

Abstract
Approximately 50% of cell wall peptidoglycan in Gram-negative bacteria is recycled with each generation. The primary substrates used for peptidoglycan biosynthesis and recycling in the cytoplasm are GlcNAc-MurNAc(anhydro)-tetrapeptide and its degradation product, the free tetrapeptide. This complex process involves ∼15 proteins, among which the cytoplasmic enzyme ld-carboxypeptidase A (LdcA) catabolizes the bond between the last two l- and d-amino acid residues in the tetrapeptide to form the tripeptide, which is then utilized as a substrate by murein peptide ligase (Mpl). LdcA has been proposed as an antibacterial target. The crystal structure of Novosphingobium aromaticivorans DSM 12444 LdcA (NaLdcA) was determined at 1.89-Å resolution. The enzyme was biochemically characterized and its interactions with the substrate modeled, identifying residues potentially involved in substrate binding. Unaccounted electron density at the dimer interface in the crystal suggested a potential site for disrupting protein-protein interactions should a dimer be required to perform its function in bacteria. Our analysis extends the identification of functional residues to several other homologs, which include enzymes from bacteria that are involved in hydrocarbon degradation and destruction of coral reefs. The NaLdcA crystal structure provides an alternate system for investigating the structure-function relationships of LdcA and increases the structural coverage of the protagonists in bacterial cell wall recycling.
INTRODUCTION
Peptidoglycan (murein) is an essential bacterial cell wall polymer consisting of long glycan chains with an alternating arrangement of N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) that are cross-linked by short peptides. The peptidoglycan barrier surrounds the cytoplasmic membrane, and its main functions are to maintain the shape and turgidity of the cell and to serve as a scaffold for anchoring other cell envelope components, such as proteins (1). In de novo peptidoglycan synthesis, the first steps of assembly involve MurA and MurB, which form UDP-GlcNAc-enolpyruvate and UDP-MurNAc, respectively. The Mur ligases, MurC, MurD, MurE, and MurF, are then involved in sequential addition of l- and d-amino acids (l-Ala, d-Glu, meso-diaminopimelate [meso-A2pm], and d-Ala-d-Ala, respectively) to form the pentapeptide derivative UDP-MurNAc-l-Ala-γ-d-Glu-meso-A2pm-d-Ala-d-Ala (2). Subsequent biosynthetic steps catalyzed by several membrane and periplasmic enzymes create the final peptidoglycan (3).
In Gram-negative bacteria, ∼30 to 60% of the cell wall peptidoglycan is recycled (4), in a process that involves numerous transglycosidases, amidases, endopeptidases, and carboxypeptidases (5). During the recycling process, ld-carboxypeptidase A (LdcA) specifically cleaves the ld-peptide bond between meso-A2pm and d-Ala in the l-Ala-γ-d-Glu-meso-A2pm-d-Ala tetrapeptide to produce the l-Ala-γ-d-Glu-meso-A2pm tripeptide, which is the substrate for the recycling enzyme, murein peptide ligase (Mpl) (6, 7), whose crystal structure was determined recently (8). Mpl then produces UDP-MurNAc-l-Ala-γ-d-Glu-meso-A2pm, which reenters the peptidoglycan synthesis pathway as the substrate for MurF. Loss of LdcA activity results in cells with heightened sensitivity to lysis in the stationary phase (9). Functional characterization of LdcA includes enzymatic studies (10, 11) and inhibitor experiments (9, 12, 13).
Structural studies of LdcA started with the elucidation of the crystal structure of PaLdcA (Protein Data Bank [PDB] ID 1zl0; ordered locus name PA5198) (14), a protein of then unknown function from Pseudomonas aeruginosa, by the Midwest Center for Structural Genomics (http://www.mcsg.anl.gov/) as part of the NIH Protein Structure Initiative (PSI) (15). This work occurred almost simultaneously with the independent structural and biochemical characterization of PaLdcA (PDB IDs 1zrs, 2aum, and 2aun) (16). This study provided the first description of the peptidase activity of LdcA, with a Ser-His-Glu catalytic triad in the active site, which subsequently led to the classification of LdcA enzymes into the Peptidase_S66 (PF02016) family in Pfam (17). PF02016 contains 2,826 sequences (Pfam, version 27.0; March 2013), among which 2,765 are bacterial proteins. This family also includes the microcin C7 self-immunity protein (MccF; plasmid-encoded protein) (18). The crystal structure of an MccF protein from Bacillus anthracis was recently described (PDB ID 3gjz; Center for Structural Genomics of Infectious Diseases [19]) and has the same fold and catalytic triad as LdcA (a genome-encoded protein).
We determined the crystal structure of the LdcA protein from Novosphingobium aromaticivorans DSM 12444 (NaLdcA) (UniProt accession no. Q2G8F3; GenBank accession no. YP_496704.1; ordered locus name, Saro_1426) and identified residues likely involved in substrate recognition. Modeling of the binding of substrate in the active site demonstrates that the catalytic residues are poised for nucleophilic attack on the scissile bond. We also tested the specific activity and substrate specificity of the enzyme and identified a novel site that could potentially be involved in protein-protein interactions. Our results serve as a basis for further structure-based biochemistry analyses of LdcA enzymes.
MATERIALS AND METHODS
Protein production and crystallization.
Clones were generated using the polymerase incomplete primer extension (PIPE) cloning method (20). The gene encoding NaLdcA was amplified by PCR from N. aromaticivorans DSM 12444 genomic DNA by using Pfu Turbo DNA polymerase (Stratagene) and I-PIPE (insert) primers that included sequences for the predicted 5′ and 3′ ends. The Joint Center for Structural Genomics (JCSG) NaLdcA expression plasmid was deposited in the PSI Biology Materials Repository (DNASU ID NaCD00087837) (21). The expression vector pSpeedET, which encodes an amino-terminal tobacco etch virus (TEV) protease-cleavable expression and purification tag (MGSDKIHHHHHHENLYFQ/G), was PCR amplified with V-PIPE (vector) primers (forward primer, 5′-TAACGCGACTTAATTAACTCGTTTAAACGGTCTCCAGC-3′; and reverse primer, 5′-GCCCTGGAAGTACAGGTTTTCGTGATGATGATGATGATG-3′). V-PIPE and I-PIPE PCR products were mixed to anneal the amplified DNA fragments together. Escherichia coli GeneHogs (Invitrogen) competent cells were transformed with the I-PIPE–V-PIPE mixture and dispensed on selective LB-agar plates. The cloning junctions were confirmed by DNA sequencing. Expression was performed in a selenomethionine-containing medium at 37°C. Selenomethionine was incorporated via inhibition of methionine biosynthesis (22), which does not require a methionine-auxotrophic strain. At the end of fermentation, lysozyme was added to the culture to a final concentration of 250 μg/ml, and the cells were harvested and frozen. After one freeze-thaw cycle, the cells were homogenized in lysis buffer [50 mM HEPES, pH 8.0, 50 mM NaCl, 10 mM imidazole, 1 mM Tris(2-carboxyethyl)phosphine-HCl (TCEP)] and passed through a microfluidizer. The lysate was clarified by centrifugation at 32,500 × g for 30 min and loaded onto a nickel-chelating affinity column (GE Healthcare) preequilibrated with lysis buffer, the column was washed with wash buffer (50 mM HEPES, pH 8.0, 300 mM NaCl, 40 mM imidazole, 10% [vol/vol] glycerol, 1 mM TCEP), and the protein was eluted with elution buffer (20 mM HEPES, pH 8.0, 300 mM imidazole, 10% [vol/vol] glycerol, 1 mM TCEP). The eluate was buffer exchanged with TEV buffer (20 mM HEPES, pH 8.0, 200 mM NaCl, 40 mM imidazole, 1 mM TCEP) by use of a PD-10 column (GE Healthcare) and was incubated with 1 mg of TEV protease per 15 mg of eluted protein for 2 h at room temperature (20°C to 25°C) and then overnight at 4°C. The protease-treated eluate was passed over nickel-chelating resin (GE Healthcare) preequilibrated with HEPES crystallization buffer (20 mM HEPES, pH 8.0, 200 mM NaCl, 40 mM imidazole, 1 mM TCEP), and the column was washed with the same buffer. The flowthrough and wash fractions were combined and concentrated to 9.9 mg/ml by centrifugal ultrafiltration (Millipore) for crystallization trials. NaLdcA was crystallized using the nanodrop vapor diffusion method (23) with standard JCSG crystallization protocols (24, 25). Drops composed of 200 nl protein solution mixed with 200 nl crystallization solution in a sitting-drop format were equilibrated against a 50-μl reservoir at 277 K for 28 days prior to harvest. The crystallization reagent consisted of 1.6 M (NH4)2SO4 and 0.1 M Tris-HCl, pH 8.0. Glycerol was added to a final concentration of 20% (vol/vol) as a cryoprotectant. Initial screening for diffraction was carried out using the Stanford automated mounting (SAM) system (26) and an X-ray microsource (27) installed at the Stanford Synchrotron Radiation Lightsource (SSRL; SLAC National Accelerator Laboratory, Menlo Park, CA). The diffraction data were indexed in orthorhombic space group P212121. The oligomeric state of NaLdcA in solution was determined by analytical size-exclusion chromatography, using a 0.8-cm by 30-cm Shodex protein KW-803 size-exclusion column (Thomson Instruments) (20).
X-ray data collection, structure determination, and refinement.
Multiwavelength anomalous diffraction (MAD) data were collected at SSRL on beamline 9-2 at wavelengths corresponding to the high-energy remote (λ1), inflection point (λ2), and peak (λ3) of a selenium MAD experiment using the BLU-ICE (28) data collection environment. The data sets were collected at 100 K, using a MarMosaic 325 charge-coupled device (CCD) detector (Rayonix). The MAD data were integrated and reduced using MOSFLM (29) and scaled with the program SCALA from the CCP4 suite (30). The heavy atom substructure was determined with SHELXD (31). Phasing was performed with autoSHARP (32), SOLOMON (33) (implemented in autoSHARP) was used for density modification, and ARP/wARP (34) was used for automatic model building to 1.89-Å resolution. Model completion and crystallographic refinement were performed with the λ1 data set, using COOT (35) and REFMAC5 (36). The refinement protocol included the experimental phase restraints in the form of Hendrickson-Lattman coefficients from autoSHARP and translation, libration, and screw (TLS) refinement with one TLS group per molecule in the crystallographic asymmetric unit (asu). Data and refinement statistics are summarized in Table 1 (37–40).
Table 1.
Summary of crystal parameters, data collection, and refinement statistics for NaLdcA (PDB entry 3g23)
| Parameter | Valuea |
||
|---|---|---|---|
| λ1 MAD-Se | λ2 MAD-Se | λ3 MAD-Se | |
| Data collection statistics | |||
| Space group | P212121 | ||
| Unit cell parameters (Å) (a, b, c) | 43.63, 91.04, 145.74 | ||
| Wavelength (Å) | 0.91162 | 0.97985 | 0.97971 |
| Resolution range (Å) | 29.7–1.89 (1.94–1.89) | 28.9–1.90 (1.95–1.90) | 29.0–2.07 (2.12–2.07) |
| No. of observations | 202,346 | 165,632 | 127,539 |
| No. of unique reflections | 47,086 | 46,218 | 36,015 |
| Completeness (%) | 99.5 (94.4) | 99.5 (100.0) | 98.8 (92.4) |
| Mean I/σ (I) | 10.3 (1.7) | 8.8 (1.7) | 8.3 (1.6) |
| Rmerge on Ib (%) | 12.9 (87.1) | 14.1 (83.9) | 14.8 (72.1) |
| Rmeas on Ib (%) | 14.7 (101.8) | 16.6 (98.4) | 17.3 (86.4) |
| Rpim on Ib (%) | 7.0 (51.3) | 8.4 (50.1) | 8.7 (46.8) |
| Model and refinement statistics | |||
| Resolution range (Å) | 1.89–29.7 | ||
| No. of reflections (total) | 47,036c | ||
| No. of reflections (test) | 2,381 | ||
| Completeness (%) | 99.5 | ||
| Data set used in refinement | λ1 | ||
| Cutoff criteria | |F| > 0 | ||
| Rcrystald | 0.171 | ||
| Rfreed | 0.207 | ||
| Stereochemical parameters | |||
| Restraints (RMSD observed) | |||
| Bond angles (°) | 1.49 | ||
| Bond lengths (Å) | 0.017 | ||
| Avg protein isotropic B valuee (Å2)/Wilson plot B value (Å2) | 27.3/18.8 | ||
| ESUf based on Rfree (Å) | 0.13 | ||
| No. of protein residues/no. of atoms | 540/4024 | ||
| No. of waters/no. of UNL/no. of sulfates/no. of glycerol molecules | 671/1/17/7 | ||
| Residues (%) in favored/allowed regions in Ramachandran plot | 98.9/100.0 | ||
Values in parentheses are for the highest-resolution shell.
Rmerge = ΣhklΣi|Ii(hkl) − <I(hkl)>|/ΣhklΣi(hkl); Rmeas = Σhkl[N/(N − 1)]1/2Σi|Ii(hkl) − <I(hkl)>|/ΣhklΣiIi(hkl) (37); Rpim (precision-indicating Rmerge) = Σhkl[(1/(N − 1)]1/2Σi|Ii(hkl) − <I(hkl)>|/ΣhklΣiIi(hkl) (38, 39).
Typically, the number of unique reflections used in refinement is slightly less than the total number that were integrated and scaled. Reflections are excluded owing to systematic absences, negative intensities, and rounding errors in the resolution limits and unit cell parameters.
Rcrystal = Σhkl‖Fobs| − |Fcalc‖/Σhkl|Fobs|, where Fcalc and Fobs are the calculated and observed structure factor amplitudes, respectively. Rfree is the same as Rcrystal, but with 5.1% of the total reflections chosen at random and omitted from refinement.
Represents the total B value, which includes TLS and residual B components.
Estimated overall coordinate error (40).
Validation and deposition.
The quality of the crystal structure was analyzed using the JCSG quality control server (http://smb.slac.stanford.edu/jcsg/QC). This server verifies the stereochemical quality of a model by using AutoDepInputTool (41), MolProbity (42), and Phenix (43), the agreement between the atomic model and the data by using RESOLVE (44), the protein sequence by using CLUSTALW (45), the ADP distribution by using Phenix, and differences in Rcrystal/Rfree, expected Rfree/Rcrystal, and various other items, including atom occupancies, consistency of noncrystallographic symmetry (NCS) pairs, ligand interactions, special positions, etc., by using in-house scripts to analyze the refinement log file and PDB header. Protein quaternary structure analysis was performed using the PISA server (46). Figure 1B was adapted from an analysis using PDBsum (47), and other figures were prepared with PyMOL (48).
Fig 1.

Crystal structure of NaLdcA. (A) Stereo ribbon diagram of NaLdcA colored from blue (N terminus) to red (C terminus). Residues 159 and 160, which are part of the linker connecting the N- and C-terminal flavodoxin-like and “swiveling” portions of each monomer, were not included in the model due to the absence of electron density. (B) Diagram showing the secondary structure elements of NaLdcA superimposed on its sequence, adapted from PDBsum (http://www.ebi.ac.uk/pdbsum), where α-helices and 310-helices are labeled sequentially (H1, H2, H3, etc.), β-strands are labeled (β1, β2, β3, etc.), and β- and γ-turns are indicated with “β” and “γ.”
Sequence analysis.
The analysis of conserved residues (see Fig. 3) was performed using CONSURF (49–51) with default parameters, with homologs collected from UniProt (http://www.uniprot.org) and PSI-BLAST (52), the E value set to 0.001, the number of PSI-BLAST iterations set to 1, the sequence identity range set to 35 to 95%, and the maximum number of homologs set to 100 based on conservation scores, with a Bayesian method of calculation and using the JTT model for substitution of proteins (53, 54). For Fig. 4, the multiple-sequence alignment was generated using CLUSTAL W (45).
Fig 3.

Residue conservation analysis. The surface representation of NaLdcA is color coded (magenta, most conserved; cyan, most variable) according to amino acid conservation based on comparison to 9 other proteins annotated as either putative LdcA enzymes or proteins of unknown function, with sequence identities of 35 to 95% compared to NaLdcA. (A) The left panel (same view as in Fig. 1) shows that one side of NaLdcA has more highly conserved residues in and around the putative active site than the other side (right panel, viewed at 180° around a vertical axis from the left panel). (B) Stick representation of the most conserved residues, which include Asp52, Arg55, Tyr78, Arg82, Ser104, Asp105, Asn167, Leu174, Thr177, Glu191, Val193, Glu195, Tyr198, Asp201, Arg202, His261, and Arg248. The Ser104-Glu191-His261 catalytic triad is shown in blue.
Fig 4.

Model of protein-ligand complex and conservation of putative functional residues. (A) The binding of NaLdcA to the ligand GlcNAc-MurNAc-l-Ala-γ-d-Glu-meso-A2pm-d-Ala (PDB ligand ID MLD, from PDB ID 2cb3) was modeled such that the carboxy-terminal d-Ala is positioned for catalysis near the active site Ser-His-Glu triad. The molecular orientation is similar to that in Fig. 3, and the helices are labeled. (B) Magnified view of the binding site. Oxygen atoms from the carboxylate groups of A2pm and d-Glu of the ligand are superimposed on the two sulfate ions (SO4-274 and SO4-276, respectively) present in the substrate-binding pocket and present similar interactions to the active site residues Arg75 and Glu195, thereby suggesting that the sulfate ions mimic portions of the substrate. (C) Sequence logo representation of relative frequencies of occurrence of residues in NaLdcA and other homologs, obtained using Weblogo (68; http://weblogo.berkeley.edu/). The same set of proteins and multiple-sequence alignment listed in Fig. 3 were used in this analysis. Panels A, B, and C represent different sequence ranges in the enzyme.
Modeling of ligand-bound NaLdcA.
The model of ligand-bound NaLdcA was obtained by manually positioning the coordinates of MLD (PDB ligand ID MLD, from PDB ID 2cb3) in the substrate-binding cleft by using COOT. The main constraints imposed during this docking were the position of the peptide bond to be cleaved facing the active site residues and the lowest number of clashes with protein residues. The manual docking of MLD was further validated computationally by using Glide (Schrödinger, LLC). This procedure was done in 3 stages. First, the MLD model was subjected to geometry optimization using the Maestro interface and to optimization by Macromodel v9.1 (Schrödinger, LLC), using the optimized potentials for liquid simulations–all-atom (OPLS-AA) force field (55) with the truncated Newton conjugate gradient protocol. Partial atomic charges were computed using the OPLS-AA force field. Second, the protein structure was prepared for calculations. Crystallographic water molecules within 5 Å of the MLD cocrystallized ligand from PDB ID 2cb3 were kept, and the NaLdcA structure was optimized for docking using the protein preparation and refinement utility provided in the Schrödinger suite. Partial atomic charges were assigned according to the OPLS-AA force field. Third, docking calculation was performed using the “extra precision” (XP) mode of Glide (56, 57). The receptor grid was generated with a box size of 20 Å around MLD and the potential for the nonpolar part of the receptor softened, keeping a van der Waals radius scaling factor of 1.0 and a partial charge cutoff of 0.25. For the ligand, the scaling factor was set to 0.8 and the partial charge cutoff to 0.15. The root mean square deviation (RMSD) between the docked conformations and the manual docking of MLD was 4.5 Å (see Fig. 5).
Fig 5.

Comparison of manual and computational ligand docking. In order to validate the manual docking shown in Fig. 4, the ligand was also subjected to computational docking using Glide (version 5.7; Schrödinger, LLC, New York, NY). Of the 8 docked ligand conformations, 6 were compatible with manual docking in terms of general location and orientation. A stereoview of the manually docked ligand (red) and the 4 closest matches (magenta, yellow, blue, and green) from the Glide docking results is shown in stick representation. The orientation of the protein (gray) is similar to that in Fig. 4.
Protein production for biochemical characterization. (i) NaLdcA.
DH5α(pSpeedET::NaldcA) cells were grown in 2YT medium (16 g/liter tryptone, 10 g/liter yeast extract, and 5 g/liter NaCl; MP Biomedicals) supplemented with kanamycin (2-liter cultures), using the JCSG expression plasmid (DNASU ID NaCD00087837) from the PSI Materials Repository. When the optical density at 600 nm reached 0.9, l-arabinose was added (final concentration, 0.02%), and incubation was continued for 3 h at 37°C. Cells were harvested at 4°C, and the pellet was washed with cold 20 mM phosphate buffer, pH 7.4, containing 0.1% 2-mercaptoethanol (buffer A). Cells were suspended in buffer A (12 ml) and disrupted by sonication in the cold, using a Bioblock Vibracell 72412 sonicator. The resulting suspension was centrifuged at 4°C for 30 min at 200,000 × g with a Beckman TL100 apparatus, and the pellet was discarded.
(ii) EcLdcA.
DH5α(pTrcHis30::EcldcA) cells were grown in 2YT medium supplemented with ampicillin (2-liter cultures). When the optical density at 600 nm reached 0.2, isopropyl-β-d-thiogalactopyranoside (IPTG) was added (1 mM), and incubation was continued overnight at 20°C. Cells were harvested at 4°C, and the pellet was washed with buffer A. Cells were suspended in buffer A (30 ml) and disrupted by sonication, and the resulting suspension was centrifuged at 4°C for 30 min at 200,000 × g. The His6-tagged LdcA proteins were purified from the soluble fractions on Ni2+-nitrilotriacetic acid (Ni2+-NTA)-agarose following the manufacturer's recommendations (Qiagen). Washing and elution steps were performed with a discontinuous gradient of imidazole (20 to 400 mM) in buffer A containing 300 mM KCl. Protein contents were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and relevant fractions were pooled and dialyzed against 100 volumes of buffer A. Glycerol (10% final concentration) was added for storage of the protein at −20°C. Protein concentrations were determined by quantitative amino acid analysis with a Hitachi L8800 analyzer (ScienceTec) after hydrolysis of samples in 6 M HCl at 105°C for 24 h.
Assays for biochemical characterization.
The standard ld-carboxypeptidase activity assay consisted of following the hydrolysis of the tetrapeptide l-Ala-γ-d-Glu-meso-A2pm-d-Ala in a reaction mixture (25 μl) containing 100 mM Tris-HCl buffer, pH 8, 0.2 mM tetrapeptide, and LdcA enzyme (0.2 ng of protein in 5 μl of buffer A). After 30 min of incubation at 37°C, the reaction was stopped by adding 5 μl of glacial acetic acid. The substrate and reaction product (the tripeptide l-Ala-γ-d-Glu-meso-A2pm) were separated by high-pressure liquid chromatography (HPLC) on a Hypersil 3μ column (250 × 4.6 mm; Alltech France), using 0.05% trifluoroacetic acid (TFA) and 2% acetonitrile as the elution buffer, at a flow rate of 0.6 ml/min. Peaks were detected by measuring the absorbance at 214 nm (under these conditions, the tetrapeptide and tripeptide were eluted in 18 and 11 min, respectively). For determination of the kinetic constants, the same assay was performed with various concentrations of tetrapeptide. In all cases, the substrate consumption was <20%, and the linearity was ensured within this interval even at the lowest substrate concentration. The data were fitted to the equation v = VmaxS/(Km + S) using the MDFitt software developed by M. Desmadril (IBBMC, Orsay, France). When other compounds were tested as substrates, the assay conditions were similar, except that the amount of LdcA enzyme used was appropriately adjusted (from 0.2 ng to 5 μg). Also, HPLC conditions were modified so that the separation of product and substrate was optimal: 50 mM ammonium formate, pH 3.9, was used for the separation of UDP-MurNAc-tetrapeptide and UDP-MurNAc-tripeptide (retention times of 24 and 12 min, respectively); the same buffer at pH 4.4 was used to separate UDP-MurNAc-pentapeptide (21 min) and UDP-MurNAc-tripeptide (8 min), as well as the A2pm-containing MurNAc-tetrapeptide (retention times for the two anomers [α/β], 14/26 min) and MurNAc-tripeptide (α/β retention times, 7/11 min). Ammonium acetate, pH 6.0 (50 mM), was used instead for the separation of Lys-containing MurNAc-tetrapeptide (α/β retention times, 14/28 min) and MurNAc-tripeptide (α/β retention times, 10/16 min). For the separation of GlcNAc-MurNAc(anhydro)-tetrapeptide from its tripeptide derivative, 0.05% TFA was used as the elution buffer, together with a gradient of acetonitrile (from 0 to 20%) applied between 0 and 40 min; the latter compounds were eluted in 34 and 29 min, respectively.
Protein structure accession number.
Atomic coordinates and experimental structure factors for NaLdcA to 1.89-Å resolution have been deposited in the Protein Data Bank (www.wwpdb.org) under PDB ID 3g23.
RESULTS
Crystal structure of NaLdcA.
The cloning, expression, purification, and crystallization of NaLdcA were carried out using standard protocols at the Joint Center for Structural Genomics (http://www.jcsg.org), as detailed in Materials and Methods. The crystal structure of NaLdcA was determined by multiwavelength anomalous diffraction (MAD) phasing to a resolution of 1.89 Å. Data collection, model, and refinement statistics are summarized in Table 1 (37–40). There are two NaLdcA molecules in the asu. Each molecule (Fig. 1) contains residues 1 to 271 of the 273-residue full-length protein, in addition to Gly0, which remains after cleavage of the expression and purification tag. Residues 159 and 160 and residues 272 and 273 in both molecules were omitted from the model due to the lack of interpretable electron density. Seventeen sulfate ions from the crystallization condition, 8 glycerol molecules from the cryoprotectant, 671 water molecules, and 1 unidentified ligand (UNL) were also identified in the asu. The Matthews coefficient (58) is ∼2.4 Å3/Da, with an estimated solvent content of ∼50%. A Ramachandran plot produced by MolProbity (42) shows that 98.9% of the residues are in the favored regions and none are in disallowed regions. The catalytic Ser104 residue in both copies in the asu is in an energetically unfavorable conformation and appears in the additionally allowed regions (lower right quadrant) of the Ramachandran plot, which is typical for serine proteases.
The NaLdcA monomer is composed of α-helices H1 to H11 and β-strands β1 to β10 (Fig. 1). Both molecules in the asu are very similar in structure: chain A can be superimposed onto chain B, with an RMSD of 0.2 Å over 270 Cα atoms. According to SCOP (59), the LdcA architecture has an N-terminal domain with a flavodoxin-like fold (residues ∼3 to 169 in PaLdcA) and a C-terminal domain with a “swiveling” β/β/α fold (residues ∼170 to 307 in PaLdcA). The active site comprises a Ser104-His261-Glu191 catalytic triad located in a cleft between the two domains.
Structural comparison with PaLdcA and MccF.
The overall structure of NaLdcA is similar to those of PaLdcA and MccF. NaLdcA can be superimposed on PaLdcA (PDB ID 1zrs; 20% sequence identity), with an RMSD of 2.4 Å over 254 Cα atoms, and on MccF (PDB ID 3gjz; 16% sequence identity), with an RMSD of 2.5 Å over 242 Cα atoms (Fig. 2). However, there are several notable differences. The counterpart of one of the regions that is disordered in the NaLdcA structure (residues 159 and 160) contains an insertion in both PaLdcA (residues 173 to 185) and MccF (residues 166 to 204) that forms a loop which, in the case of MccF, extends out toward the active site and forms a lid (residues 168 to 195). This lid partially covers and restricts access to the substrate-binding cavity and could have important functional consequences. In addition, both PaLdcA and MccF have longer C-terminal extensions than that of NaLdcA.
Fig 2.
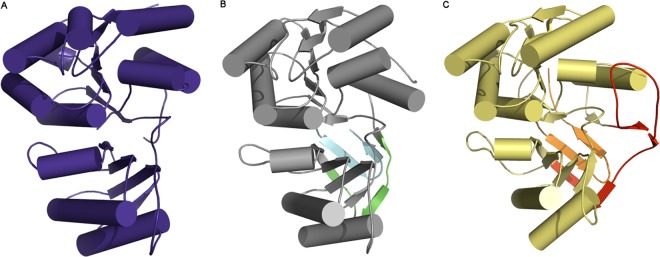
Structure comparisons. NaLdcA (A) superimposes on PaLdcA (PDB ID 1zrs; sequence identity of ∼20%) (B) with an RMSD of ∼2.4 Å over 254 Cα atoms. NaLdcA superimposes on MccF (PDB ID 3gjz; sequence identity of ∼16%) (C) with an RMSD of ∼2.5 Å over 242 Cα atoms. The small gap from residues 158 to 161 in NaLdcA corresponds to a long insertion in PaLdcA (residues 173 to 185; green) and a longer insertion in MccF (residues 166 to 204; red). A part of this insertion in MccF (residues 168 to 195) forms a flap that partially covers and restricts access to the substrate-binding cavity, which could have important consequences. Both PaLdcA and MccF also have longer C-terminal extensions (cyan and orange, respectively) than that of NaLdcA.
Sequence and structure analysis.
The majority (2,084 proteins) of the members of Pfam PF02016 contain a single Peptidase_S66 domain of ∼300 residues, which encompasses both the N-terminal and C-terminal domains of LdcA. The crystal structure of NaLdcA is only the third structure determined from this family (note that while the manuscript was in preparation, structures of three other MccF proteins from different bacterial species were determined, with PDB codes 3tla, 4e5s, and 4e94). In order to identify conserved regions of the protein, sequences of the following 9 proteins, annotated as either putative LdcA proteins or proteins of unknown function, with sequence identities ranging from 35 to 95% compared to NaLdcA as identified in a PSI-BLAST search, were selected for comparison (UniProt [60] IDs are given): B4W8U2 from Brevundimonas sp. BAL3, D9QNU0 from Brevundimonas subvibrioides, A3WBK2 from Erythrobacter sp. NAP1, Q2N9T0 from Erythrobacter litoris, A5P850 from Erythrobacter sp. SD-21, Q1GRW2 from Sphingopyxis alaskensis RB2256, Q2WB04 from Magnetospirillum magneticum, Q0C337 from Hyphomonas neptunium, and B8FC74 from Desulfatibacillum alkenivorans AK-01 (see Materials and Methods for more details). The residue conservation was mapped onto the NaLdcA structure to identify potential areas of functional importance. One side of NaLdcA displays significantly more conserved residues than the other, and as expected, these highly conserved regions include putative substrate-binding site residues (Asp52, Arg55, Tyr78, Arg82, Asp105, Asn167, Leu174, Thr177, Val193, Tyr198, Asp201, and Arg248), the catalytic triad (Ser104-His261-Glu191), and residues involved in dimer interactions (Glu195 and Arg202) (Fig. 3). The Ser (nucleophile)-His (general base)-Glu catalytic triad is characteristic of other LdcA proteins (16), thereby placing LdcA into the category of nonclassical serine proteases with a catalytic triad that is also seen in lipases and esterases (61). LdcA binds a large substrate that has a specific peptide portion and a less-specific sugar moiety. The residues that bind the peptide moiety of the substrate are the most conserved, and residues in the sugar-binding pocket are more variable. This distribution is consistent with data reported previously (62) and here (see “Substrate specificity”) showing that the enzyme prefers tetrapeptide substrates and can discriminate between tetrapeptide and pentapeptide substrates (Table 2, rows 2 and 4) but is less selective toward the N-terminal sugar moiety, i.e., GlcNAc-MurNAc, UDP-MurNAc, or MurNAc (Table 2, compare row 2 to rows 3, 5, and 6).
Table 2.
Specific activities and substrate specificities of N. aromaticivorans and E. coli ld-carboxypeptidasesa
| Substrate | Sp act (nmol/min/mg) |
|
|---|---|---|
| NaLdcA | EcLdcA | |
| l-Ala-γ-d-Glu-meso-A2pm-d-Ala | 230,000 | 250,000 |
| UDP-MurNAc-l-Ala-γ-d-Glu-meso-A2pm-d-Ala | 24,500 | 65,500 |
| UDP-MurNAc-l-Ala-γ-d-Glu-meso-A2pm-d-Ala-d-Ala | 7 | 51 |
| GlcNAc-MurNAc(anhydro)-l-Ala-γ-d-Glu-meso-A2pm-d-Ala | 14,300 | 26,500 |
| MurNAc-l-Ala-γ-d-Glu-meso-A2pm-d-Ala | 30,000 | 35,500 |
| MurNAc-l-Ala-γ-d-Glu-l-Lys-d-Ala | 1,000 | 450 |
The standard enzyme assay conditions described in Materials and Methods were used. The amount of enzyme varied from 0.2 ng to 0.5 μg, depending on the substrate used. To ensure linearity, the consumption of substrate was <20% in all cases. Values represent the means for three experiments; the standard deviation was <10% in all cases.
Substrate recognition residues and active site mechanism.
To elucidate protein-ligand interactions, GlcNAc-MurNAc-l-Ala-γ-d-Glu-meso-A2pm-d-Ala (PDB ligand ID MLD, from PDB ID 2cb3), which represents one of the substrates of LdcA, was manually docked into the substrate-binding site such that the C-terminal d-Ala of the tetrapeptide is positioned for attack by the catalytic Ser-His-Glu triad (Fig. 4). In general, the MLD substrate, with an approximate calculated volume of 374 Å3 (http://www.scfbio-iitd.res.in/software/utility/VolumeCalculator.jsp), fits quite well into the substrate-binding site, which has an approximate volume of 400 Å3 (calculated using the CASTp server [63]). Interestingly, the oxygen atoms from the carboxylate groups of the A2pm and d-Glu groups of MLD superimpose onto two SO4 ions present in the NaLdcA substrate-binding pocket, near active site residues Glu195 and Arg75. This observation supports the docking and suggests that the bound sulfate ions mimic portions of the natural substrate. The manual docking was validated computationally using Glide (version 5.7, Schrödinger, LLC, New York, NY). The Glide docking procedure resulted in 8 top-scoring ligand conformations, with energies ranging from −9.2 to −4.0 kcal/mol. Six of these top-scoring conformations were very similar to the manually docked ligand in general location and orientation (Fig. 5).
From this modeling, the NaLdcA residues that interact with substrates were identified as Thr12, Pro13, Thr15, Asp18, Ser44, His47, Arg75, Gly76, Gly77, Tyr78, Tyr103, Ser104, Asp105, Arg131, Arg132, Asn167, Met169, Thr170, Glu191, Glu195, and His261 (Fig. 4). In addition to the catalytic residues, i.e., Ser104, Glu191, and His261 (16), Tyr78, Asp105, Asn167, and Glu195 are among the most highly conserved residues in LdcA enzymes (Fig. 3 and 4) and are likely involved in interactions that lock the peptide in place for action by the catalytic triad (Tyr78 and Glu195 interact with A2pm, whereas Asn167 and Asp105 interact with Ala). Our modeling of ligand bound to NaLdcA is compatible with the proposed mechanism (16, 61) and provides a structural view of how the nucleophilic Ser104 residue is positioned to attack the carbonyl carbon of the peptide bond, while His261 acts as the general base and Glu191 provides additional stabilizing interactions by influencing the positioning of His261. Sufficient space is available for a water molecule to enter for the second step of the reaction. In addition, the nitrogen atoms from the main-chain amide bonds of Gly102 and Ser104 likely provide the oxyanion hole for stabilizing the reaction intermediate, which is typical of serine proteases. It is assumed that the variation in the nature of the other putative substrate-binding residues observed across the LdcA homologs bestows specific activity or substrate specificity to the individual enzymes. Other highly conserved residues, including Asp52, Arg55, Arg82, Leu174, Thr177, Val193, Tyr198, Asp201, Arg202 (in the vicinity of the UNL at the dimer interface), and Arg248 (Fig. 3 and 4), which do not interact with substrates directly, are likely to affect other aspects of enzyme function that might include some discrimination of the substrates' sugar moieties. Structural characterization and the identification of putative important residues present a basis for future studies of panels of single-substitution enzyme mutants and their roles in function.
Oligomeric assembly and ligand at the dimer interface.
Analytical size-exclusion chromatography indicated that NaLdcA is a monomer in solution; however, the crystallographic data indicate that it forms a stable dimer. The NaLdcA dimer in the crystal asu has a buried surface area of ∼2,600 Å2 excluding the UNL and ∼3,000 Å2 with the UNL included, as calculated by PISA (46), suggesting a significant dimer interaction. Analysis of the PaLdcA crystal structure with PISA also indicated a dimer as the probable oligomeric state (PDB IDs 1zl0 and 1zrs; buried areas of ∼3,000 Å2). MccF also forms a dimer in the crystal structure, with a buried area of ∼3,600 Å2. In all of these crystal structures, the dimeric assembly and interface are similar to those observed in NaLdcA, although the residues in the dimer interface are not highly conserved. Size-exclusion chromatography analysis of PaLdcA (PDB ID 1zrs) supports the presence of a dimer in solution; however, the apparent size of the dimer was lower than expected (56 kDa versus 70 kDa) (16). E. coli LdcA was a monomer in solution (13), similar to our results for NaLdcA. Thus, there may be some interconversion between monomeric and dimeric forms of LdcA, which, due to the location of the active site, may be influenced by substrate binding.
A relatively flat, triangular UNL with sides measuring ∼4 by 4 by 5 Å was modeled at the dimer interface, based on significant electron density in an Fo-Fc difference density map contoured at the 3.0 σ level (Fig. 6). Residues around the UNL include Met169, His 206, Leu203, Arg202, Glu195, Ala172, and SO4-274/275. Of these, Met169 and Glu195 may also be involved in substrate interactions, and Glu195 and Arg202 are highly conserved residues (Fig. 4). Interestingly, a glycerol molecule is bound to this region in the PaLdcA structure (PDB ID 1zl0), indicating that this may have some functional relevance.
Fig 6.

Potential site for modulating protein-protein interaction. An unidentified ligand (UNL) at the NaLdcA dimer interface (monomers are shown in yellow and beige) reveals a novel binding site that could be targeted for modulating oligomerization and activity of LdcA enzymes. A close-up of this site shows the residues that were found to interact with the ligand. Glu195 is highly conserved in other homologs as displayed in Fig. 3. Arg202 and His206 are also conserved as displayed in Fig. 5. A combination of highly conserved and less-conserved residues at this site suggests that it could be explored for the design of a general LdcA dimer inhibitor whose properties could be modified for individual LdcA proteins.
Enzymatic activity.
The expression and purification of NaLdcA and EcLdcA for biochemical analysis were carried out as detailed in Materials and Methods. Both proteins appeared to be >95% pure, as judged by SDS-PAGE analysis, with yields of 12 and 4 mg of purified protein/liter of culture, respectively. The specific activities of purified NaLdcA and EcLdcA, determined under standard assay conditions and using the tetrapeptide l-Ala-γ-d-Glu-meso-A2pm-d-Ala as the reference substrate, had similar values of 230 and 250 μmol/min/mg, respectively. Analogous to the studies performed on PaLdcA to test whether the enzyme possessed metallopeptidase activity, the effect of cations was also tested on NaLdcA. Neither enzyme required a cation (Mg2+ and Zn2+ were tested). EDTA at 2.5 mM did not inhibit the reaction. However, addition of 2 mM ZnSO4 dramatically decreased the activity of NaLdcA (3.8 versus 230 μmol/min/mg). The optimal pH for activity of NaLdcA was ∼7.4 to 7.6. The Km for the tetrapeptide of NaLdcA was estimated to be 0.17 ± 0.01 mM, and the kcat was 160 ± 25 s−1.
Substrate specificity.
The activity of the NaLdcA and EcLdcA enzymes was tested on a series of potential substrates, including peptidoglycan precursors and intermediates generated during maturation of the cell wall polymer (Table 2). The free tetrapeptide was clearly the best substrate. The presence of either a MurNAc, UDP-MurNAc, or GlcNAc-MurNAc(anhydro) moiety at the N terminus of the peptide significantly reduced the activity (8- to 16-fold). The replacement of diaminopimelate (A2pm) by Lys at position 3 of the peptide reduced the enzyme activity 30-fold (tested with MurNAc-peptide substrates), suggesting that the ε-carboxyl group at the d center of A2pm is important for substrate binding within the catalytic site as a result of stabilizing H-bonding interactions between the ε-carboxyl group and Glu195. UDP-MurNAc-pentapeptide also appeared to be a substrate, although at a much lower rate than that with UDP-MurNAc-tetrapeptide (0.007 versus 24.5 μmol/min/mg), indicating that the LdcA enzyme also exhibits a very low ld-endopeptidase activity. Comparison of the two LdcA (NaLdcA/EcLdcA) orthologs shows that their properties are quite similar in terms of both specific activity and substrate specificity (Table 2).
DISCUSSION
Substrates originating from peptidoglycan biosynthesis or recycling that are normally present in the cytoplasm are primarily GlcNAc-MurNAc(anhydro)-tetrapeptide and its degradation product, the free tetrapeptide. If the tetrapeptide is not rapidly cleaved by LdcA, it is added to UDP-MurNAc by the Mpl enzyme, and the UDP-MurNAc-tetrapeptide generated accumulates (as an “end product,” which is not a normal intermediate in this pathway) until it is processed by LdcA (7, 9). In this study, we confirmed that these compounds are substrates for NaLdcA, with a preference for the free tetrapeptide. An ld-endopeptidase secondary activity of NaLdcA was also observed with UDP-MurNAc-pentapeptide, which was very low and therefore should not have any deleterious effect (or regulatory function) on peptidoglycan metabolism (note that EcLdcA activity on pentapeptide compounds was previously reported as “not detectable” [11, 13]). Some data on the kinetic properties and substrate specificity of EcLdcA were available from earlier studies (11). However, the molecular mass estimated by these authors for EcLdcA was 12,000 Da, whereas it is actually above 33,000 Da. In addition, the specific activity determined earlier for EcLdcA was only 12 nmol/min/mg protein with UDP-MurNAc-tetrapeptide as the substrate, i.e., a value 2,000-fold lower than the one established here. Metz et al. (11) also found inhibition by Zn2+, but they did not detect any endopeptidase activity on UDP-MurNAc-pentapeptide. A 32-kDa ld-carboxypeptidase species (likely LdcA) was studied, which cleaved UDP-MurNAc-tetrapeptide (Km = 10−4 M) but not UDP-MurNAc-pentapeptide (13). Later, when the ldcA gene was identified by the same group (9), these authors produced and isolated the enzyme and investigated its properties to some extent. In particular, they confirmed similar activities on free tetrapeptide, UDP-MurNAc-tetrapeptide, and MurNAc-tetrapeptide, as well as (to a lesser extent, ca. 25%) on disaccharide tetrapeptide and its anhydro derivative.
Our results extend previous biochemical and structural work on LdcA proteins by providing insights into how the substrate is likely to bind and the residues that may be involved in substrate interactions. Functional characterization reveals how the activity of NaLdcA compares with that of EcLdcA. Prior work on LdcA has indicated that it may be a promising antibacterial target, with LdcA inhibitors being useful in the late stages of replication in bacteria or in combination therapy with drugs acting in earlier stages (12). Although crystal structures of LdcA show a consistent dimer, it is not known whether LdcA is functionally active as a dimer and, if so, whether dimers are more or less active than monomers. Our structure reveals a putative binding site for molecules that may stabilize or disrupt protein-protein interactions in dimer formation, and future studies will be needed to address this question. Modulation of LdcA activity by ligands could find applications in medicine for destabilizing pathogenic Gram-negative bacteria, in general, and NaLdcA in particular. For example, N. aromaticivorans has been linked to primary biliary cirrhosis (64, 65), and Novosphingobium or Sphingomonas species have been associated with environmental damage resulting from the death of coral reefs (66). Applications in industrial biotechnology may also be feasible, such as hydrocarbon bioremediation, where B8FC74 from Desulfatibacillum alkenivorans AK-01 has been proposed as a model organism for anaerobic alkane biodegradation (67).
ACKNOWLEDGMENTS
This work was conducted at the JCSG and the CNRS (UMR 8619). The JCSG is supported by the NIH, National Institutes of General Medical Sciences, Protein Structure Initiative (grant U54 GM094586). R.U.K. is grateful to the Swiss National Science Foundation for a postdoctoral fellowship.
Genomic DNA from Novosphingobium aromaticivorans DSM 12444 (ATCC 700278D) was obtained from the American Type Culture Collection (ATCC). We thank all members of the JCSG for their contribution to the development and operation of our HTP structural biology pipeline and for bioinformatic analyses, protein production, and structure determination.
Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a directorate of the SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393).
The content of this study is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
Footnotes
Published ahead of print 11 October 2013
For this virtual institution, see http://www.jcsg.org.
REFERENCES
- 1.Vollmer W, Blanot D, de Pedro MA. 2008. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 32:149–167 [DOI] [PubMed] [Google Scholar]
- 2.Barreteau H, Kovač A, Boniface A, Sova M, Gobec S, Blanot D. 2008. Cytoplasmic steps of peptidoglycan biosynthesis. FEMS Microbiol. Rev. 32:168–207 [DOI] [PubMed] [Google Scholar]
- 3.Bouhss A, Trunkfield AE, Bugg TD, Mengin-Lecreulx D. 2008. The biosynthesis of peptidoglycan lipid-linked intermediates. FEMS Microbiol. Rev. 32:208–233 [DOI] [PubMed] [Google Scholar]
- 4.Park JT, Uehara T. 2008. How bacteria consume their own exoskeletons (turnover and recycling of cell wall peptidoglycan). Microbiol. Mol. Biol. Rev. 72:211–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vollmer W, Joris B, Charlier P, Foster S. 2008. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol. Rev. 32:259–286 [DOI] [PubMed] [Google Scholar]
- 6.Mengin-Lecreulx D, van Heijenoort J, Park JT. 1996. Identification of the mpl gene encoding UDP-N-acetylmuramate:l-alanyl-γ-d-glutamyl-meso-diaminopimelate ligase in Escherichia coli and its role in recycling of cell wall peptidoglycan. J. Bacteriol. 178:5347–5352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hervé M, Boniface A, Gobec S, Blanot D, Mengin-Lecreulx D. 2007. Biochemical characterization and physiological properties of Escherichia coli UDP-N-acetylmuramate:l-alanyl-γ-d-glutamyl-meso-diaminopimelate ligase. J. Bacteriol. 189:3987–3995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Das D, Hervé M, Feuerhelm J, Farr CL, Chiu HJ, Elsliger MA, Knuth MW, Klock HE, Miller MD, Godzik A, Lesley SA, Deacon AM, Mengin-Lecreulx D, Wilson IA. 2011. Structure and function of the first full-length murein peptide ligase (Mpl) cell wall recycling protein. PLoS One 6:e17624. 10.1371/journal.pone.0017624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Templin MF, Ursinus A, Holtje JV. 1999. A defect in cell wall recycling triggers autolysis during the stationary growth phase of Escherichia coli. EMBO J. 18:4108–4117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Metz R, Henning S, Hammes WP. 1986. ld-Carboxypeptidase activity in Escherichia coli. I. The ld-carboxypeptidase activity in ether treated cells. Arch. Microbiol. 144:175–180 [DOI] [PubMed] [Google Scholar]
- 11.Metz R, Henning S, Hammes WP. 1986. ld-Carboxypeptidase activity in Escherichia coli. II. Isolation, purification and characterization of the enzyme from E. coli K 12. Arch. Microbiol. 144:181–186 [DOI] [PubMed] [Google Scholar]
- 12.Baum EZ, Crespo-Carbone SM, Foleno B, Peng S, Hilliard JJ, Abbanat D, Goldschmidt R, Bush K. 2005. Identification of a dithiazoline inhibitor of Escherichia coli l,d-carboxypeptidase A. Antimicrob. Agents Chemother. 49:4500–4507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ursinus A, Steinhaus H, Holtje JV. 1992. Purification of a nocardicin A-sensitive ld-carboxypeptidase from Escherichia coli by affinity chromatography. J. Bacteriol. 174:441–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. 2000. The Protein Data Bank. Nucleic Acids Res. 28:235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Norvell JC, Berg JM. 2007. Update on the protein structure initiative. Structure 15:1519–1522 [DOI] [PubMed] [Google Scholar]
- 16.Korza HJ, Bochtler M. 2005. Pseudomonas aeruginosa ld-carboxypeptidase, a serine peptidase with a Ser-His-Glu triad and a nucleophilic elbow. J. Biol. Chem. 280:40802–40812 [DOI] [PubMed] [Google Scholar]
- 17.Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL, Bateman A. 2008. The Pfam protein families database. Nucleic Acids Res. 36:D281–D288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tikhonov A, Kazakov T, Semenova E, Serebryakova M, Vondenhoff G, Van Aerschot A, Reader JS, Govorun VM, Severinov K. 2010. The mechanism of microcin C resistance provided by the MccF peptidase. J. Biol. Chem. 285:37944–37952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nocek B, Tikhonov A, Babnigg G, Gu M, Zhou M, Makarova KS, Vondenhoff G, Van Aerschot A, Kwon K, Anderson WF, Severinov K, Joachimiak A. 2012. Structural and functional characterization of microcin C resistance peptidase MccF from Bacillus anthracis. J. Mol. Biol. 420:366–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klock HE, Koesema EJ, Knuth MW, Lesley SA. 2008. Combining the polymerase incomplete primer extension method for cloning and mutagenesis with microscreening to accelerate structural genomics efforts. Proteins 71:982–994 [DOI] [PubMed] [Google Scholar]
- 21.Cormier CY, Park JG, Fiacco M, Steel J, Hunter P, Kramer J, Singla R, LaBaer J. 2011. PSI:Biology-materials repository: a biologist's resource for protein expression plasmids. J. Struct. Funct. Genomics 12:55–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. 1993. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 229:105–124 [DOI] [PubMed] [Google Scholar]
- 23.Santarsiero BD, Yegian DT, Lee CC, Spraggon G, Gu J, Scheibe D, Uber DC, Cornell EW, Nordmeyer RA, Kolbe WF, Jin J, Jones AL, Jaklevic JM, Schultz PG, Stevens RC. 2002. An approach to rapid protein crystallization using nanodroplets. J. Appl. Crystallogr. 35:278–281 [Google Scholar]
- 24.Lesley SA, Kuhn P, Godzik A, Deacon AM, Mathews I, Kreusch A, Spraggon G, Klock HE, McMullan D, Shin T, Vincent J, Robb A, Brinen LS, Miller MD, McPhillips TM, Miller MA, Scheibe D, Canaves JM, Guda C, Jaroszewski L, Selby TL, Elsliger MA, Wooley J, Taylor SS, Hodgson KO, Wilson IA, Schultz PG, Stevens RC. 2002. Structural genomics of the Thermotoga maritima proteome implemented in a high-throughput structure determination pipeline. Proc. Natl. Acad. Sci. U. S. A. 99:11664–11669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elsliger MA, Deacon AM, Godzik A, Lesley SA, Wooley J, Wuthrich K, Wilson IA. 2010. The JCSG high-throughput structural biology pipeline. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66:1137–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cohen AE, Ellis PJ, Miller MD, Deacon AM, Phizackerley RP. 2002. An automated system to mount cryo-cooled protein crystals on a synchrotron beamline, using compact sample cassettes and a small-scale robot. J. Appl. Crystallogr. 2002:720–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller MD, Deacon AM. 2007. An X-ray microsource based system for crystal screening and beamline development during synchrotron shutdown periods. Nucl. Instrum. Methods Phys. Res. A 582:233–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. 2002. Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat. 9:401–406 [DOI] [PubMed] [Google Scholar]
- 29.Leslie AGW, Powell HR. 2007. Processing diffraction data with Mosflm, p 41–51 In Read RJ, Sussman JL. (ed), Evolving methods for macromolecular crystallography. NATO Science Series, vol 245 Springer, Dordrecht, Netherlands [Google Scholar]
- 30.Collaborative Computing Project 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 50:760–763 [DOI] [PubMed] [Google Scholar]
- 31.Sheldrick GM. 2008. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 64:112–122 [DOI] [PubMed] [Google Scholar]
- 32.Vonrhein C, Blanc E, Roversi P, Bricogne G. 2007. Automated structure solution with autoSHARP. Methods Mol. Biol. 364:215–230 [DOI] [PubMed] [Google Scholar]
- 33.Abrahams JP, Leslie AG. 1996. Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr. Sect. D Biol. Crystallogr. 52:30–42 [DOI] [PubMed] [Google Scholar]
- 34.Langer G, Cohen SX, Lamzin VS, Perrakis A. 2008. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3:1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 36.Winn MD, Murshudov GN, Papiz MZ. 2003. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 374:300–321 [DOI] [PubMed] [Google Scholar]
- 37.Diederichs K, Karplus PA. 1997. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat. Struct. Biol. 4:269–275 [DOI] [PubMed] [Google Scholar]
- 38.Weiss MS, Hilgenfeld R. 1997. On the use of the merging R factor as a quality indicator for X-ray data. J. Appl. Crystallogr. 30:203–205 [Google Scholar]
- 39.Weiss MS, Metzner HJ, Hilgenfeld R. 1998. Two non-proline cis peptide bonds may be important for factor XIII function. FEBS Lett. 423:291–296 [DOI] [PubMed] [Google Scholar]
- 40.Cruickshank DW. 1999. Remarks about protein structure precision. Acta Crystallogr. Sect. D Biol. Crystallogr. 55:583–601 [DOI] [PubMed] [Google Scholar]
- 41.Yang H, Guranovic V, Dutta S, Feng Z, Berman HM, Westbrook JD. 2004. Automated and accurate deposition of structures solved by X-ray diffraction to the Protein Data Bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 60:1833–1839 [DOI] [PubMed] [Google Scholar]
- 42.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. 2007. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35:W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 66:213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Terwilliger TC. 2000. Maximum-likelihood density modification. Acta Crystallogr. Sect. D Biol. Crystallogr. 56:965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948 [DOI] [PubMed] [Google Scholar]
- 46.Krissinel E, Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372:774–797 [DOI] [PubMed] [Google Scholar]
- 47.Laskowski RA, Chistyakov VV, Thornton JM. 2005. PDBsum more: new summaries and analyses of the known 3D structures of proteins and nucleic acids. Nucleic Acids Res. 33:D266–D268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.DeLano WL. 2008. The PyMOL molecular graphics system. DeLano Scientific LLC, Palo Alto, CA [Google Scholar]
- 49.Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. 2010. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38:W529–W533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T, Ben-Tal N. 2005. ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 33:W299–W302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Glaser F, Pupko T, Paz I, Bell RE, Bechor-Shental D, Martz E, Ben-Tal N. 2003. ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 19:163–164 [DOI] [PubMed] [Google Scholar]
- 52.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gonnet GH, Cohen MA, Benner SA. 1992. Exhaustive matching of the entire protein sequence database. Science 256:1443–1445 [DOI] [PubMed] [Google Scholar]
- 54.Jones DT, Taylor WR, Thornton JM. 1992. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8:275–282 [DOI] [PubMed] [Google Scholar]
- 55.Jorgensen WL, Maxwell DS, Tirado-Rives J. 1996. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118:11225–11236 [Google Scholar]
- 56.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. 2004. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47:1739–1749 [DOI] [PubMed] [Google Scholar]
- 57.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. 2004. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 47:1750–1759 [DOI] [PubMed] [Google Scholar]
- 58.Matthews BW. 1968. Solvent content of protein crystals. J. Mol. Biol. 33:491–497 [DOI] [PubMed] [Google Scholar]
- 59.Murzin AG, Brenner SE, Hubbard T, Chothia C. 1995. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247:536–540 [DOI] [PubMed] [Google Scholar]
- 60.UniProt Consortium 2011. Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Res. 39:D214–D219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ekici OD, Paetzel M, Dalbey RE. 2008. Unconventional serine proteases: variations on the catalytic Ser/His/Asp triad configuration. Protein Sci. 17:2023–2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Leguina JI, Quintela JC, de Pedro MA. 1994. Substrate specificity of Escherichia coli ld-carboxypeptidase on biosynthetically modified muropeptides. FEBS Lett. 339:249–252 [DOI] [PubMed] [Google Scholar]
- 63.Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. 2006. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 34:W116–W118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bogdanos DP, Vergani D. 2009. Bacteria and primary biliary cirrhosis. Clin. Rev. Allergy Immunol. 36:30–39 [DOI] [PubMed] [Google Scholar]
- 65.Mattner J, Savage PB, Leung P, Oertelt SS, Wang V, Trivedi O, Scanlon ST, Pendem K, Teyton L, Hart J, Ridgway WM, Wicker LS, Gershwin ME, Bendelac A. 2008. Liver autoimmunity triggered by microbial activation of natural killer T cells. Cell Host Microbe 3:304–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Richardson LL, Goldberg WM, Kuta KG, Aronson RB, Smith GW, Ritchie KB, Halas JC, Feingold JS, Miller SL. 1998. Florida's mystery coral-killer identified. Nature 392:557–558 [Google Scholar]
- 67.Callaghan AV, Morris BE, Pereira IA, McInerney MJ, Austin RN, Groves JT, Kukor JJ, Suflita JM, Young LY, Zylstra GJ, Wawrik B. 2012. The genome sequence of Desulfatibacillum alkenivorans AK-01: a blueprint for anaerobic alkane oxidation. Environ. Microbiol. 14:101–113 [DOI] [PubMed] [Google Scholar]
- 68.Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]