In support of this conclusion, the binding properties of a monomeric LcrFm variant resembled those of ExsA in that both shift products 1 and 2 were readily formed (Fig. 5A). Furthermore, the mobilities of the LcrF-PexsC and ExsA-PexsC shift product 2 complexes were similar (Fig. 2A and B), indicating binding of two molecules of LcrF or ExsA. Binding of LcrF to the PexoT, PexsD, PyopN, PyscN, and PlcrG promoter probes, however, resulted in the formation of complexes with reduced mobility relative to those formed by ExsA. Although the lower mobility complexes generated by LcrF could reflect binding of multiple LcrF dimers, our data indicate that the basis for reduced mobility is differential DNA bending. The significance of ExsA- and LcrF-induced bending is unclear, but it is interesting to note that LcrF was better than ExsA in activation of each transcriptional reporter examined in this study. ExsA and LcrF induced PexsC promoter bending to a similar extent (Fig. 2A and B), and there was only a 30% difference in PexsC-lacZ reporter activity (Fig. 1B). With the other promoter probes (PexoT, PexsD, PyopN, PyscN, and PlcrG), however, LcrF resulted in a higher degree of bending and a significant increase in activity relative to ExsA (ranging from 2.5-fold higher for the PexsD-lacZ reporter to nearly 20-fold higher for the PexoT-lacZ reporter). Increased bending, therefore, positively correlates with promoter activity and may partially account for the observation that LcrF activates transcription better than ExsA.
DNase I footprints of LcrF/VirF and ExsA bound to the P. aeruginosa PexsC and yersiniae PlcrG promoter regions identified a similar region of protection (Fig. 6; see also Fig. S5 in the supplemental material) (28, 48). In each case, the protected area was centered on the ExsA consensus site. Fe-Babe footprinting studies with ExsA found that the protected region consists of two adjacent binding sites for monomeric ExsA and that both monomers are bound in the same orientation (i.e., head to tail) (31). The ExsA monomer bound to site 1 makes base-specific contacts with the conserved GnC and TGnnA sequences through amino acids located in the first (L198, T199, and K202) and second HTH (Y250) motifs, respectively (31). Since the highest degree of sequence conservation between ExsA- and LcrF-dependent promoters occurs at binding site 1, we focused on the interaction of LcrF with site 1 and conclude that ExsA and LcrF bind to site 1 in the same manner. Supportive of this, in vivo data using the P. aeruginosa PexoT and yersiniae PyscN transcriptional reporters found that disruption of the GnC and TGnnA determinants resulted in a significant reduction in activation by both ExsA and LcrF (Fig. 4B and 8B). Paradoxically, however, single nucleotide substitutions in the PexoT GnC and TGnnA determinants had no obvious effect on LcrF binding (Fig. 4D), presumably owing to the stabilizing effect of binding as a dimer. Nevertheless, the finding that LcrFm binding is sensitive to GnC and TGnA substitutions in site 1 suggests that the same substitutions perturb the interaction of dimeric LcrF at site 1. One model to account for the discrepancy, therefore, is that the GnC and TGnnA substitutions alter the binding of the LcrF molecule at site 1 in a manner that prevents recruitment of RNAP but overall have no observable effect on binding of the LcrF dimer. Alternatively, it is possible that the in vitro binding data are not reflective of the in vivo situation (i.e., the GnC and TGnnA substitutions actually do disrupt wild-type LcrF binding in vivo).
ExsA-dependent activation is controlled through a direct interaction with the ExsD antiactivator (10). Y. pestis lacks an obvious ExsD homolog. Nevertheless, we tested LcrF sensitivity to ExsD by examining PexoT-lacZ reporter activity in an exsD deletion strain. In the absence of ExsD, there was virtually no increase in LcrF-dependent reporter activity (Fig. 1A). Second, we tested LcrF-dependent activation while overexpressing ExsD from a second plasmid and found that PexoT-lacZ reporter activity was only modestly reduced (data not shown). Taken together, these data suggest that ExsD is a poor inhibitor of LcrF activity, a property that likely contributes to increased LcrF activity in P. aeruginosa. The dimeric state of LcrF in solution may also partially explain increased transcriptional activation by LcrF. Whereas ExsA-dependent activation requires ordered binding of two monomers, dimeric LcrF binds to promoter DNA in a single step that may be kinetically favored. It is curious that purified ExsA is monomeric in solution when most other characterized members of the AraC/XylS family are dimeric. ExsA is also unusual in that it forms a 1:1 complex with the ExsD antiactivator. Because self-association and ExsA-ExsD complex formation are mutually exclusive interactions (30, 33), high-affinity self-association of ExsA might be incompatible with ExsA-ExsD complex formation. The weaker self-association properties of ExsA, therefore, may strike a balance between the requirements to both self-associate and interact with ExsD.
Photorhabdus, Aeromonas, and Vibrio parahaemolyticus all have ExsD homologs, and we have previously shown that ExsA from Photorhabdus and Aeromonas can complement a P. aeruginosa exsA mutant for T3SS gene expression (27). Our in vivo findings demonstrated that the ExsA consensus binding sequence is a primary determinant for promoter recognition by each activator (Fig. 10). Although biochemical assays will be necessary to characterize the binding characteristics of these activators, we speculate that each is monomeric in solution and will exhibit DNA binding properties similar to ExsA, because the activity of each is controlled by an ExsD homolog.
Supplementary Material
ACKNOWLEDGMENTS
We thank Linda McCarter for kindly providing V. parahaemolyticus genomic DNA.
This study was supported by the National Institutes of Health (RO1-AI055042 to T.L.Y.).
Footnotes
Published ahead of print 18 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00990-13.
REFERENCES
- 1.Hauser AR. 2009. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat. Rev Microbiol. 7:654–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buttner D. 2012. Protein export according to schedule: architecture, assembly, and regulation of type III secretion systems from plant- and animal-pathogenic bacteria. Microbiol. Mol. Biol. Rev. 76:262–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matsumoto H, Young GM. 2009. Translocated effectors of Yersinia. Curr. Opin. Microbiol. 12:94–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Engel J, Balachandran P. 2009. Role of Pseudomonas aeruginosa type III effectors in disease. Curr. Opin. Microbiol. 12:61–66 [DOI] [PubMed] [Google Scholar]
- 5.Hayes CS, Aoki SK, Low DA. 2010. Bacterial contact-dependent delivery systems. Annu. Rev. Genet. 44:71–90 [DOI] [PubMed] [Google Scholar]
- 6.Vallis AJ, Yahr TL, Barbieri JT, Frank DW. 1999. Regulation of ExoS production and secretion by Pseudomonas aeruginosa in response to tissue culture conditions. Infect. Immun. 67:914–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brutinel ED, Yahr TL. 2008. Control of gene expression by type III secretory activity. Curr. Opin. Microbiol. 11:128–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frank DW, Iglewski BH. 1991. Cloning and sequence analysis of a trans-regulatory locus required for exoenzyme S synthesis in Pseudomonas aeruginosa. J. Bacteriol. 173:6460–6468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brutinel ED, Vakulskas CA, Yahr TL. 2010. ExsD inhibits expression of the Pseudomonas aeruginosa type III secretion system by disrupting ExsA self-association and DNA binding activity. J. Bacteriol. 192:1479–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCaw ML, Lykken GL, Singh PK, Yahr TL. 2002. ExsD is a negative regulator of the Pseudomonas aeruginosa type III secretion regulon. Mol. Microbiol. 46:1123–1133 [DOI] [PubMed] [Google Scholar]
- 11.Zheng Z, Chen G, Joshi S, Brutinel ED, Yahr TL, Chen L. 2007. Biochemical characterization of a regulatory cascade controlling transcription of the Pseudomonas aeruginosa type III secretion system. J. Biol. Chem. 282:6136–6142 [DOI] [PubMed] [Google Scholar]
- 12.Dasgupta N, Lykken GL, Wolfgang MC, Yahr TL. 2004. A novel anti-anti-activator mechanism regulates expression of the Pseudomonas aeruginosa type III secretion system. Mol. Microbiol. 53:297–308 [DOI] [PubMed] [Google Scholar]
- 13.Lykken GL, Chen G, Brutinel ED, Chen L, Yahr TL. 2006. Characterization of ExsC and ExsD self-association and heterocomplex formation. J. Bacteriol. 188:6832–6840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Urbanowski ML, Lykken GL, Yahr TL. 2005. A secreted regulatory protein couples transcription to the secretory activity of the Pseudomonas aeruginosa type III secretion system. Proc. Natl. Acad. Sci. U. S. A. 102:9930–9935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rietsch A, Vallet-Gely I, Dove SL, Mekalanos JJ. 2005. ExsE, a secreted regulator of type III secretion genes in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 102:8006–8011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diaz MR, King JM, Yahr TL. 2011. Intrinsic and extrinsic regulation of type III secretion gene expression in Pseudomonas aeruginosa. Front. Microbiol. 2:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cornelis GR. 1993. Role of the transcription activator virF and the histone-like protein YmoA in the thermoregulation of virulence functions in yersiniae. Zentralbl. Bakteriol. 278:149–164 [DOI] [PubMed] [Google Scholar]
- 18.Cornelis GR, Sluiters C, Delor I, Geib D, Kaniga K, Lambert de Rouvroit C, Sory MP, Vanooteghem JC, Michiels T. 1991. ymoA, a Yersinia enterocolitica chromosomal gene modulating the expression of virulence functions. Mol. Microbiol. 5:1023–1034 [DOI] [PubMed] [Google Scholar]
- 19.Bohme K, Steinmann R, Kortmann J, Seekircher S, Heroven AK, Berger E, Pisano F, Thiermann T, Wolf-Watz H, Narberhaus F, Dersch P. 2012. Concerted actions of a thermo-labile regulator and a unique intergenic RNA thermosensor control Yersinia virulence. PLoS Pathog. 8:e1002518. 10.1371/journal.ppat.1002518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jackson MW, Silva-Herzog E, Plano GV. 2004. The ATP-dependent ClpXP and Lon proteases regulate expression of the Yersinia pestis type III secretion system via regulated proteolysis of YmoA, a small histone-like protein. Mol. Microbiol. 54:1364–1378 [DOI] [PubMed] [Google Scholar]
- 21.Hoe NP, Goguen JD. 1993. Temperature sensing in Yersinia pestis—translation of the Lcrf activator protein is thermally regulated. J. Bacteriol. 175:7901–7909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pettersson J, Nordfelth R, Dubinina E, Bergman T, Gustafsson M, Magnusson KE, Wolf-Watz H. 1996. Modulation of virulence factor expression by pathogen target cell contact. Science 273:1231–1233 [DOI] [PubMed] [Google Scholar]
- 23.Williams AW, Straley SC. 1998. YopD of Yersinia pestis plays a role in negative regulation of the low-calcium response in addition to its role in translocation of Yops. J. Bacteriol. 180:350–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Y, Anderson DM. 2011. Expression hierarchy in the Yersinia type III secretion system established through YopD recognition of RNA. Mol. Microbiol. 80:966–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferracci F, Schubot FD, Waugh DS, Plano GV. 2005. Selection and characterization of Yersinia pestis YopN mutants that constitutively block Yop secretion. Mol. Microbiol. 57:970–987 [DOI] [PubMed] [Google Scholar]
- 26.Egan SM. 2002. Growing repertoire of AraC/XylS activators. J. Bacteriol. 184:5529–5532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brutinel ED, Vakulskas CA, Brady KM, Yahr TL. 2008. Characterization of ExsA and of ExsA-dependent promoters required for expression of the Pseudomonas aeruginosa type III secretion system. Mol. Microbiol. 68:657–671 [DOI] [PubMed] [Google Scholar]
- 28.Lambert de Rouvroit C, Sluiters C, Cornelis GR. 1992. Role of the transcriptional activator, VirF, and temperature in the expression of the pYV plasmid genes of Yersinia enterocolitica. Mol. Microbiol. 6:395–409 [PubMed] [Google Scholar]
- 29.Schleif R. 2010. AraC protein, regulation of the l-arabinose operon in Escherichia coli, and the light switch mechanism of AraC action. FEMS Microbiol. Rev. 34:779–796 [DOI] [PubMed] [Google Scholar]
- 30.Thibault J, Faudry E, Ebel C, Attree I, Elsen S. 2009. Anti-activator ExsD forms a 1:1 complex with ExsA to inhibit transcription of type III secretion operons. J. Biol. Chem. 284:15762–15770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.King JM, Brutinel ED, Marsden AE, Schubot FD, Yahr TL. 2012. Orientation of Pseudomonas aeruginosa ExsA monomers bound to promoter DNA and base-specific contacts with the P(exoT) promoter. J. Bacteriol. 194:2573–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brutinel ED, King JM, Marsden AE, Yahr TL. 2012. The distal ExsA-binding site in Pseudomonas aeruginosa type III secretion system promoters is the primary determinant for promoter-specific properties. J. Bacteriol. 194:2564–2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brutinel ED, Vakulskas CA, Yahr TL. 2009. Functional domains of ExsA, the transcriptional activator of the Pseudomonas aeruginosa type III secretion system. J. Bacteriol. 191:3811–3821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vakulskas CA, Brady KM, Yahr TL. 2009. Mechanism of transcriptional activation by Pseudomonas aeruginosa ExsA. J. Bacteriol. 191:6654–6664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vakulskas CA, Brutinel ED, Yahr TL. 2010. ExsA recruits RNA polymerase to an extended −10 promoter by contacting region 4.2 of sigma-70. J. Bacteriol. 192:3597–3607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vogel HJ, Bonner DM. 1956. Acetylornithinase of Escherichia coli: partial purification and some properties. J. Biol. Chem. 218:97–106 [PubMed] [Google Scholar]
- 37.Une T, Brubaker RR. 1984. In vivo comparison of avirulent Vwa− and Pgm− or Pstr phenotypes of yersiniae. Infect. Immun. 43:895–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fetherston JD, Lillard JW, Jr, Perry RD. 1995. Analysis of the pesticin receptor from Yersinia pestis: role in iron-deficient growth and possible regulation by its siderophore. J. Bacteriol. 177:1824–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14 [DOI] [PubMed] [Google Scholar]
- 41.Perry RD, Straley SC, Fetherston JD, Rose DJ, Gregor J, Blattner FR. 1998. DNA sequencing and analysis of the low-Ca2+-response plasmid pCD1 of Yersinia pestis KIM5. Infect. Immun. 66:4611–4623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoang TT, Kutchma AJ, Becher A, Schweizer HP. 2000. Integration-proficient plasmids for Pseudomonas aeruginosa: site-specific integration and use for engineering of reporter and expression strains. Plasmid 43:59–72 [DOI] [PubMed] [Google Scholar]
- 44.Martin RG, Rosner JL. 2001. The AraC transcriptional activators. Curr. Opin. Microbiol. 4:132–137 [DOI] [PubMed] [Google Scholar]
- 45.Frank DW. 1997. The exoenzyme S regulon of Pseudomonas aeruginosa. Mol. Microbiol. 26:621–629 [DOI] [PubMed] [Google Scholar]
- 46.Crothers DM, Gartenberg MR, Shrader TE. 1991. DNA bending in protein-DNA complexes. Methods Enzymol. 208:118–146 [DOI] [PubMed] [Google Scholar]
- 47.LaRonde-LeBlanc N, Wolberger C. 2000. Characterization of the oligomeric states of wild type and mutant AraC. Biochemistry 39:11593–11601 [DOI] [PubMed] [Google Scholar]
- 48.Wattiau P, Cornelis GR. 1994. Identification of DNA sequences recognized by VirF, the transcriptional activator of the Yersinia yop regulon. J. Bacteriol. 176:3878–3884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yahr TL, Mende-Mueller LM, Friese MB, Frank DW. 1997. Identification of type III secreted products of the Pseudomonas aeruginosa exoenzyme S regulon. J. Bacteriol. 179:7165–7168 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.