

Abstract
In Helicobacter pylori, the ferric uptake regulator (Fur) has evolved additional regulatory functions not seen in other bacteria; it can repress and activate different groups of genes in both its iron-bound and apo forms. Because little is understood about the process of apo-Fur repression and because only two apo-Fur-repressed genes (pfr and sodB) have previously been identified, we sought to expand our understanding of this type of regulation. Utilizing published genomic studies, we selected three potential new apo-Fur-regulated gene targets: serB, hydA, and the cytochrome c553 gene. Transcriptional analyses confirmed Fur-dependent repression of these genes in the absence of iron, as well as derepression in the absence of Fur. Binding studies showed that apo-Fur directly interacted with the suspected hydA and cytochrome c553 promoters but not that of serB, which was subsequently shown to be cotranscribed with pfr; apo-Fur-dependent regulation occurred at the pfr promoter. Alignments of apo-regulated promoter regions revealed a conserved, 6-bp consensus sequence (AAATGA). DNase I footprinting showed that this sequence lies within the protected regions of the pfr and hydA promoters. Moreover, mutation of the sequence in the pfr promoter abrogated Fur binding and DNase protection. Likewise, fluorescence anisotropy studies and binding studies with mutated consensus sequences showed that the sequence was important for apo-Fur binding to the pfr promoter. Together these studies expand the known apo-Fur regulon in H. pylori and characterize the first reported apo-Fur box sequence.
INTRODUCTION
Helicobacter pylori is a unique Gram-negative, microaerophilic, spiral bacterium that colonizes the gastric mucosa of humans and some nonhuman primates (1). Over half of the world's population is infected with this organism, but the vast majority of those infections are asymptomatic (2). H. pylori causes symptomatic disease in about 20% of infected individuals, and the disease spectrum ranges from gastritis and peptic ulcer disease to two distinct types of cancer, gastric adenocarcinoma and mucosa-associated lymphoid tissue lymphoma (2). At present, H. pylori is the only bacterium known to be a carcinogen (3).
The gastric niche in which H. pylori resides is both a dynamic and yet caustic environment. In the stomach, the bacterium encounters a wide variety of stressors, including oxidative stress, nutrient limitation, large fluctuations in pH, and osmotic stress. Gastric pH levels can fall to as low as 2. In order to survive, H. pylori, which is not acidophilic, must be able to cope with these pH changes. Furthermore, the influx and subsequent digestion of food materials alter ion concentrations and osmotic conditions, as well as the availability of various nutrients. Among the ions that are critical for survival is iron, a nutrient that is essential for nearly all living organisms; however, too much iron results in the formation of potentially damaging hydroxyl radicals through Fenton chemistry. H. pylori responds and adapts to gastric stressors by altering gene expression through the use of transcriptional regulators. One such regulator, the ferric uptake regulator (Fur), is utilized to maintain iron homeostasis (4–6) but also to respond to pH (7–9), nitrosative, and oxidative stressors (10–12). Given the paucity of transcriptional regulators found in H. pylori compared to other bacterial organisms (13), it is perhaps not surprising that through the course of evolution, H. pylori Fur has taken on a more global role in gene regulation beyond iron uptake and storage.
Fur is a small (15- to 17-kDa) regulatory protein that is known to regulate iron homeostasis in a wide variety of bacterial species. This regulation typically occurs when Fur, which is bound by its ferrous iron cofactor (Fe-Fur), binds to specific DNA sequences (Fur boxes) in the promoters of genes involved in iron uptake and represses their expression. These Fur boxes tend to lie close to the core promoter elements (14), which is logical given that repression occurs because Fe-Fur occludes the binding of RNA polymerase. In H. pylori, the iron-bound Fur box consists of a 15-bp DNA sequence with 7-1-7 motif and dyad symmetry, 5′-TAATAATnATTATTA-3′ (14). While Fe-Fur repression is the best-characterized type of Fur regulation both in H. pylori and broadly among other bacteria, it is by no means the only way in which Fur regulates gene expression. For instance, H. pylori Fe-Fur has been shown to function as an activator for the expression of oorDABC (7, 15), nifS (12), and cagA (14).
Less traditional means of Fur regulation, which involves utilization of Fur in the absence of its iron cofactor (in its apo form), have also been reported. This type of apo regulation can take the form of activation or repression. For example, in H. pylori and Staphylococcus aureus, apo-Fur has been shown to activate expression of fur (16) and norA (17), respectively. Conversely, apo-Fur is known to repress expression of sodB and pfr in H. pylori. Indeed, apo-Fur repression was first characterized in H. pylori (18) but has also been shown to occur in Campylobacter jejuni (19–21) and proposed to occur in Desulfovibrio vulgaris Hildenborough (22). In addition to the two characterized apo-Fur repressed genes that have been described for C. jejuni, the crystal structure for apo-Fur from this organism was recently resolved (20). Interestingly, this structure showed that while the apo form of the protein retained the classic V-shaped dimer, the DNA binding domain was rotated by 180° compared to the previously resolved holo-dimeric structures of Fur from other organisms (20). These structural differences between the apo and holo forms of the protein could facilitate recognition of different DNA sequences within the promoters of apo and iron-bound target genes.
Despite knowledge of the existence of apo-Fur regulation in H. pylori for over 10 years and a predicted regulon of about 16 genes (18, 23), there are currently only two definitive H. pylori apo-Fur-repressed targets: pfr and sodB. pfr encodes an iron storage molecule (24) whose expression is repressed by Fur when iron levels are low. Similarly, sodB, which is the only superoxide dismutase found in this organism (13), is repressed by Fur when iron levels are low. With only these two characterized gene targets in H. pylori and the few targets found in other organisms, there are presently more questions regarding apo-Fur repression than there are answers. For example, because of the few known apo-Fur targets, it has been impossible to identify the apo-Fur box sequence required for apo-Fur binding. Therefore, in this work, we sought to expand the characterized apo-Fur regulon of H. pylori and to use this information to better define a consensus binding sequence for apo-Fur in this organism. Herein, we show that hydA and the cytochrome c553 gene are both regulated by apo-Fur in H. pylori. Moreover, with the addition of these two genes to the apo-Fur regulon, we define and show the importance of a 6-bp sequence, AAATGA, in the binding of apo-Fur to these target promoters using DNase I footprinting, fluorescence anisotropy (FA), and binding assays.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
All plasmids and bacterial strains utilized in these studies are listed in Table 1, and all oligonucleotides utilized in these studies are listed in Table S1 in the supplemental material. H. pylori strains were maintained as frozen stocks at −80°C in brain heart infusion broth (Becton Dickinson) with 10% fetal bovine serum (FBS) (Gibco) and 20% glycerol. H. pylori was grown on horse blood agar plates composed of 4% Columbia agar base (Neogen Corp.), 5% defibrinated horse blood (HemoStat Laboratories, Dixon, CA), 0.2% β-cyclodextrin (Sigma), 10 μg/ml of vancomycin (Amresco), 2.5 U/ml of polymyxin B (Sigma), 5 μg/ml of trimethoprim (Sigma), and 5 μg/ml of amphotericin B (Amresco) or in liquid medium composed of brucella broth (Neogen Corp.) with 10% FBS and 10 μg/ml of vancomycin. All H. pylori cultures were grown at 37°C under microaerobic conditions (5% O2, 10% CO2, and 85% N2) generated with an Anoxomat gas evacuation and replacement system (Spiral Biotech). Liquid cultures were grown with shaking at 100 rpm. Escherichia coli strains were maintained at −80°C in LB medium with 40% glycerol, and E. coli cultures were grown at 37°C either on LB agar plates or in LB broth with 225-rpm shaking.
Table 1.
Plasmids and strains used in this study
| Plasmid or strain | Description | Reference |
|---|---|---|
| Plasmid | ||
| pDSM430 | pET21A:: fur | 10 |
| H. pylori strains | ||
| G27 | WT H. pylori | 51 |
| DSM300 | G27 Δfur::cat; Cmr | 25 |
| E. coli strains | ||
| DSM431 | BL21(DE3) Rosetta/pLys Δfur (pDSM430); Ampr Cmr Kanr | 10 |
| DSM1215 | Top10 (pGEM-T Easy::HP0653 promoter region AAATGA apo-Fur box I ACACAC); Ampr | This study |
| DSM1384 | Top10 (pGEM-T Easy::HP0653 promoter region AAATGA apo-Fur box II ACACAC); Ampr | This study |
| DSM1385 | Top10 (pGEM-T Easy::HP0653 promoter region AAATGA apo-Fur boxes I and II ACACAC); Ampr | This study |
| DSM1386 | Top10 (pGEM-T Easy::HP0653 promoter region AAATGA apo-Fur box III ACACAC); Ampr | This study |
| DSM1387 | Top10 (pGEM-T Easy::HP0653 promoter region AAATGA apo-Fur boxes I, II, and III ACACAC); Ampr | This study |
RPAs.
RNase protection assays (RPAs) were performed essentially as previously described (10, 25, 26). In brief, liquid cultures of wild-type (WT) and Δfur G27 were grown to exponential phase, and one half of each culture was removed for RNA isolation. To the remaining half of each culture, a 200 μM concentration of the iron chelator 2,2′-dipryidyl (DPP; Sigma), was added, and the cultures were maintained for an additional 1 h prior to RNA isolation. RNA was isolated as previously described (27). Agarose gels were used to visualize the RNA and to determine integrity. Riboprobes were generated using the primer pairs shown in Table S1 along with a Maxiscript kit (Applied Biosystems) and 50 μCi of [32P]UTP (Perkin-Elmer). Two micrograms of RNA was used in the RPAs for pfr, hydA, and the cytochrome c553 gene, and 10 μg of RNA was used in the RPAs for serB. The RPA III kit (Applied Biosystems) along with the specified amount of RNA was used to generate the RPA reactions. These reactions were resolved on 5% acrylamide–1× Tris-borate-EDTA–8 M urea denaturing gels. Gels were subsequently exposed to phosphor screens and visualized by scanning with an FLA-5100 multifunctional scanner (Fuji-Film). Multi-Gauge software (version 3.0; Fuji-Film) was used to analyze and quantitate the data. Four biologically independent repeats of each experiment were performed.
cDNA synthesis and RT-PCR.
Reverse transcriptase PCR (RT-PCR) was also used to evaluate expression of apo-Fur target genes. Bacterial cultures were grown and treated with DPP, and RNA was isolated for the RT-PCR experiments in the same manner as described above for the RPA studies. cDNA synthesis and quantitative real-time PCRs (qPCR) were performed as previously described (15). Briefly, the Quantitect reverse transcriptase kit (Qiagen) was used to generate first-strand cDNA by following the manufacturer's instructions; a corresponding control reaction in which no reverse transcriptase (no-RT) enzyme was added was also included for each cDNA synthesis reaction. The Roto-gene Q instrument (Qiagen) was used to perform qPCR with the primer pairs listed in Table S1. Reaction mixtures contained the following in a total volume of 20 μl: 1 μl of cDNA or no-RT control reaction to be used as the template, 1× Roto-Gene SYBR green RT-PCR master mix (Qiagen), and 3 pmol each of the forward and reverse primer pairs as listed in Table S1. Cycling conditions were as follows: 5 min at 95°C (initial activation) and 35 cycles of 95°C for 10 s (denaturing), 50°C for 20 s (annealing), and 72°C for 30 s (extension), with SYBR green fluorescence being measured at each extension step. 16S was used as the internal reference gene, and the comparative threshold cycle method (2−ΔΔCT) was used to calculate the relative gene expression. To ensure specificity of amplification, postrun melt curve analyses were carried out. Three biologically independent replicates of these experiments were conducted.
rFur purification.
Recombinant H. pylori Fur (rFur) was purified from the E. coli expression strain DSM431 as previously described (10). In brief, expression of rFur was induced in mid-exponential-phase growing cultures of DSM431 through the addition of 1 mM isopropyl-d-thiogalactopyranoside (IPTG; Sigma). Induced cultures were grown overnight at 30°C with shaking, and then cells were collected the following morning through centrifugation. The cells were lysed with a French pressure cell (Amicon), and rFur was purified from the lysates using fast-protein liquid chromatography (FPLC) in a two-phase manner. rFur was purified using cation exchange, followed by size exclusion. The peak fractions were pooled and stored at 4°C in buffer containing 50 mM sodium phosphate and 500 mM NaCl at pH 8.0. rFur was subsequently diluted into the appropriate buffers for downstream experiments.
EMSA.
Electrophoretic mobility shift assays (EMSAs) were performed to evaluate apo-Fur binding to the promoters of serB, hydA, and the cytochrome c553 gene. All promoter fragments were amplified from WT G27 genomic DNA using the primer pairs given in Table S1. One promoter fragment was selected for hydA (222 bp) and the cytochrome c553 gene (299 bp), while two fragments of various sizes were selected for serB (serB1, 288 bp, and serB2, 173 bp); two fragments were chosen due to the possibility of serB sitting in an operon with pfr. The promoter fragments from pfr (233 bp) and rpoB (142 bp) were used as the positive and negative controls, respectively (10, 26). rpoB expression is not regulated by Fur in either its iron-bound or apo form, and thus, it is routinely used as a negative control in these assays. Promoter fragments were amplified, purified, and end labeled with 32P as previously described (7, 10, 26). EMSAs were performed under iron-free conditions as previously described (10, 11, 26) using apo-binding buffer, 1 ng of labeled promoter fragment, and rFur concentrations of 0.93 μg/ml, 0.186 μg/ml, and 0.093 μg/ml. No-protein control reactions were also performed for each promoter fragment, as well as cold DNA competition reactions using 200 ng of unlabeled promoter DNA and the highest concentration of rFur. Following a 30-min incubation at 37°C, reaction mixtures were separated on polyacrylamide gels containing 5% 19:1 acrylamide, 1× Tris-glycine-EDTA (1× TGE) buffer, and 2.5% glycerol. Electrophoresis was performed in 1× TGE buffer at 70 V for 2 to 3 h depending on fragment size. Gels were then exposed to phosphor screens and analyzed in the same manner as described above for the RPAs. Additionally, to further ensure that any interaction observed between the promoter fragments and rFur occurred only in the absence of iron (apo form of the protein), each of these EMSA reactions was also performed under iron substitution conditions using MnCl2. These iron-bound EMSAs were conducted as previously described (26) using MnCl2-binding buffer, 1 ng of labeled promoter fragment, and rFur concentrations of 0.372 μg/ml, 0.186 μg/ml, and 0.093 μg/ml. No-protein control and cold DNA competition reactions (using 200 ng of unlabeled DNA) were also performed under the iron substitution conditions. Reaction mixtures were separated on polyacrylamide gels composed of 5% 19:1 acrylamide, 1× Tris-glycine (1× TG) buffer, 2.5% glycerol, and 0.133 mM MnCl2, and electrophoresis was performed in 1× TG buffer at 70 V for 2 to 3 h. Gels were exposed to phosphor screens and analyzed as described above.
EMSAs were also performed on the pfr promoter in which the apo-Fur box sequence was scrambled (see section on cloning of mutated pfr promoter sequences). WT pfr was used as the positive control and rpoB as the negative control. The WT pfr promoter fragment and the versions of the promoter in which apo-Fur boxes I, II, and I and II combined were mutated were amplified from WT G27 genomic DNA and DSM1215, DSM1384, and DSM1385 plasmid DNA, respectively, using the pfr EMSA-F and pfr EMSA-R primer pair. The scrambled apo-Fur box III and I, II, and III combined scrambled sequences were amplified from DSM1386 and DSM1387 plasmid DNA, respectively, using the pfr EMSA MUT3-F and pfr EMSA-R primer pair. rpoB was amplified as described above. EMSAs were performed on these promoter fragments under iron-free conditions as detailed above but with the following concentrations of rFur: 1.406 μg/ml, 0.703 μg/ml, and 0.141 μg/ml. The percentage of unbound, labeled pfr WT or scrambled promoter was quantitated for each EMSA replicate using densitometry in a manner analogous to that employed for the RPA studies. The average percentage of unbound promoter was also calculated to serve as a means of comparing the effect of the various scramble mutations on binding to apo-Fur.
hydABCDE and pfr-serB-fucT gene junction PCR.
Qualitative reverse transcription-PCR was conducted on the hydABCDE and pfr-serB-fucT gene junctions to determine whether hydA and serB were expressed as independent genes or as part of a larger operon. These junctional PCRs were performed as previously described (15). Briefly, cDNA and no-RT control reactions were synthesized from RNA isolated from exponential-phase cultures of WT G27 as described above for the RT-PCR analysis. One microgram of cDNA (or the no-RT reaction) was combined with 3 pmol each of the forward and reverse primer pairs as given in Table S1 in the supplemental material in a GoTaq green master mix (Promega). PCR cycling, product separation, and visualization were conducted exactly as previously described (15). Using biologically independent cDNA templates, each junctional PCR was conducted twice, with similar results.
Primer extension and TSS mapping of new apo-Fur gene targets.
Primer extension and transcriptional start site (TSS) mapping were performed on the promoter regions of hydA, serB, and the cytochrome c553 gene from H. pylori G27. These experiments were conducted as previously described (15). RNA was isolated from WT G27 H. pylori in exponential phase as described above for the RPA analyses. Five micrograms of RNA was combined with 10 pmol of γ-32P-labeled RT primers, as specified in Table S1 in the supplemental material, and the avian myeloblastosis virus (AMV) reverse transcription kit (Promega) by following the manufacturer's instructions. Using the Sequenase PCR amplicon sequencing kit and the labeled gene-specific RT primers, dideoxy sequencing reactions were performed according to the manufacturer's instructions. Polyacrylamide gel electrophoresis was utilized to resolve the primer extension and sequencing reactions. Gels were subsequently exposed to phosphor screens and analyzed as detailed above for the RPA analysis. These experiments were conducted a minimum of two times, with comparable results.
Identification of a conserved sequence in apo-Fur-regulated targets.
The promoter sequences of pfr, sodB, hydA, and the cytochrome c553 gene were aligned and analyzed for regions of homology that might constitute an apo-Fur box. A sequence logo was generated using an online Weblogo tool (http://weblogo.berkeley.edu/) according to the methods of Crooks et al. (28). Genamics Expression software (Genamics) was utilized to scan the genome for the frequency of the conserved AAATGA sequence.
Cloning of mutated pfr promoter sequences.
The AAATGA sequence in the first apo-Fur box of the pfr promoter was changed to the scrambled sequence, ACACAC, using splicing-by-overlap-extension (SOE) PCR. Primers PFR_EMSA_F and PFR_ApoMT_SOE_R were used to amplify from the 5′ end of the fragment through the desired mutation; primers PFR_ApoMT_SOE_F and PFR_PE were used to amplify from the desired mutation through the 3′ end of the fragment. Each PCR product was gel purified using a QIAquick gel extraction kit (Qiagen). Fifty nanograms of the clean PCR products was combined with the PFR_EMSA_F and PFR_PE primers in a SOE reaction mixture. The resulting PCR products were again gel purified and cloned into the pGEM-T Easy cloning vector (Promega). Proper insertion of the desired fragment was confirmed by sequencing with the PFR_EMSA_F and PFR_PE primers; the E. coli strain bearing this construct was named DSM1215. Using the same procedure, the second AAATGA sequence in the apo-Fur box of the pfr promoter was changed to the ACACAC scrambled sequence using the following primer pairs: PFR_EMSA_F and PFR_ApoMT2_SOE_R and PFR_ApoMT2_SOE_F and PFR_PE. These primers were used in conjunction with WT G27 genomic DNA and pDSM1215 as templates in order to create pfr promoters in which only the second apo-Fur box was mutated or both boxes I and II were mutated, respectively. The E. coli strain carrying the apo-Fur box II mutation only was DSM1384, while strain DSM1385 carried the apo-Fur box I and II scramble sequences. These mutations were confirmed by sequencing with the PFR_EMSA_F and PFR_PE primers. Mutated pfr promoter fragments, which contained the scrambled sequence (ACACAC) in place of the third apo-Fur box as well as scrambled sequences in place of all three apo-Fur boxes, were synthesized as gBlock gene fragments by Integrated DNA Technologies (IDT;Coralville, IA). These fragments were also cloned into pGEM-T Easy and confirmed using SP6 and T7 universal primers. DSM1386 and DSM1387 are the E. coli strains bearing the apo-Fur box III only and apo-Fur box I, II, and III mutations, respectively.
DNase I footprinting.
DNase I footprinting reactions were performed on labeled fragments of the target promoter regions using rFur. pfr WT and scrambled promoter fragments were amplified from WT genomic G27 DNA and pDSM1215 DNA, respectively, using forward primer, pfr_EMSA_F, and the 6-carboxyfluorescein (FAM)-labeled reverse primer, pfr_PE_FAM, and the GoTaq green master mix (Promega). A longer fragment of the WT pfr promoter (pfr large) was also amplified with primers PFR_large_ftptng_F and pfr_PE_FAM using GoTaq green master mix. The hydA promoter fragment was amplified using WT genomic G27 DNA, the FAM-labeled forward primer, hydA_FAM_F, the reverse primer, hydA_PE, and FideliTaq PCR master mix (Affymetrix). All labeled PCR promoter fragments were purified using a QIAquick PCR purification kit (Qiagen) and eluted in 20 μl of the elution buffer (EB) contained in the kit. Fifteen to 25 μg of labeled WT or scrambled pfr fragment, 50 μg of labeled pfr large, and 50 μg hydA fragment were incubated with 20 μl of 4.9-μg/ml rFur, 7.0-μg/ml rFur, and 24.5-μg/ml rFur, respectively, and 20 μl of DNase I footprinting binding buffer (10 mM Tris-HCl [pH 8.0], 50 mM NaCl, 10 mM KCl, 1 mM dithiothreitol [DTT], 0.1% NP-40, 10% glycerol, 0.1 mg/ml of sheared salmon sperm DNA, and 200 μM EDTA) in a 50-μl total volume reaction. These reaction mixtures were incubated at 37°C for 30 min prior to DNase I digestion treatment in order to allow for binding of the protein to the DNA template. Following this incubation, 0.5 μl of 250 mM CaCl2 was added to the reactions prior to the addition of 0.125 U of DNase I (New England BioLabs) for the WT or scrambled pfr fragment and the hydA fragment. Footprinting reactions with the pfr large fragment utilized 0.250 U of DNase I. DNase I reaction mixtures were incubated at room temperature for 80 s for pfr WT, scrambled, and pfr large templates and 120 s for the hydA template. DNase I digestion reactions were halted through the addition of 150 μl of DNase I stop solution (5) and placed immediately on ice. Digestion reaction mixtures were purified using a MinElute reaction cleanup kit (Qiagen) per the manufacturer's instructions, and cleaned digestion fragments were eluted in 20 μl of EB. To prepare the digestion fragments for fragment analysis (29), the purified samples were dried using a SpeedVac concentrator (Savant) and resuspended in 9.7 μl of HiDi formamide (Life Technologies), and 0.3 μl of GENEScan-600 LIZ size standards (Life Technologies) was added to the samples. The samples (10 μl) were subsequently analyzed using fragment analysis on an ABI 3500XL analyzer (Applied Biosystems) at the Biomedical Instrumentation Center at the Uniformed Services University of the Health Sciences. GeneMapper software, version 4.1 (Applied Biosystems), was used to visualize the results.
Fluorescence anisotropy.
A competitive binding fluorescence anisotropy assay was used to measure the specificity of binding of WT Fur to the pfr promoter. In a quartz cuvette, a 50 nM concentration of a 5′ FluorT-labeled pfr promoter DNA (pfr-FA_LABEL) was mixed with 130 nM WT Fur (65 nM Fur dimers) in buffer containing 40 mM Tris, 150 mM KCl, 2 mM DTT, 600 μg/ml of bovine serum albumin (BSA), 200 μM EDTA, 24% glycerol, and 0.1 mg/ml of sheared salmon sperm DNA at pH 8.0. This solution was maintained at 20°C in the sample chamber of an ISS PC-1 spectrofluorimeter (ISS, Inc., Champaign, IL). Anisotropy was recorded with the excitation and emission polarizers in L-format. The excitation monochromator was set to 495 nm with 8-nm band-pass, and the emission monochromator was set to 526 nm with 8-nm band-pass. Anisotropy measurements were determined from the average of 30 measurements recorded with an integration time of 3 s. The fraction of labeled pfr DNA bound to Fur (fB) was determined from the anisotropy (r) using the expression
where rfree and rbound are the anisotropies of the free and bound DNA, respectively. The quantum yield ratio (Q) was determined to be 0.7 from the ratio of intensities of free and bound DNA (30).
The Fur-labeled DNA complex was titrated with scrambled apo-Fur box DNA sequence (pfr-FA_SCRAM-1) or unlabeled pfr promoter DNA, as a control, and competition was monitored as a decrease in the anisotropy toward saturation (rfree). The raw data (fB as a function of the total concentration of unlabeled DNA, L2t) were simulated using a modified version of a previously described curve-fitting algorithm (31). The fitting algorithm, written in Mathematica (Wolfram Research, Inc.), requires input values for the dissociation constant of the labeled DNA-Fur complex (Kd1), the concentration of labeled DNA (L1t), and the concentration of Fur (Pt, expressed as dimer). Given these input values, the algorithm fits the real roots of a cubic expression to the data and varies the dissociation constant for the unlabeled DNA-Fur complex (Kd2) in order to find the best-fit value that minimizes the residual sum of squares. The error in Kd2 is determined from the variation in this parameter that causes the best-fit residual error to increase by 20%.
Statistical analysis.
In order to determine if there was a significant difference in the fold decrease of expression upon iron chelation of pfr, serB, hydA, and cytochrome c553 between the WT and fur deletion strain backgrounds, Student's t tests were employed. If the fold change differed 2-fold or greater and the P value was ≤0.05, the changes were considered to be significant. Geometric means are reported for the fold changes in the RPA data because the data are comparisons of ratios.
RESULTS
Identification of candidate apo-Fur-regulated gene targets.
We utilized the numerous previous transcriptional and proteomic studies regarding H. pylori Fur regulation (7, 8, 23, 32–34) to identify potential uncharacterized apo-Fur-repressed gene targets. Analysis of those studies showed considerable variability; thus, we imposed the following criteria to narrow the list of genes for study: (I) changes in expression (mRNA or protein) were consistent with apo Fur regulation (i.e., a Fur-dependent decrease in expression in the WT background upon iron chelation); (ii) the genes were present in strains G27 (35), 26695 (13), J99 (36), and HPAG1 (37), which are each well-characterized H. pylori strains; (iii) the genes did not appear to be essential (38); and (iv) the changes in expression exhibited in response to iron chelation and/or fur deletion were substantial (defined as a minimum 2-fold change). Based on these criteria, the cytochrome c553 gene (HP1227) (23), hydA (HP0631) (7, 23, 32, 34), and serB (HP0652) (23, 32, 33) were chosen for further study.
apo-Fur repression of selected gene targets.
To determine whether the cytochrome c553 gene, hydA, and serB were targets for apo-Fur regulation, we first sought to establish that these genes exhibited the changes in expression expected for apo-Fur-repressed genes. Total RNA that had been isolated from WT G27 and an isogenic fur mutant (Δfur) that had been exposed to iron-replete and -depleted conditions was used to monitor expression of the cytochrome c553 gene, hydA, and serB via RNase protection assay. Additionally, pfr was included as a well-characterized, apo-Fur-repressed control gene. Densitometric analysis was used to compare the amount of transcript for each gene under iron chelation conditions to the amount present under normal (iron-abundant) conditions; the fold decrease in expression for four biological replicates is shown in Fig. 1A, in which each experiment is plotted as a point on the graph and the geometric mean fold decrease is plotted as a bar. As expected (5, 10, 11, 25, 26), pfr expression was decreased in the WT strain upon iron chelation, and this decrease was lost in the Δfur strain. Similarly, cytochrome c553 gene, hydA, and serB expression all showed a marked decrease upon iron chelation in the WT background, and this decrease was abrogated in the absence of Fur. Though the changes in serB expression did not reach statistical significance, the differences in the fold changes between the WT and Δfur strain were statistically significant for the cytochrome c553 gene, hydA, and pfr: P values of <0.0001, <0.01, and <0.001, respectively.
Fig 1.
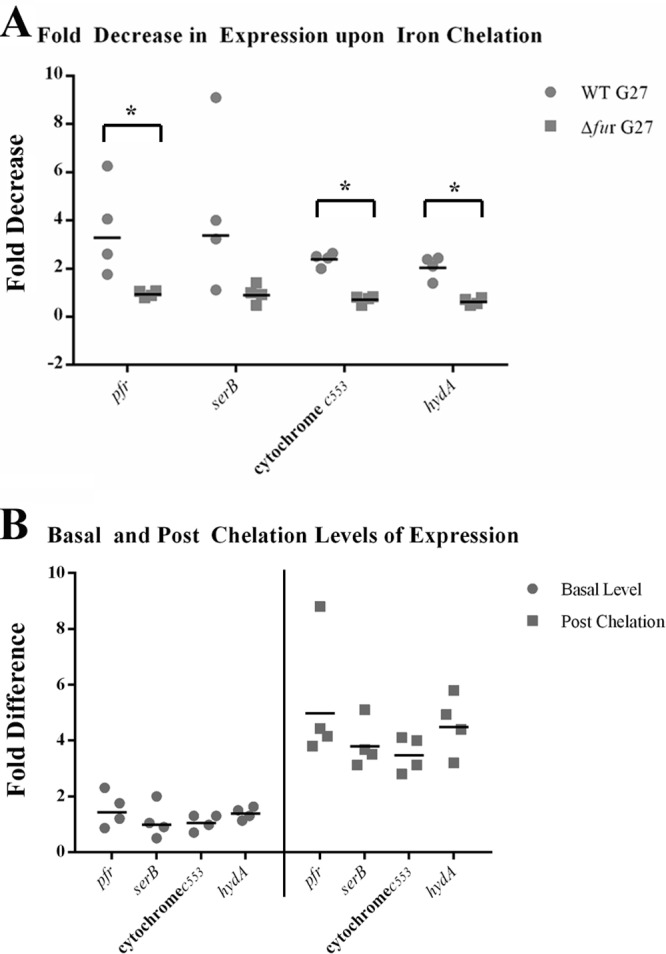
apo-Fur regulation of the cytochrome c553 gene, hydA, and serB. RNA was isolated from exponential-phase cultures of WT and Δfur strains of H. pylori under iron-replete and iron chelation conditions as detailed in Materials and Methods. Riboprobes for the cytochrome c553 gene, hydA, serB, and the control promoter, pfr, were used to evaluate changes in expression of these genes. The fold decrease in expression upon iron limitation is shown in panel A. The fold difference in the basal levels of gene expression between the Δfur and WT strains is shown on the left side of panel B. The fold difference in the postchelation levels of gene expression between the Δfur and WT strains is shown on the right side of panel B. The geometric means of the fold decrease and fold difference in the relative levels of expression from the four biological replicates are shown as black lines. An asterisk above a bracket indicates that there was a statistically significant difference in the fold decrease between WT and Δfur strains upon iron chelation (P ≤ 0.01).
In addition to analyzing the fold decrease in expression for the genes upon iron chelation, we also examined the difference in basal-level gene expression between the Δfur and WT strains under normal (iron-replete) (Fig. 1B, left side) and iron chelation (Fig. 1B, right side) conditions. Change was calculated by dividing the level of expression seen in the Δfur strain by the amount of expression seen in the WT strain. As expected, since apo-Fur would be active only when iron was chelated from the media, the levels of expression of each of the genes were similar in the WT and Δfur strains when iron was present (Fig. 1B, left side). Conversely, after iron chelation, the levels of expression of each of the target gene were considerably higher in the Δfur strain (Fig. 1B, right side). Thus, in agreement with Fig. 1A, apo-Fur is required for repression of pfr, the cytochrome c553 gene, hydA, and serB under iron-limited conditions. Of note, similar results were obtained using RNA harvested from H. pylori strain 26695 (data not shown), demonstrating that apo-Fur repression of the cytochrome c553 gene, hydA, serB, and pfr is conserved across H. pylori strains.
To further verify the RPA results, we performed qRT-PCR analysis using mRNA harvested from biologically independent cultures in which the WT G27 and Δfur were exposed to normal or iron-limited environments. These results were identical to those obtained via RPA, with the exception that because qRT-PCR is more sensitive than RPA, the fold decreases observed for each of the genes were more pronounced than that observed by RPA (data not shown). Taken together, the transcriptional analyses suggest that the cytochrome c553 gene, hydA, and serB are all regulated by apo-Fur in a manner similar to what has previously been shown for pfr (5, 10, 25, 26) and sodB (10, 11).
In vitro binding of apo-Fur to the cytochrome c553 gene, hydA, and serB.
Since the transcriptional data indicated that repression of the cytochrome c553 gene, hydA, and serB under iron-limited conditions required Fur, we next asked if apo-Fur was working directly or indirectly to regulate expression of these genes. To determine if the regulation was direct or indirect, we analyzed the ability of rFur to bind to predicted promoter fragments of each of the genes via electrophoretic mobility shift assays (EMSAs). The promoter fragment of pfr was used as a positive control (10, 26), and the promoter fragment of rpoB, a constitutively expressed, non-Fur-regulated gene, was used as a negative control for nonspecific binding (7, 10, 26). If apo-Fur was directly regulating expression of these genes, we expected to see binding to the promoters and a concomitant change in the migration of the promoter fragment compared to the no-protein control reaction. Indeed, as increasing amounts of apo-Fur were added to the reactions, a shift in the migration pattern was observed for the pfr control as well as hydA and the cytochrome c553 gene (Fig. 2). The hydA promoter fragment displayed two upward shifts in migration: the first was present at all protein concentrations, while the highest level of shift was observed only at the highest protein concentration (Fig. 2). The presence of the second higher shift could indicate the presence of more than one binding site for apo-Fur. A single shift was observed for the cytochrome c553 promoter (Fig. 2). As expected from previous EMSA analysis (26) and footprinting experiments (5) that showed three protected regions for this promoter, the pfr promoter fragment underwent three distinct changes in migration as the concentration of apo-Fur was increased (Fig. 2). The interaction between apo-Fur and these promoters was specific because it could be competed away by the addition of excess unlabeled promoter fragment to the reaction with the highest apo-Fur concentration, thus returning the banding pattern of the labeled fragment to that of the no protein control (Fig. 2, lanes C). Together, these data indicated that apo-Fur is capable of binding to the promoters of hydA and the cytochrome c553 gene. In contrast, and similar to what was seen for the rpoB negative control (Fig. 2), apo-Fur did not appear to interact with two separate potential serB promoter fragments of various sizes; even at the highest concentration of apo-Fur, there was no distinct alteration in the banding pattern of the labeled serB promoter fragments (Fig. 2).
Fig 2.
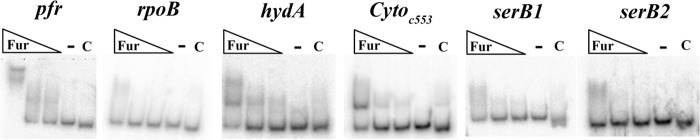
Determination of apo-Fur binding to the cytochrome c553 gene, hydA, and serB promoters. EMSAs were performed using purified rFur protein and end-labeled PCR promoter fragments for the cytochrome c553 gene, hydA, and serB. Labeled pfr and rpoB promoter fragments were used as the positive and negative controls, respectively. Increasing concentrations of protein are indicated by the triangles, the no-protein control reactions are indicated by a minus signs, and the cold (unlabeled) competition reactions are indicated by the letter C. EMSAs were performed with apo-Fur (0.093 μg/ml, 0.186 μg/ml, and 0.93 μg/ml of protein), and data are representative of 2 or 3 experimental replicates.
To ensure that the interaction between apo-Fur and the promoters of the predicted apo-Fur-regulated genes was specific to the apo form of Fur and not just the consequence of the general functionality of Fur as a DNA binding protein, we repeated the EMSAs using iron substitution conditions to mimic iron-bound Fur. Even at the highest concentrations of rFur, there was no distinct shift of the banding patterns for any of the examined genes (data not shown). Given the alteration in the migration of the labeled promoter fragments for the cytochrome c553 gene and hydA only in the presence of apo-Fur (Fig. 2 and data not shown), and Fur-dependent changes in expression seen upon iron chelation (Fig. 1A), we concluded that these genes are directly regulated by apo-Fur. Conversely, while serB showed the appropriate changes in expression for an apo-Fur-regulated gene (Fig. 1A and B), the regulation by apo-Fur appears to be indirect, since there was no change in the migration of the labeled serB promoter fragment in the presence of apo-Fur.
Operon analysis of serB and hydA.
Given that serB expression followed the basic pattern for an apo-Fur-regulated gene, but apo-Fur does not directly bind to the promoter for this gene (Fig. 2), we reasoned that apo regulation of serB could be due to cotranscription of serB with its neighboring gene, pfr, which is a known apo-Fur-repressed gene. Indeed, analysis of the genome sequence of G27 (35) showed that pfr-serB-fucT were arranged in such a fashion as to suggest that the genes were expressed as an operon under the control of the pfr promoter (Fig. 3A). Furthermore, in H. pylori strain 26695, these genes have been suggested to be cotranscribed as an operon (39). However, whether these genes constitute an operon in G27 or any other strain had not yet been directly determined. Therefore, gene junction PCR was performed on cDNA using primers that spanned the junctions between pfr and serB as well as serB and fucT. As shown in Fig. 3A and B, PCR product was generated from cDNA for each of these junctions only when reverse transcriptase (RT) was added to the reaction mixtures, indicating that these genes are all cotranscribed. These results suggest that apo-Fur regulation of serB occurs indirectly through regulation of the pfr promoter.
Fig 3.
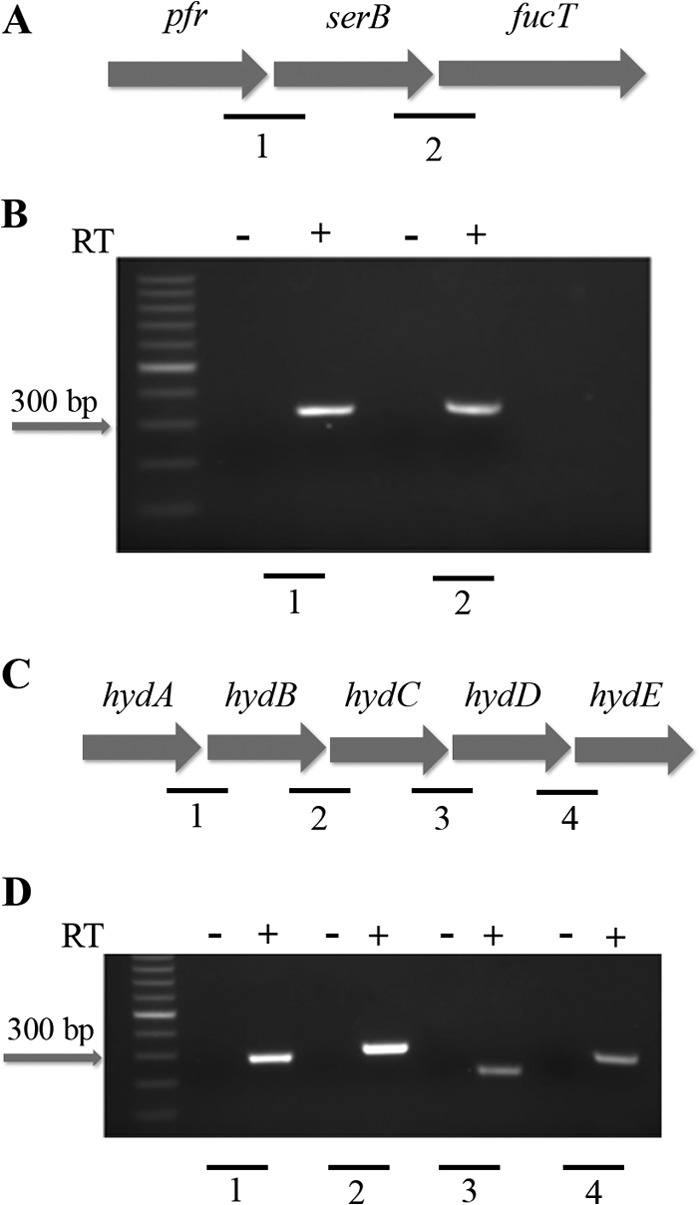
Organization of the pfr and hydA operons. The genes encoding pfr, serB, and fucT are organized into an operon as shown in panel A. Panel B shows the PCR amplification of the gene junctions between pfr and serB and serB and fucT using cDNA (+) and no-RT (−) control reactions as templates. The organization of the hydACBDE operon is shown in panel C. Panel D shows the PCR amplification of the hydAB, hydBC, hydCD, and hydDE gene junctions using cDNA (+) and no-RT (−) control reactions as templates. The individual junctions are indicated by numbers (A and C), which correspond to the numbers shown below the gel images (B and D). Each junctional PCR was conducted twice using biologically independent cDNA templates, with similar results.
Additionally, based on the genomic organization in H. pylori G27 (Fig. 3C), we were also interested in determining if hydA was the first gene in an operon with the remaining hyd genes, which are all necessary for the formation of the functional quinine-reactive Ni-Fe hydrogenase. Though not as pronounced as the changes seen for hydA, hydB and hydC have also been shown to exhibit changes in expression that are consistent with apo-Fur repression (23). hydA has been suggested to be the first gene in the hydABCDE operon in H. pylori strain 26695 (39), but whether or not these genes constitute an operon had not been directly examined in any H. pylori strain. Once again, junctional PCR was performed as indicated in Fig. 3C. Products were generated only when RT was added to the reaction mixtures for the junctions between hydA and hydB, hydB and hydC, hydC and hydD, and hydD and hydE (Fig. 3D). Together, these data indicate that these genes are all cotranscribed and constitute an operon in H. pylori.
Primer extension and TSS mapping.
Given our evidence that these genes are directly regulated by apo-Fur, we next sought to better define the promoter elements for the cytochrome c553 gene and hydA. To this end, primer extension analysis was performed on RNA isolated from WT G27 to identify the transcriptional start site (TSS) for both genes. As shown in in Fig. 4A, primer extension of the cytochrome c553 gene yielded a single cDNA product. The TSS for the cytochrome c553 gene was identified as the adenosine nucleotide located 29 bases upstream from the predicted start codon (Fig. 4B). Further upstream, a well-conserved −10 (TATCAT) and less well-conserved −35 promoter element (TAAGCA) were also identified (Fig. 4B). These data agree with TSS and promoter elements predicted for the cytochrome c553 gene in strain 26695 (39). A single cDNA product was also observed for the hydA primer extension reaction (Fig. 4C). The hydA TSS was determined to be the guanine nucleotide located 25 bases upstream from the predicted start codon (Fig. 4D), which differs by a single nucleotide from the predicted TSS for hydA in 26695 (39). A well-conserved −10 (TATGTT) and less well-conserved −35 (TAATCA) promoter element were also identified (Fig. 4D).
Fig 4.
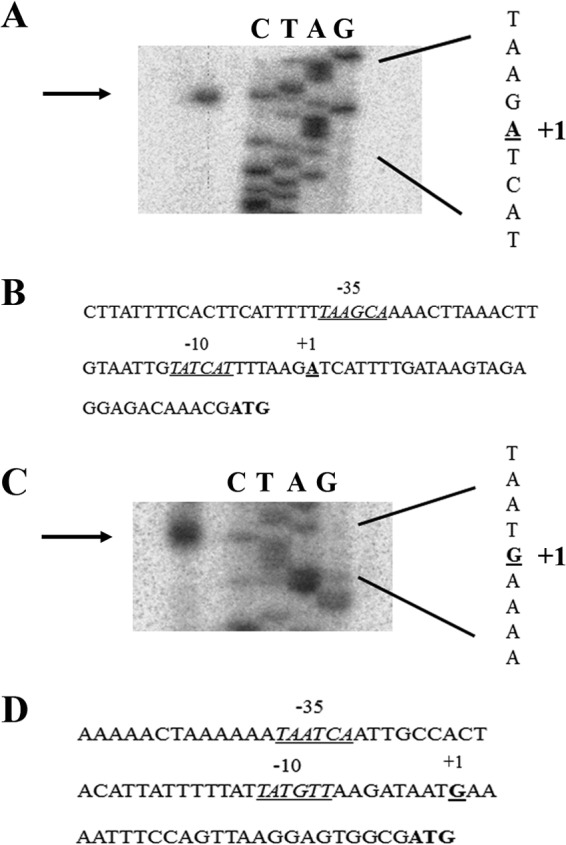
Mapping of the cytochrome c553 gene and hydA TSSs. The TSS of the cytochrome c553 gene was mapped using WT H. pylori RNA. Using the cyto_c553_PE primer, which lies within the cytochrome c553 coding region, first-strand cDNA was synthesized. Only a single band was detected, indicating a single TSS for this gene as shown in panel A. Panel B shows the cytochrome c553 gene promoter region, with the core promoter elements indicated. The TSS of the hydA promoter was mapped in a similar manner using the hydA_PE primer. A single band was detected for this promoter, which indicated a single TSS for hydA as shown in panel C. The hydA promoter region with the core promoter elements indicated is shown in panel D. The TSS is indicated by “+1”; the −10 and −35 promoter elements are underlined and labeled accordingly. The translational start codon is given in bold. Data shown are representative of a minimum of two experimental replicates.
Identification of a putative apo-Fur box consensus sequence.
Given that previous attempts to identify a conserved DNA sequence that is recognized by apo-Fur (apo-Fur box) have been largely unsuccessful due to the small number of previously known apo-Fur-regulated genes (5, 10, 11), we reasoned that the addition of the cytochrome c553 gene and hydA, both of which are suggested to be directly apo-Fur repressed by our data, could help to identify such a conserved apo-Fur box. Therefore, the promoter regions of the cytochrome c553 gene and hydA along with the previously characterized apo-Fur repressed genes, pfr (5) and sodB (10, 11), were analyzed to identify any regions of homology. This analysis revealed a 6-bp sequence, AAATGA, which was found at least once in all of the apo-Fur-repressed gene promoter regions. Some of the promoters contained more than one of these conserved sequences; the pfr and hydA promoters contained two, while the cytochrome c553 gene promoter contained three. Unlike the consensus sequence for Fe-Fur, this sequence is not palindromic and as a result was identified on only one strand of the DNA sequence and always reading AAATGA in the 5′ to 3′ orientation. Of note, the AAATGA sequence is located within the previously identified apo-Fur DNase I-protected regions of the pfr and sodB promoters (5, 11), which suggests that this sequence is bound by apo-Fur. Furthermore, the conserved sequence was found to overlap the −10 and/or −35 region for pfr, sodB, and the cytochrome c553 gene, which is consistent with apo-Fur repression; binding of apo-Fur would block the ability of the RNA polymerase (RNAP) to interact with the promoter. Based on the presence of the conserved sequence, we next used the DNA sequences surrounding the AAATGA to generate a sequence alignment (Fig. 5A) as well as a Weblogo (28) to identify any further common sequence elements in the promoter regions for all four of these genes (Fig. 5B). As shown in Fig. 5B, there appears to be very little further sequence conservation surrounding the AAATGA sequence. Combined, these data may suggest that this 6-bp sequence constitutes the core of the apo-Fur box.
Fig 5.
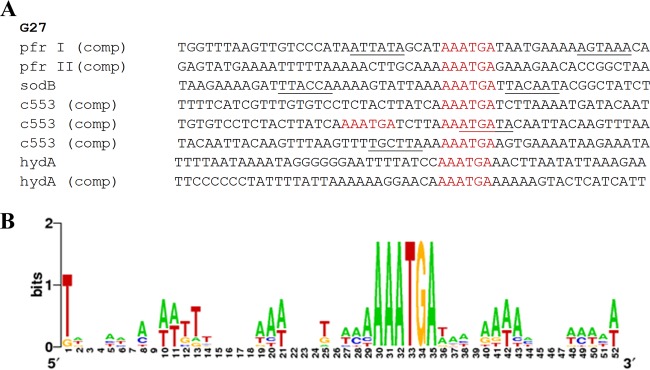
Identification of the apo-Fur box consensus sequence. The promoter regions of the four characterized H. pylori apo-Fur repressed genes, pfr, sodB, the cytochrome c553 gene (c553), and hydA, were aligned (A), and a sequence logo (28) was generated (B). The logo is composed of stacks of letters, one for each position of the sequence. Sequence conservation at each position is indicated by the height of the letter stack (measured in bits). The apo-Fur box sequence(s) found in each promoter is colored in red. Other nearby promoter elements (−10 and −35 conserved sequences) are underlined. “comp” indicates that the complementary strand of DNA was used in the alignment. Designations of pfr I and pfr II are from the previously described protected regions (5).
DNase I footprinting.
As mentioned above, the conserved AAATGA sequence is located within the apo-Fur DNase I-protected regions of the pfr and sodB promoters (5, 11). We therefore wondered if this region would similarly be protected in the cytochrome c553 gene and hydA promoters. To determine this, DNase I footprinting was conducted on fluorescently labeled promoter fragments of the cytochrome c553 gene and hydA, along with the control, pfr. Digestion reactions were performed in the presence or absence of apo-Fur, and digestion products were visualized by fragment analysis (29).
Since the previous footprinting studies of pfr (5) were conducted using older technologies that utilized radioactively labeled fragments and sequencing gels, we first analyzed the pfr control fragment to confirm that nonradioactive fragment analysis gave results similar to those in the published studies. When we used a 3′-end-labeled pfr promoter fragment, we observed two distinct regions of protection that spanned from approximately 68 to 100 and 144 to 167 bases (Fig. 6A). In the first protected region, there was a prominent hypersensitivity peak found at 83 bases. This protected region, which lies within the −10 and −35 promoter elements (Fig. 6C), encompassed the first AAATGA sequence (bases 95 to 100) found in the pfr promoter. The second protected region, though less pronounced than the first, encompasses the second AAATGA sequence (bases 148 to 153) (Fig. 6C). We repeated the footprinting reactions using a 5′-end-labeled pfr promoter fragment, and we found similar protected regions surrounding the first and second AAATGA sequences within this promoter (data not shown). This suggests that apo-Fur is able to bind to the pfr promoter on both strands of the DNA molecule. As shown in Fig. 6C, both of the identified protected regions for the pfr promoter region overlap at least part (second region) or nearly all (first region) of the protected regions previously identified for this promoter (5), which suggests that fragment analysis can reproducibly identify areas bound by apo-Fur. Given that the original footprinting of the pfr promoter showed three protected regions (5) and our results showed only two, we hypothesized that we did not detect the third protected region due to its location at the end of our footprinting fragment (Fig. 6C). Therefore, we generated a larger labeled pfr promoter fragment and repeated the analysis. As shown in Fig. S1A in the supplemental material, using the large pfr fragment, we now identified a third region of protection in the presence of apo-Fur. Using this fragment, the protected regions span bases 63 to 109 (with the hypersensitivity peak at 83 bp), 140 to 153, and 182 to 221 (see Fig. S1A). Interestingly, the third protected region contains a sequence, TAATGA, which is similar to the apo-Fur box consensus sequence, with only a single mismatch (see Fig. S1B).
Fig 6.
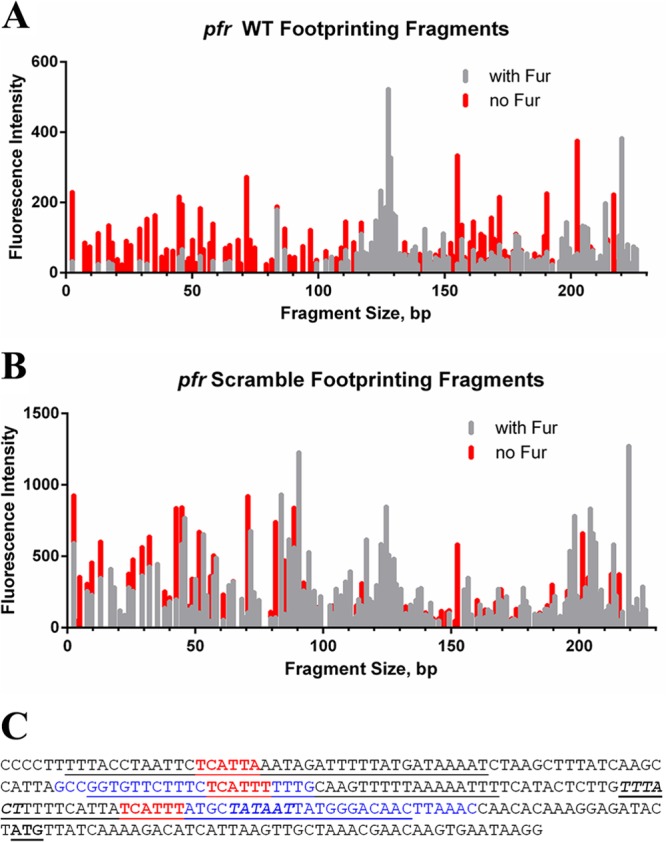
DNase I footprinting of the pfr promoter. A fragment of the pfr promoter fluorescently labeled at the 3′ end was subjected to DNase I digestion in the absence and presence of apo-Fur (A). A fragment of the pfr promoter fluorescently labeled at the 3′ end and containing ACACAC scrambled sequence in place of AAATGA in the first apo-Fur box was subjected to DNase I digestion in the absence and presence of apo-Fur (B). Protected regions are those with reduced peak height and/or entirely missing peaks in the presence of apo-Fur (gray lines) compared to digestion fragments in the absence of Fur (red lines). (C) Sequence of the pfr promoter fragment utilized in the footprinting experiments. The conserved apo-Fur box sequences are in red, and the protected regions as identified through DNase I footprinting are in blue. The −10 and −35 promoter elements are shown in bold italics; the ATG start codon is in bold and underlined. The previously reported protected regions for the pfr promoter (5) are underlined.
Given these results, we next analyzed a 5′-end-labeled hydA promoter fragment. Fragment analysis revealed a protected region spanning bases 28 to 44 of the fragment that was marked by peaks that were either entirely missing or drastically diminished in the presence of apo-Fur (Fig. 7A). An additional region with greatly diminished peak size and some missing peaks was also identified in the hydA promoter between bases 74 and 100 (Fig. 7A). One of the predicted AAATGA sequences lies within the protected region that spans bases 74 to 100, which is closer to the core promoter elements (Fig. 7B). The other predicted sequence, which is further upstream from the core promoter elements, is located immediately downstream from and adjacent to the protected region, which spans bases 28 to 44; this sequence is encoded on the opposite DNA strand (Fig. 7B). These data suggest that apo-Fur might bind to both strands of the DNA in this region. Thus, we also conducted footprinting analysis using a 3′-labeled hydA promoter fragment. Unlike what was seen with the pfr promoter, we identified no clearly defined protected regions using the 3′-labeled fragment (data not shown). This suggests that apo-Fur can bind to both strands of DNA at some, but not all, promoters and that sequence orientation of the apo-Fur box with regard to the direction of transcription may be important for apo-Fur function.
Fig 7.
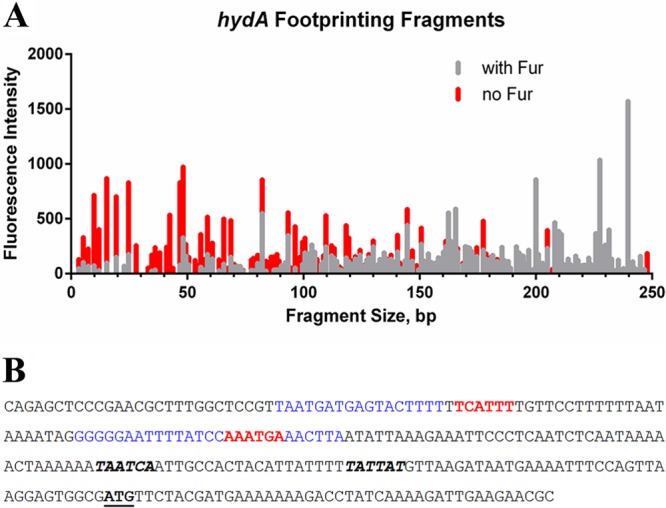
DNase I footprinting of the hydA promoter. A fragment of the hydA promoter fluorescently labeled at the 5′ end was subjected to DNase I digestion in the absence and presence of apo-Fur (A). Protected regions are those with reduced peak height and/or entirely missing peaks in the presence of apo-Fur (gray lines) compared to digestion fragments in the absence of Fur (red lines). (B) Sequence of the hydA promoter fragment utilized in the footprinting experiments. The conserved apo-Fur box sequences are in red, and the protected regions as identified through DNase I footprinting are in blue. The −10 and −35 promoter elements are shown in bold italics; the ATG start codon is in bold and underlined.
Given the differing results for pfr and hydA in terms of the ability of apo-Fur to bind both strands, we utilized both 3′- and 5′-labeled cytochrome c553 gene promoter fragments for DNase I footprinting and fragment analysis. Unexpectedly, despite the fact that there are three predicted apo-Fur boxes within this promoter, no clear protected regions could be distinguished for either cytochrome c553 gene fragment (data not shown). The reason for this inability to demonstrate any protection is currently unclear. This is particularly true given the fact that the EMSA data show a very distinct change in the banding pattern in the presence of the apo-Fur protein (Fig. 2).
Importance of the AAATGA sequence in apo-Fur binding.
Given that the AAATGA sequence is found within the apo-Fur-protected regions of hydA and pfr, we next wanted to directly explore the role that this sequence plays in apo-Fur regulation. To this end, we first attempted to utilize pfr promoter green fluorescent protein (GFP) transcriptional fusions (25) that either were WT or contained mutations where the two AAATGA sequences in the pfr promoter were changed to AAACGC. Unfortunately, qRT-PCR analysis of gfp expression for the strains containing the promoter mutations showed that mutation of the conserved sequence resulted in a negligible amount of gfp expression compared to the WT pfr promoter fusion (data not shown). Due to the proximity of the first AAATGA sequence to the −10 promoter element, it is very likely that mutations within this region disrupted transcription initiation (14, 39, 40). Thus, we concluded that we would not be able to address the role of the AAATGA sequence within the context of transcriptional fusions. Therefore, we opted to study the importance of the AAATGA sequence using DNase I footprinting, fluorescence anisotropy (FA), and EMSAs.
DNase I footprinting was performed on a 3′-end-labeled pfr promoter fragment that was identical to what was utilized above except that the first AAATGA sequence (box I, closest to the −10 promoter element) was mutated to ACACAC. Footprinting reactions and fragment analysis were conducted in the same manner as for the previous assays with the WT promoter fragments. As shown in Fig. 6B, the fragment pattern of the scrambled pfr promoter with apo-Fur (gray lines) is nearly identical to that of the scrambled pfr promoter in the absence of protein (Fig. 6A, red lines). In comparison to results with the WT pfr promoter, mutation of the AAATGA sequence resulted in a loss of protection in the first region (bases 68 to 100) (Fig. 6B). Furthermore, the second protected region that spanned bases 144 to 167 in the WT pfr footprint was truncated in the scrambled pfr promoter fragment; a very small region of protection from bases 146 to 152 (Fig. 6B) was observed. These data suggest that the AAATGA sequence is important for apo-Fur binding to its target promoters and indicate that there may be a hierarchy among the binding regions and/or a cooperative effect; lack of apo-Fur binding at the first (mutated) site resulted in diminished binding and protection at the second, unaltered site.
Next, FA experiments were also performed using a small labeled portion of the pfr promoter that contained the first AAATGA sequence. Previous FA data from our group showed that apo-Fur was able to bind to this segment of the pfr promoter (J. J. Gilbreath and D. S. Merrell, unpublished data). We therefore investigated the ability of an unlabeled WT pfr promoter fragment or a fragment in which the AAATGA sequence was mutated to ACACAC to compete for binding with the labeled fragment. As shown in Fig. 8, as increasing amounts of unlabeled WT pfr promoter fragment were added to the labeled-pfr promoter fragment–apo-Fur mixture, the unlabeled fragment competed for binding to the protein and titrated out the labeled fragment; the Kd (dissociation constant) of apo-Fur for WT pfr was determined to be 14 ± 5 nM. In contrast, the fragment in which AAATGA was changed to ACACAC was unable to compete for binding (Fig. 8). The inability of the mutated promoter fragment to titrate the labeled pfr promoter fragment further supports the notion that the AAATGA sequence is required for apo-Fur binding.
Fig 8.
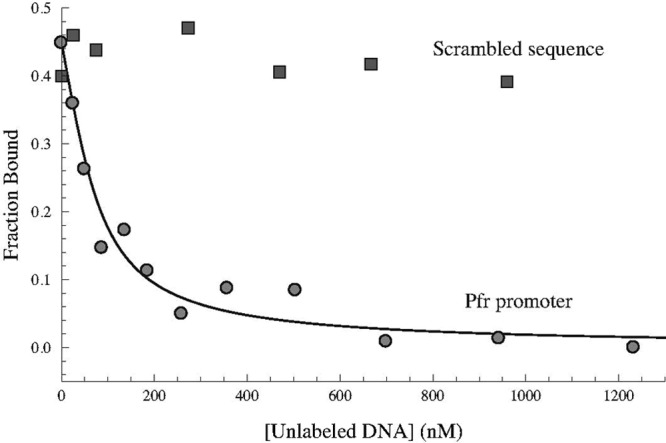
Specific interaction between apo-Fur and the apo-Fur box consensus sequence. Unlabeled WT and scrambled pfr promoter sequences were used to titrate labeled WT pfr promoter bound to apo-Fur in fluorescence anisotropy studies. As the concentration of unlabeled WT pfr promoter was increased (circles), it competed with the labeled promoter for binding to apo-Fur and successfully titrated the labeled promoter fragment. The Kd was determined to be 14 ± 5 nM, and the R-squared value for the best fit line was 0.981. Increasing concentrations of unlabeled scrambled pfr promoter fragment (squares) were unable to titrate the labeled WT pfr fragments from apo-Fur.
While the FA experiments gave us valuable insight into the role of the AAATGA sequence in apo-Fur binding to the promoter, this type of analysis only afforded us the opportunity to examine a single apo-Fur box in isolation from the rest of the promoter. Therefore, we also performed EMSAs on mutated pfr promoter fragments in which each predicted apo-Fur box was scrambled (ACACAC) alone or in combination with the others. As shown in Fig. 9, when apo-Fur boxes I and II were independently scrambled, the observed overall shifts in the banding pattern were similar to the results for the WT pfr control (a low, middle, and high shift). However, the efficiency of the shifts and overall banding pattern were not nearly as distinct as those of the WT control. For example, for the box II scramble, the low-shift band was barely discernible at the lowest concentration of apo-Fur. Conversely for the apo-Fur box III scramble, the low and middle shifts in banding pattern were distinct, but not the highest shift; this band was lost even at the highest concentration of apo-Fur (Fig. 9). When the apo-Fur box I and II scrambled mutations were combined, the low shift was observed at the lowest concentration of protein, but the middle shift was less distinct and the high shift was not observed as the protein concentration was increased (Fig. 9). Lastly, when the pfr promoter fragment was synthesized to contain scrambled sequence in all three predicted apo-Fur boxes, no distinct shift in the banding pattern was observed; there was some nonspecific interaction between this fragment and the protein that resulted in a smear above the unbound bands in the reactions with the two highest concentrations of protein (Fig. 9).
Fig 9.

Determination of the role of the AAATGA sequences in apo-Fur binding to the pfr promoter. EMSAs were performed using purified rFur protein and end-labeled promoter fragments for pfr. Box I, box II, and box III indicate fragments where the AAATGA sequence has been scrambled alone or in various combinations. Labeled WT pfr and rpoB promoter fragments were used as the positive and negative controls, respectively. Increasing concentrations of protein are indicated by the triangles, the no-protein control reactions are indicated by minus signs, and the cold (unlabeled) competition reactions are indicated by the letter C. EMSAs were performed with apo-Fur (0.406 μg/ml, 0.703 μg/ml, and 0.141 μg/ml of protein), and data are representative of 2 or 3 experimental replicates. The average percentage of unbound labeled promoter from all of the experimental replicates is given below each reaction lane.
For each of the various scrambled mutations (singles and combinations), the shifting patterns are less clear than those of the WT pfr promoter control (Fig. 9). This suggests less specific interaction between the protein and the scrambled promoter fragment. Thus, we also attempted to quantitate the level of interaction through determination of the percentage of unbound promoter fragment in each reaction; decreased interaction should increase the amount of unbound probe. Quantitation showed that as a general trend, there was more unbound material in the reaction mixtures containing the various scrambled mutations than in the corresponding reaction with the WT pfr promoter fragment. This effect was most pronounced when comparing the middle concentration of protein. At this concentration of apo-Fur, 3.9% of the WT pfr promoter was unbound (i.e., not shifted), but the scrambled mutations in box I, box II, and box III show larger amounts of unbound promoter fragment, 9.8%, 20.8%, and 38.1%, respectively (Fig. 9). This trend holds true with the combined box I and II scrambled promoter fragments (16.9%) as well as the box I, II, and III combined scrambled fragment (44.3%) (Fig. 9). Given that the box I, II, and III combined scrambled promoter fragment showed no distinct change in the banding pattern at any protein concentration, the percentage of unbound material in each of these reactions was higher than the corresponding reactions in virtually all of the other promoter fragments (scrambled and WT) (Fig. 9).
Combined, the visual and quantitative analyses suggest that within the confines of the single box mutations, the loss of box III displayed the greatest effect on the ability of apo-Fur to bind to the pfr promoter. Indeed, box III appears sufficient to promote apo-Fur binding, since the low shift band was visible in the box I and II combined scrambled fragment. However, introducing the box III scramble results in a loss of all shifting for the triple box mutant. These data suggest that the third predicted apo-Fur box plays a significant role in facilitating the DNA-protein interaction. En Masse, the data presented herein show that the cytochrome c553 gene and hydA are members of the apo-Fur regulon, and the data suggest that the identified AAATGA sequence constitutes the first identified core apo-Fur box consensus sequence.
DISCUSSION
H. pylori has relatively few transcriptional regulators for a genome of its size (13), so the regulatory factors that it does encode are utilized more broadly than their counterparts in other organisms. This is true of Fur. Indeed, Fur regulation in H. pylori is arguably the most complex form of Fur regulation identified in the bacterial realm. Not only does Fur function as a repressor and activator but also it does so with and without its iron cofactor. This capacity to regulate different sets of genes in four distinct manners (iron-bound repression, iron-bound activation, apo repression, and apo activation) means that the regulon of genes controlled by Fur in H. pylori is extensive (4–7, 10–12, 14–16, 23, 33, 41, 42). Therefore, it is often considered to be a “master regulator” in this organism.
Our understanding of nonclassical Fur regulation (i.e., iron-free or apo) is still limited at best. Therefore, we sought to expand the number of characterized gene targets that are repressed by apo-Fur in H. pylori and to begin to characterize the binding sequence for apo-Fur. Herein we show that serB, the cytochrome c553 gene, and hydA are all repressed by apo-Fur, though the mechanisms of this repression are direct only for the cytochrome c553 gene and hydA. While serB, a phosphoserine phosphatase gene, showed transcriptional changes indicative of an apo-Fur repressed gene, serB was cotranscribed with pfr and fucT (Fig. 3A and B), and apo-Fur did not bind to the fragment of DNA that would be predicted to contain an independent promoter for serB if one existed (Fig. 2). Thus, apo-Fur-dependent regulation of serB occurs indirectly via control of the pfr promoter.
In contrast to the indirect regulation observed for serB, we found that the cytochrome c553 gene and hydA were both directly regulated by apo-Fur (Fig. 1 and Fig. 2). Cytochrome c553 is a small c-type cytochrome which is responsible for transferring electrons from cytochrome reductase to a cbb3-type terminal oxidase (43). Given that iron is a component of the single heme C motif found in cytochrome c553 (43), it seems logical that expression of this gene is regulated by Fur. Due to the critical role of cytochrome c553 in the electron transport chain, it makes sense that H. pylori would want to express its gene under favorable conditions and then repress expression under unfavorable conditions in which the iron cofactor was unavailable. hydA is the first gene in the hydABCDE operon (Fig. 3C and D), whose components constitute a Ni/Fe hydrogenase. This Ni/Fe hydrogenase was first identified in H. pylori in 1996 (44) and was subsequently shown to allow H. pylori to utilize molecular H2 as an energy source and to enhance colonization in mice (45). Given that this enzyme is cofactored by both iron and nickel, it is perhaps not surprising that the hydABCDE operon is regulated by Fur. Repression by apo-Fur would ensure that expression of the hydrogenase occurs only under conditions in which the iron cofactor is available. hydB and hydC were both previously predicted to be part of the apo-Fur regulon (23, 33), although our data suggest that this regulation most likely occurs through interaction at the hydA promoter (Fig. 3C and D).
In addition to their regulation by apo-Fur, hydA, and consequently the entire hyd operon, is also repressed by NikR (46, 47); NikR is a nickel-cofactored transcriptional regulator. Given the importance of nickel as a hydrogenase cofactor, regulation of hydA expression via nickel availability seems logical. Indeed, coregulation of the hyd operon by Fur as well as NikR likely allows for very precise modulation of the expression of this operon. The interplay between nickel and iron in gene regulation seems to be a common theme in H. pylori; the Fur and NikR regulons share several genes, including pfr, fur, nikR, and exbB (14, 46–49). Thus, control of expression of key genes via multiple regulators is likely a mechanism utilized by H. pylori to maximize the effectiveness of the few transcriptional regulators encoded in the genome.
The addition of the cytochrome c553 gene and hydA to the list of apo-Fur-regulated genes allowed us to identify a 6-bp sequence (AAATGA) that appears at least once in each of the promoters of the characterized apo-Fur repressed genes, pfr, sodB, the cytochrome c553 gene, and hydA (Fig. 5). This sequence is also conserved in the promoters of these genes in H. pylori strain 26695 (data not shown). DNase I footprinting studies showed that this sequence was found in protected regions of hydA (Fig. 7A), pfr (Fig. 6A and reference 5; see also Fig. S1 in the supplemental material), and sodB (11) and showed that mutation of the sequence in the first apo-Fur box of the pfr promoter resulted in a loss of protection at that site as well as decreased protection at a downstream second site (Fig. 6B). Furthermore, mutation of the sequence resulted in an inability of the pfr fragment to titrate the WT pfr promoter bound to apo-Fur in fluorescence anisotropy experiments (Fig. 8). Using EMSAs, we were able to observe that mutation of each of the apo-Fur boxes alone or in combination within the pfr promoter also resulted in changes in apo-Fur binding to the promoter fragments, as shown in the altered shifting patterns (Fig. 9). The combination of the fluorescence anisotropy, footprinting, and EMSA studies with WT and scrambled promoter fragments showed that the AAATGA sequence is essential for apo-Fur binding to its target promoters. Thus, we propose that this sequence constitutes an essential element of the apo-Fur box and is necessary for apo-Fur binding to its target promoters.
In light of what is known concerning iron-bound Fur regulation in other organisms (reviewed in reference 50), and our recent elucidation of the iron-bound Fur box in H. pylori (14), it is likely not surprising that the apo-Fur box sequence is located in close proximity to the −10 and −35 regions in the characterized promoters. As with iron-bound Fur, binding of apo-Fur in the vicinity of the core promoter elements would physically occlude the binding site for RNA polymerase. However, aside from sharing a similar binding location with respect to the promoter, the apo-Fur box is rather dissimilar from the iron-bound Fur box. The H. pylori iron-bound Fur box is composed of a 15-bp sequence which is arranged in a 7-1-7 motif exhibiting dyad symmetry (5′-TAATAATnATTATTA-3′) (14). In contrast, the apo-Fur box is only 6 bp in length and shows no symmetry. Thus, the apo-Fur box sequence is less than half the size of the iron-bound Fur box and bears no sequence homology to its iron-bound counterpart. The question of how such a short consensus sequence could direct binding of apo-Fur box is intriguing. A scan of the G27 genome (35) showed that the AAATGA sequence was present 1,916 times: 878 times on the positive strand and 1,038 times on the negative strand (data not shown). It is very unlikely that each of these sequences is involved in apo-Fur regulation, especially given the high percentage of A/Ts within this genome (13, 35). Given the size of the sequence as well as its frequency within the genome, we acknowledge that it is possible that the apo-Fur box is longer than what we have identified, and this may become evident as more apo-Fur repression targets are identified. However, among the four current targets, there do not appear to be additional conserved residues immediately upstream or downstream (Fig. 5). Perhaps it is not surprising that the apo-Fur box consensus binding sequence is distinctly different from the iron-bound consensus sequence given the fact that apo-Fur itself has a significant rearrangement of the DNA binding domain compared to the holo protein (20). While the apo-Fur dimer maintains the classical V shape, the DNA binding domain is rotated 180° compared to the holo-Fur structure (20). The difference in the position of the DNA binding domain would allow for different amino acid residues to come into contact with the DNA. Together, these changes in the structural configuration of apo-Fur enable the protein to recognize a different DNA sequence from that of iron-bound Fur.
It is also worth noting that in H. pylori, apo-Fur has also been shown to have the ability to function as an activator as part of the autoregulation of fur. Intriguingly, the apo-Fur box we have identified is not found within the regions of the fur promoter where apo-Fur has been shown to bind and stimulate expression (16). Thus, it appears that the apo-Fur binding consensus sequence that we have identified is unique and specific for apo-Fur repression. Given that apo-Fur repression has been shown to modulate expression of two genes in C. jejuni (20, 21), we wondered if the identified apo-Fur box sequence could be found within the promoters of these genes. Indeed, for both genes, there is at least one AAATGA sequence that lies in close proximity to the −10 and −35 promoter elements, suggesting that the apo-Fur box may be conserved among the organisms that utilize Fur in this manner.
The data presented here likely suggest that there are various affinities of apo-Fur for its target genes. For example, our transcriptional data (both RPA and qRT-PCR) show that the most pronounced changes in gene expression that were observed upon iron chelation occurred for pfr and the cotranscribed serB (Fig. 1A and data not shown). hydA and the cytochrome c553 gene do not achieve the same degree of repression in the absence of iron, suggesting that the affinity of apo-Fur for pfr is higher than for the other targets. To further support this notion, in order to see protection of the hydA promoter fragment in the footprinting assays, five times the concentration of apo-Fur was required compared to the pfr reactions. Given that the 6-base sequence is completely conserved in each of the promoters we investigated, this suggests that other unidentified DNA sequences or structural components likely affect apo-Fur binding. We do note that the consensus sequence for hydA is located much further upstream from the core promoter elements than for pfr. Thus, perhaps DNA structural constraints affect apo-Fur binding. Furthermore, our data suggest that there may be a cooperative binding effect for apo-Fur on some promoters. For example, the footprinting assays utilizing the mutated pfr fragment show a loss of protection at the mutation site as well as decreased protection at the second apo-Fur binding site (Fig. 6A and B). Thus, binding of apo-Fur to the high-affinity site appears to help facilitate binding at subsequent sites.
The notion of a cooperative binding effect is also supported by the EMSA data generated using the mutated pfr promoter fragments. Mutation of any one of the three apo-Fur boxes alone results in altered interaction with the protein, and the effect of the mutations is even more pronounced when they are combined (Fig. 9). Mutation of box III had the most pronounced effect on the ability of Fur to bind to the pfr promoter. Alteration of this binding site alone led to increased amounts of unbound promoter at all concentrations of apo-Fur compared to the WT or box I and box II individual mutations (Fig. 9). In fact, there appeared to be a hierarchy in terms of the importance of each of the individual boxes for the initial binding event; box III was more important than box II, which was more important than box I. The decreased importance for box I in the initial binding event is furthered supported by the observation that the box I and II combined mutations exhibited alteration in binding similar to that of the box II-only mutation (Fig. 9). When the box III mutation was added in conjunction with box I and box II mutations, apo-Fur binding was virtually abrogated (Fig. 9), suggesting that this third and most distal (from the core promoter elements) binding site plays an important role in apo-Fur binding to the pfr promoter. This was a somewhat unexpected finding, since Delany et al. previously used DNase footprinting of the pfr promoter to show that box III had the lowest affinity for apo-Fur (5); the region closest to the −10 region (box I) was bound first, followed by binding to the next-closest region to the −10 region (box II), and the region farthest from the −10 region was bound last (box III) (5). While our EMSA data also reveal three distinct shifts (Fig. 2), suggesting three separate binding events, it is currently unclear in which order each of the apo-Fur boxes is bound. Regardless, it is clear that each one of the apo-Fur binding sites within the pfr promoter is important for protein-DNA interaction, as alteration of even one of them results in decreased binding (Fig. 9).
Based on our data, it is likely that out of the promoters we examined, apo-Fur has the least affinity for the cytochrome c553 gene promoter. There are three predicted apo-Fur boxes within this promoter, yet only one shift in binding was observed by EMSA (Fig. 2). Additionally, though we attempted to do so numerous times with various concentrations of apo-Fur, we were unable to successfully footprint this promoter. Taken together, these data suggest that while apo-Fur interacts with the cytochrome c553 gene promoter (Fig. 2), it does so less avidly than the other promoters. Interestingly, while the apo-Fur box consensus sequences lie upstream of the −10 promoter element for the pfr, sodB, and hydA promoters, the AAATGA sequences found within the cytochrome c553 gene promoter do not show this exact orientation. Instead, the first sequence lies between the translational start site (ATG) and the −10 promoter region, the second sequence overlaps the −10 promoter region, and the third region lies just upstream of the −35 promoter region. It is possible that the close proximity of sites one and two (only 5 bp separates them) makes it difficult for apo-Fur to bind efficiently at either site. Binding at one of the two sites may prohibit binding to the other due to their very close proximity to each other compared to the apo-Fur binding sites found in the pfr promoter, which have considerably more distance between them (Fig. 6C). Also, inefficient binding at the first two sites would likely result in a decreased cooperative effect for binding at the third site. In all, our data indicate that apo-Fur has a wide range of affinities for its target promoters.
Among the promoters for the apo-Fur regulated genes, the AAATGA sequence is found in two different orientations (Fig. 5). In the first, the sequence reads AAATGA on the coding strand and in the 5′ to 3′ direction, as is the case for the sodB and hydA promoters. In the second, the AAATGA sequence is located in the 5′ to 3′ orientation on the noncoding strand (3′ to 5′ on the coding strand). This is the orientation found in the pfr and cytochrome c553 gene promoters as well as the apo-Fur box found adjacent to the second protected region in the hydA promoter (Fig. 5 to 7). At present it is unclear what these differences entail for apo-Fur regulation. As more apo-Fur-repressed genes are identified and characterized, it will be easier to determine the role the orientation of the consensus sequence plays in apo-Fur regulation.
Despite our knowledge of the existence of apo-Fur regulation in H. pylori for over a decade, the details of this type of regulation have remained mostly unknown. In this work, we have characterized two genes as being repressed by apo-Fur. Through the addition of these gene targets, the first apo-Fur box consensus sequence was identified. This consensus sequence is found in both the two previously characterized apo-Fur-repressed genes (pfr and sodB) and the two newly characterized gene targets (hydA and the cytochrome c553 gene). The AAATGA consensus sequence was also shown to be important for apo-Fur binding to its target promoters. Although there are still lingering questions (e.g., what role does the orientation of this sequence play in regulation), the information presented here will allow for more efficient identification and characterization of apo-Fur-regulated genes not just in H. pylori but also in those organisms, like C. jejuni and D. vulgaris, in which apo-Fur regulation is also poorly understood.
Supplementary Material
ACKNOWLEDGMENT
Research in the laboratory of D. Scott Merrell is made possible by grant AI065529 from the NIAID.
Footnotes
Published ahead of print 4 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01026-13.
REFERENCES
- 1.Dunn BE, Cohen H, Blaser MJ. 1997. Helicobacter pylori. Clin. Microbiol. Rev. 10:720–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blaser MJ. 1990. Helicobacter pylori and the pathogenesis of gastroduodenal inflammation. J. Infect. Dis. 161:626–633 [DOI] [PubMed] [Google Scholar]
- 3.Asaka M, Kudo M, Kato M, Sugiyama T, Takeda H. 1998. Review article: long-term Helicobacter pylori infection—from gastritis to gastric cancer. Aliment. Pharmacol. Ther. 12(Suppl 1):9–15 [DOI] [PubMed] [Google Scholar]
- 4.Delany I, Pacheco AB, Spohn G, Rappuoli R, Scarlato V. 2001. Iron-dependent transcription of the frpB gene of Helicobacter pylori is controlled by the Fur repressor protein. J. Bacteriol. 183:4932–4937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Delany I, Spohn G, Rappuoli R, Scarlato V. 2001. The Fur repressor controls transcription of iron-activated and -repressed genes in Helicobacter pylori. Mol. Microbiol. 42:1297–1309 [DOI] [PubMed] [Google Scholar]
- 6.van Vliet AH, Stoof J, Vlasblom R, Wainwright SA, Hughes NJ, Kelly DJ, Bereswill S, Bijlsma JJ, Hoogenboezem T, Vandenbroucke-Grauls CM, Kist M, Kuipers EJ, Kusters JG. 2002. The role of the Ferric Uptake Regulator (Fur) in regulation of Helicobacter pylori iron uptake. Helicobacter 7:237–244 [DOI] [PubMed] [Google Scholar]
- 7.Gancz H, Censini S, Merrell DS. 2006. Iron and pH homeostasis intersect at the level of Fur regulation in the gastric pathogen Helicobacter pylori. Infect. Immun. 74:602–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merrell DS, Goodrich ML, Otto G, Tompkins LS, Falkow S. 2003. pH-regulated gene expression of the gastric pathogen Helicobacter pylori. Infect. Immun. 71:3529–3539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skouloubris S, Labigne A, De Reuse H. 2001. The AmiE aliphatic amidase and AmiF formamidase of Helicobacter pylori: natural evolution of two enzyme paralogues. Mol. Microbiol. 40:596–609 [DOI] [PubMed] [Google Scholar]
- 10.Carpenter BM, Gancz H, Gonzalez-Nieves RP, West AL, Whitmire JM, Michel SL, Merrell DS. 2009. A single nucleotide change affects fur-dependent regulation of sodB in H. pylori. PLoS One 4:e5369. 10.1371/journal.pone.0005369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ernst FD, Homuth G, Stoof J, Mader U, Waidner B, Kuipers EJ, Kist M, Kusters JG, Bereswill S, van Vliet AH. 2005. Iron-responsive regulation of the Helicobacter pylori iron-cofactored superoxide dismutase SodB is mediated by Fur. J. Bacteriol. 187:3687–3692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alamuri P, Mehta N, Burk A, Maier RJ. 2006. Regulation of the Helicobacter pylori Fe-S cluster synthesis protein NifS by iron, oxidative stress conditions, and Fur. J. Bacteriol. 188:5325–5330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J, Zhou L, Kirkness EF, Peterson S, Loftus B, Richardson D, Dodson R, Khalak HG, Glodek A, McKenney K, Fitzegerald LM, Lee N, Adams MD, Hickey EK, Berg DE, Gocayne JD, Utterback TR, Peterson JD, Kelley JM, Cotton MD, Weidman JM, Fujii C, Bowman C, Watthey L, Wallin E, Hayes WS, Borodovsky M, Karp PD, Smith HO, Fraser CM, Venter JC. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539–547 [DOI] [PubMed] [Google Scholar]
- 14.Pich OQ, Carpenter BM, Gilbreath JJ, Merrell DS. 2012. Detailed analysis of Helicobacter pylori Fur-regulated promoters reveals a Fur box core sequence and novel Fur-regulated genes. Mol. Microbiol. 84:921–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilbreath JJ, West AL, Pich OQ, Carpenter BM, Michel S, Merrell DS. 2012. Fur activates expression of the 2-oxoglutarate oxidoreductase genes (oorDABC) in Helicobacter pylori. J. Bacteriol. 194:6490–6497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Delany I, Spohn G, Rappuoli R, Scarlato V. 2003. An anti-repression Fur operator upstream of the promoter is required for iron-mediated transcriptional autoregulation in Helicobacter pylori. Mol. Microbiol. 50:1329–1338 [DOI] [PubMed] [Google Scholar]
- 17.Deng X, Sun F, Ji Q, Liang H, Missiakas D, Lan L, He C. 2012. Expression of multidrug resistance efflux pump gene norA is iron responsive in Staphylococcus aureus. J. Bacteriol. 194:1753–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bereswill S, Greiner S, van Vliet AH, Waidner B, Fassbinder F, Schiltz E, Kusters JG, Kist M. 2000. Regulation of ferritin-mediated cytoplasmic iron storage by the ferric uptake regulator homolog (Fur) of Helicobacter pylori. J. Bacteriol. 182:5948–5953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holmes K, Mulholland F, Pearson BM, Pin C, McNicholl-Kennedy J, Ketley JM, Wells JM. 2005. Campylobacter jejuni gene expression in response to iron limitation and the role of Fur. Microbiology 151:243–257 [DOI] [PubMed] [Google Scholar]
- 20.Butcher J, Sarvan S, Brunzelle JS, Couture JF, Stintzi A. 2012. Structure and regulon of Campylobacter jejuni ferric uptake regulator Fur define apo-Fur regulation. Proc. Natl. Acad. Sci. U. S. A. 109:10047–10052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grabowska AD, Wandel MP, Lasica AM, Nesteruk M, Roszczenko P, Wyszynska A, Godlewska R, Jagusztyn-Krynicka EK. 2011. Campylobacter jejuni dsb gene expression is regulated by iron in a Fur-dependent manner and by a translational coupling mechanism. BMC Microbiol. 11:166. 10.1186/1471-2180-11-166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bender KS, Yen HC, Hemme CL, Yang Z, He Z, He Q, Zhou J, Huang KH, Alm EJ, Hazen TC, Arkin AP, Wall JD. 2007. Analysis of a ferric uptake regulator (Fur) mutant of Desulfovibrio vulgaris Hildenborough. Appl. Environ. Microbiol. 73:5389–5400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ernst FD, Bereswill S, Waidner B, Stoof J, Mader U, Kusters JG, Kuipers EJ, Kist M, van Vliet AH, Homuth G. 2005. Transcriptional profiling of Helicobacter pylori Fur- and iron-regulated gene expression. Microbiology 151:533–546 [DOI] [PubMed] [Google Scholar]
- 24.Doig P, Austin JW, Trust TJ. 1993. The Helicobacter pylori 19.6-kilodalton protein is an iron-containing protein resembling ferritin. J. Bacteriol. 175:557–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carpenter BM, McDaniel TK, Whitmire JM, Gancz H, Guidotti S, Censini S, Merrell DS. 2007. Expanding the Helicobacter pylori genetic toolbox: modification of an endogenous plasmid for use as a transcriptional reporter and complementation vector. Appl. Environ. Microbiol. 73:7506–7514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carpenter BM, Gancz H, Benoit SL, Evans S, Olsen CH, Michel SL, Maier RJ, Merrell DS. 2010. Mutagenesis of conserved amino acids of Helicobacter pylori Fur reveals residues important for function. J. Bacteriol. 192:5037–5052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thompson LJ, Merrell DS, Neilan BA, Mitchell H, Lee A, Falkow S. 2003. Gene expression profiling of Helicobacter pylori reveals a growth-phase-dependent switch in virulence gene expression. Infect. Immun. 71:2643–2655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu C, Genco CA. 2012. Fur-mediated activation of gene transcription in the human pathogen Neisseria gonorrhoeae. J. Bacteriol. 194:1730–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lakowicz JR. 1983. Principles of fluorescence spectroscopy. Plenum Press, New York, NY [Google Scholar]
- 31.Giri K, Scott RA, Maynard EL. 2009. Molecular structure and biochemical properties of the HCCH-Zn2+ site in HIV-1 Vif. Biochemistry 48:7969–7978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Merrell DS, Thompson LJ, Kim CC, Mitchell H, Tompkins LS, Lee A, Falkow S. 2003. Growth phase-dependent response of Helicobacter pylori to iron starvation. Infect. Immun. 71:6510–6525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Danielli A, Roncarati D, Delany I, Chiarini V, Rappuoli R, Scarlato V. 2006. In vivo dissection of the Helicobacter pylori Fur regulatory circuit by genome-wide location analysis. J. Bacteriol. 188:4654–4662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee HW, Choe YH, Kim DK, Jung SY, Lee NG. 2004. Proteomic analysis of a ferric uptake regulator mutant of Helicobacter pylori: regulation of Helicobacter pylori gene expression by ferric uptake regulator and iron. Proteomics 4:2014–2027 [DOI] [PubMed] [Google Scholar]
- 35.Baltrus DA, Amieva MR, Covacci A, Lowe TM, Merrell DS, Ottemann KM, Stein M, Salama NR, Guillemin K. 2009. The complete genome sequence of Helicobacter pylori strain G27. J. Bacteriol. 191:447–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, Smith DR, Noonan B, Guild BC, deJonge BL, Carmel G, Tummino PJ, Caruso A, Uria-Nickelsen M, Mills DM, Ives C, Gibson R, Merberg D, Mills SD, Jiang Q, Taylor DE, Vovis GF, Trust TJ. 1999. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 397:176–180 [DOI] [PubMed] [Google Scholar]
- 37.Oh JD, Kling-Backhed H, Giannakis M, Xu J, Fulton RS, Fulton LA, Cordum HS, Wang C, Elliott G, Edwards J, Mardis ER, Engstrand LG, Gordon JI. 2006. The complete genome sequence of a chronic atrophic gastritis Helicobacter pylori strain: evolution during disease progression. Proc. Natl. Acad. Sci. U. S. A. 103:9999–10004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salama NR, Shepherd B, Falkow S. 2004. Global transposon mutagenesis and essential gene analysis of Helicobacter pylori. J. Bacteriol. 186:7926–7935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, Sittka A, Chabas S, Reiche K, Hackermuller J, Reinhardt R, Stadler PF, Vogel J. 2010. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464:250–255 [DOI] [PubMed] [Google Scholar]
- 40.Petersen L, Larsen TS, Ussery DW, On SL, Krogh A. 2003. RpoD promoters in Campylobacter jejuni exhibit a strong periodic signal instead of a −35 box. J. Mol. Biol. 326:1361–1372 [DOI] [PubMed] [Google Scholar]
- 41.Delany I, Spohn G, Pacheco AB, Ieva R, Alaimo C, Rappuoli R, Scarlato V. 2002. Autoregulation of Helicobacter pylori Fur revealed by functional analysis of the iron-binding site. Mol. Microbiol. 46:1107–1122 [DOI] [PubMed] [Google Scholar]
- 42.van Vliet AH, Stoof J, Poppelaars SW, Bereswill S, Homuth G, Kist M, Kuipers EJ, Kusters JG. 2003. Differential regulation of amidase- and formamidase-mediated ammonia production by the Helicobacter pylori Fur repressor. J. Biol. Chem. 278:9052–9057 [DOI] [PubMed] [Google Scholar]
- 43.Koyanagi S, Nagata K, Tamura T, Tsukita S, Sone N. 2000. Purification and characterization of cytochrome c-553 from Helicobacter pylori. J. Biochem. 128:371–375 [DOI] [PubMed] [Google Scholar]
- 44.Maier RJ, Fu C, Gilbert J, Moshiri F, Olson J, Plaut AG. 1996. Hydrogen uptake hydrogenase in Helicobacter pylori. FEMS Microbiol. Lett. 141:71–76 [DOI] [PubMed] [Google Scholar]
- 45.Olson JW, Maier RJ. 2002. Molecular hydrogen as an energy source for Helicobacter pylori. Science 298:1788–1790 [DOI] [PubMed] [Google Scholar]
- 46.Contreras M, Thiberge JM, Mandrand-Berthelot MA, Labigne A. 2003. Characterization of the roles of NikR, a nickel-responsive pleiotropic autoregulator of Helicobacter pylori. Mol. Microbiol. 49:947–963 [DOI] [PubMed] [Google Scholar]
- 47.Muller C, Bahlawane C, Aubert S, Delay CM, Schauer K, Michaud-Soret I, De Reuse H. 2011. Hierarchical regulation of the NikR-mediated nickel response in Helicobacter pylori. Nucleic Acids Res. 39:7564–7575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Delany I, Ieva R, Soragni A, Hilleringmann M, Rappuoli R, Scarlato V. 2005. In vitro analysis of protein-operator interactions of the NikR and Fur metal-responsive regulators of coregulated genes in Helicobacter pylori. J. Bacteriol. 187:7703–7715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dosanjh NS, West AL, Michel SL. 2009. Helicobacter pylori NikR's interaction with DNA: a two-tiered mode of recognition. Biochemistry 48:527–536 [DOI] [PubMed] [Google Scholar]
- 50.Carpenter BM, Whitmire JM, Merrell DS. 2009. This is not your mother's repressor: the complex role of Fur in pathogenesis. Infect. Immun. 77:2590–2601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Covacci A, Censini S, Bugnoli M, Petracca R, Burroni D, Macchia G, Massone A, Papini E, Xiang Z, Figura N. 1993. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc. Natl. Acad. Sci. U. S. A. 90:5791–5795 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.