

Abstract
NBU1 is a mobilizable transposon found in Bacteroides spp. Mobilizable transposons require gene products from coresident conjugative transposons for excision and transfer to recipient cells. The integration of NBU1 requires IntN1, which has been identified as a tyrosine recombinase, as well as Bacteroides host factor BHFa. Excision of NBU1 is a more complicated process, involving five element-encoded proteins (IntN1, Orf2, Orf2x, Orf3, and PrmN1) as well as a Bacteroides host factor and a cis-acting DNA sequence. Little has been known about what role the proteins play in excision, although IntN1 and Orf2x have been shown to be the only proteins absolutely required for detectable excision. To determine where IntN1 and Orf2x bind during the excision of NBU1, both proteins were partially purified and tested in DNase I footprinting experiments with the excisive attachment sites attL and attR. The results demonstrate that IntN1 binds to four core-type sites that flank the region of cleavage and strand exchange, as well as six arm-type sites. A unique feature of the system is the location of DR2a and DR2b arm-type sites immediately downstream of the attL core. The DR1a, DR1b, DR3a, and DR3b arm-type sites were shown to be required for in vitro integration of NBU1. In addition, we have identified one Orf2x binding site (O1) on attL as well as a dA+dT-rich upstream element that is required for Orf2x interactions with O1.
INTRODUCTION
Mobilizable transposons are genetic elements that can catalyze their own integration into host chromosomes but are dependent on coresident conjugative transposons (CTns) or plasmids for excision and transfer into recipient cells (1, 2). They have been implicated in contributing to the increase in antibiotic resistances observed in Bacteroides spp. (3, 4). The best-characterized mobilizable transposon in Bacteroides spp. is NBU1. NBU1 encodes an integrase (IntN1) that is required for both integration into and excision from the bacterial chromosome. IntN1 is a tyrosine recombinase that contains the signature RKHRHY motif in its C terminus (5, 6). A Bacteroides-encoded host factor is also required for the site-specific integration into the chromosomal attBT1-1 site located at the 3′ end of a leucyl-tRNA gene. An identical 14-bp sequence known as the “common core” is located on attBT1-1 and NBU1 (attN1). This sequence is duplicated upon integration (7, 8). The mechanism that IntN1 employs during integration appears to differ from those of other well-studied tyrosine recombinases like lambda Int. A hallmark of tyrosine recombinases is the requirement for sequence identity between the two sites of strand exchange (9). Surprisingly, a previous study demonstrated that creating specific mismatches between the NBU1 attN1 and attBT1-1 overlap sequences increased the integration frequency 100-fold (10). This is the only system identified thus far where mismatches in the overlap region enhance recombination frequency.
NBU1 excision has not been as extensively characterized as the integration reaction. A previous study demonstrated that in addition to IntN1, four NBU1-encoded proteins are involved in excision: Orf2x, Orf2, Orf3, and PrmN1 (Fig. 1A) (11). All five proteins were required when a Southern blot assay was used to detect excision from the Bacteroides chromosome. However, some excision was detected using a more sensitive PCR-based assay to detect the joined ends containing the attN1 site of NBU1 when in-frame deletions were constructed in orf2, orf3, or prmN1. No excision was detected when intN1 or orf2x was disrupted, demonstrating that IntN1 and Orf2x are the only two proteins absolutely required for excision (N. Shoemaker, unpublished results). Efficient excision also requires a cis-acting DNA sequence located downstream of prmN1 that includes oriT and two-thirds of the mobN1 gene (11). This region has been named the excision required sequence, or XRS (Fig. 1A). It is possible that one or more excision proteins bind to oriT and prevent it from being nicked until NBU1 fully excises from the chromosome (11).
Fig 1.

(A) Region of NBU1 involved in excision. Black arrows indicate genes absolutely required for excision. Gray arrows denote genes involved in excision. The oriT is indicated by the gray circle, and the gray rectangle represents two-thirds of the mobN1 gene; both are part of the “excision required sequence” or XRS. The XRS is necessary for efficient excision. The attN1 site is not located in the region shown in the figure. The entire region is approximately 6.4 kb in length. (B) Schematic showing the construction of DNA fragment attN1, attL, or attR for DNase I footprinting. One primer containing a 6-carboxyfluorescein phosphoramidate (FAM) label was paired with an unlabeled primer to generate DNA fragments containing either a top- or bottom-strand label. Arrows represent the locations where primers anneal during PCRs. The overlap sequence is indicated by a green box. Direct repeats identified in a previous study (10) are shown as purple boxes. The number “0” indicates the central base in the overlap sequence. Bases to the left of the overlap sequence are given negative numbers, and bases to the right are given positive numbers.
NBU1 also requires proteins encoded by an endogenous CTn such as CTnDOT for transfer to a recipient cell. CTnDOT encodes a two-component regulatory system consisting of the rteA and rteB genes. rteA encodes the sensor kinase, and rteB encodes the response regulator (12). RteB activates expression of the operon containing orf2 and orf2x on NBU1 (K. Moon, unpublished results). Since expression of rteA and rteB is dependent on the presence of tetracycline, NBU1 does not excise unless tetracycline is present (1, 2).
Orf2x is a 104-amino-acid protein and contains a putative helix-turn-helix motif. Because Orf2x is a small, basic protein that is required for excision, it likely belongs to a class of proteins called recombination directionality factors (RDFs). In systems that show directionality, integrase proteins are required for both integration and excision. RDFs are responsible for promoting excisive recombination and often inhibit integration (13). One well-studied RDF is Xis, a protein required for the excision of phage lambda. Xis binds to attR and bends the DNA, which facilitates Int binding and the formation of higher-order nucleoprotein complexes called intasomes. Xis also interacts with Int through direct protein-protein interactions (14–17). It is possible that Orf2x may perform similar functions during the excision of NBU1. The binding site(s) of Orf2x had not been identified.
Presumably, IntN1 is similar to lambda Int in that it contains three different DNA binding domains. The core-binding (CB) and catalytic (CAT) domains interact with core-type sites that are immediately adjacent to the overlap sequence where cleavage and strand exchange occurs. The N-terminal arm-binding (N) domain binds arm-type sites that are distal to the overlap sequence (13, 18). However, the exact binding sites of IntN1 were unknown. In this study, gel shift assays, DNase I footprinting experiments, and mutagenesis were utilized to characterize IntN1 and Orf2x interactions during excision.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and reagents.
All oligonucleotides were obtained from IDT and are listed in Table S1 in the supplemental material. Escherichia coli strain BL21(DE3) was purchased from Promega. BL21(DE3) ihfA was constructed by P1 transduction of a himA Δ82 deletion (19, 20) linked with Tn10 (Tetr) (J. Gardner, unpublished results). All E. coli strains were grown in Luria-Bertani broth (LB) (21). DNase I was supplied by Worthington. Antibiotics and bovine serum albumin (BSA) were supplied by Sigma. T4 polynucleotide kinase (T4 PNK) was obtained from Fermentas, and [γ-32P]ATP was obtained from Perkin-Elmer. Dithiothreitol (DTT) and isopropyl-β-d-1-thiogalactopyranoside (IPTG) were purchased from RPI. Antibiotics were used at the concentrations indicated: ampicillin (Amp), 100 μg/ml; kanamycin (Kan), 50 μg/ml; tetracycline, 10 μg/ml.
Purification of IntN1.
The intN1 gene was cloned into pET28a as described previously (5) to construct pET28a-intN1 (see Table S1 in the supplemental material). The plasmid was transformed into BL21(DE3) infA for expression, and IntN1 was purified using a previously described protocol (10). Peaks corresponding to absorbance at 280 nm were collected and analyzed using SDS-PAGE to identify fractions containing a 53-kDa protein, which is the size of IntN1. Fractions were screened for activity using DNA cleavage assays with attBT1-1 substrate as described previously (5). Active fractions were pooled and dialyzed into SU-75 buffer (50 mM sodium phosphate, pH 8, 150 mM NaCl, 5% glycerol, 2 mM DTT). The dialyzed IntN1 preparation was then loaded onto a HiLoad 16/60 Superdex 75 preparative-grade column (GE Healthcare). IntN1 eluted approximately 40 ml after loading as an approximately 50-kDa monomer. After confirmation that IntN1 was still active following gel filtration chromatography, fractions were pooled and dialyzed into storage buffer (50 mM sodium phosphate, pH 8, 150 mM NaCl, 1 mM DTT, 40% glycerol) and stored at −80°C. SDS-PAGE analysis of IntN1 following purification is shown in Fig. S1 in the supplemental material.
Partial purification of Orf2x.
The wild-type orf2x gene was cloned into the NdeI and EcoRI sites of pET27b using primers NdeI-orf2x and orf2x-EcoRI, creating plasmid pLR21 (see Table S1 in the supplemental material) (L. Rajeev, unpublished results). Expression of the orf2x gene was dependent on T7 RNA polymerase. The plasmid was sequenced by the UIUC Core Sequencing Facility to ensure that there were no mutations in the orf2x coding sequence. pLR21 was transformed into BL21(DE3) ihfA. To overexpress Orf2x, 5-ml cultures were initially grown overnight in LB plus Kan at 37°C and then subcultured 1:100 in 2 liters of LB plus Kan and grown at 30°C to mid-log phase. IPTG was then added to the culture to a final concentration of 0.1 mM, and growth was shifted to room temperature for 4 h. Cells were harvested by centrifugation and stored at −80°C. Cells were thawed on ice and then resuspended in 40 ml of low-salt lysis buffer (50 mM sodium phosphate, pH 8, 400 mM NaCl, 1 mM EDTA, 5% glycerol, 1 mM DTT, 0.02 mg/ml lysozyme). The suspension was lysed by sonication, and cell debris was collected by centrifugation at 10,000 × g for 60 min. The crude extract was loaded onto four 5-ml-volume heparin agarose columns (GE Healthcare) attached in tandem and eluted with elution buffer (50 mM sodium phosphate, pH 8, 1 mM EDTA, 5% glycerol, and 1 mM DTT) with a linear salt gradient of 0 to 2 M NaCl. Fractions corresponding to peaks showing absorbance at 280 nm were subjected to electrophoresis on SDS-PAGE gels to ascertain the presence of a 12-kDa band. Liquid chromatography-mass spectroscopy was performed by the UIUC Protein Sciences Facility to confirm the identity of the 12-kDa band as Orf2x. Previous experiments had demonstrated that Orf2x binds attL (Rajeev, unpublished), so protein activity was evaluated using gel shift assays with attL DNA. Active fractions were pooled and dialyzed against low-salt SP column buffer (50 mM sodium phosphate, pH 8, 50 mM NaCl, 1 mM EDTA, 5% glycerol, 1 mM DTT). The dialyzed Orf2x preparation was then loaded onto one 5-ml SP Sepharose column (GE Healthcare) and eluted with a linear salt gradient of 0 to 2 M NaCl. Fractions showing gel shift activity were pooled and dialyzed overnight into low-salt storage buffer (50 mM sodium phosphate, 50 mM NaCl, 1 mM EDTA, 40% glycerol, 1 mM DTT).
Gel shift assays.
A 258-bp attL fragment was PCR amplified from pLR20 using primers LR207 and LR200 (see Table S1 in the supplemental material) and purified using a Qiagen PCR purification kit. The DNA was 5′ radiolabeled with [γ-32P]ATP and T4 PNK, gel extracted, and precipitated as described previously (22). A 228-bp attR gel shift substrate was PCR amplified from pLR20 with primers LR212 and LR193 (see Table S1) and similarly prepared. Gel shift assays were performed in GSBA75 buffer (50 mM Tris-HCl, pH 8, 1 mM EDTA, 50 mM NaCl, 10% glycerol, and 0.075 μg/μl herring sperm DNA). Crude extract or partially purified Orf2x was incubated with 10 nM attL or attR DNA for 20 min at room temperature and then loaded on a prerun 5% polyacrylamide gel and subjected to electrophoresis at 20 mA for 2 h. The gel was then vacuum dried, exposed to an imaging plate, and visualized using a FujiFilm FLA-3000 phosphorimager and FujiFilm Image Gauge software (version 3.4 for Macintosh).
DNase I footprinting of Orf2x and IntN1.
A DNA fragment containing attL was used in DNase I footprinting experiments with Orf2x, and DNA fragments containing attL, attR, and attN1 were used in DNase I footprinting experiments with IntN1. To amplify the fragments, a forward primer containing 6-carboxyfluorescein phosphoramidate (6-FAM) was paired with an unlabeled reverse primer to create a DNA fragment with only one 5′-6-carboxyfluorescein phosphoramidate-labeled strand (Fig. 1B). Primers LR207 FAM and LR200 were used to amplify top-strand-labeled attL, and primers LR200 FAM and LR207 were used to amplify bottom-strand-labeled attL from pLR20 (see Table S1 in the supplemental material). Primers LR193 FAM and LR212 were used to amplify top-strand-labeled attR, and LR212 FAM and LR193 were used to amplify bottom-strand-labeled attR from pLR20 (see Table S1). To generate attN1 footprinting fragments from plasmid template pJWS200, LR193 FAM and LR200 were used to amplify top-strand-labeled fragments and LR200 FAM and LR193 were used to amplify bottom-strand-labeled fragments (see Table S1). DNA containing labeled attachment sites was extracted from agarose gels using a Qiagen gel extraction kit as described by the manufacturer.
IntN1 and Orf2x were diluted to the appropriate final concentrations in integration host factor (IHF) dilution buffer (50 mM Tris-HCl, pH 7.4, 10% glycerol, 800 mM KCl, 2 mg/ml BSA), and then 2 μl of protein was incubated with 1 pmol (0.05 μM) of DNA in a mixture containing 3 mM CaCl2, 7 mM MgCl2, 9.5% glycerol, 50 mM Tris-HCl, pH 7.4, 25 μg/ml BSA for 30 min at room temperature. DNase I (Worthington) diluted in DNase I dilution buffer (2.5 mM MgCl2, 0.5 mM CaCl2, 10 mM Tris-HCl, pH 7.6, 0.1 mg/ml BSA) to a final concentration of 0.0625 μg/ml was incubated with reaction mixtures for 1 min. The final volumes of the DNase I cleavage reactions were 20 μl. The reactions were quenched by the addition of 20 μl of 0.5 M EDTA. DNA fragments were purified using a Qiagen PCR purification kit and analyzed by the UIUC Core Sequencing Center using Applied Biosystems GeneMapper software (version 3.7).
To generate sequencing ladders so that exact regions of DNase I protection could be identified, a Thermo Sequenase Dye Primer manual cycle sequencing kit by USB was utilized essentially according to the manufacturer's instructions.
In vitro integration assays with substrates containing arm-type site mutants.
IntN1 arm-type sites identified using DNase I footprinting were mutated by introducing AatII restriction sites into each arm-type site. A derivative of pGEM-T containing the attN1 sequence, pJWS200, was used as a template for the mutagenesis reactions. Primers containing each of the mutations are listed in Table S1 in the supplemental material. Mutations were constructed using the QuikChange II mutagenesis kit (Agilent), and potential mutants were identified and screened as described previously (22).
In vitro integration assays were utilized to determine the effects of the arm-type mutations on integration frequency. Assays with wild-type or mutant attN1 sites were performed in 20-μl reaction volumes as described previously (8), except that reaction mixtures were incubated overnight at 37°C. Ten microliters of each reaction mixture was loaded onto a 1% agarose gel and electrophoresed at 120 V for 2 h, and then the gel was dried for 1 h and exposed to imaging plates for 4 h. The gels were visualized using FujiFilm FLA-3000 PhosphorImager and FujiFilm Image Gauge software (version 3.4 for Macintosh). Percent recombination was calculated by dividing the number of counts in the linear recombinant product by the total number of counts for a given reaction. Wild-type levels of in vitro integration were approximately 35%.
RESULTS
Purification of IntN1.
In a previous study, IntN1 was partially purified using heparin-agarose chromatography and used in in vitro recombination assays to study NBU1 integration (5, 8). The BL21(DE3) strain used to overexpress intN1 also expressed wild-type integration host factor (IHF). IHF is a DNA binding protein that interacts with specific sites in DNA and can also bind nonspecifically to DNA (23–25). We were concerned that IHF might copurify with IntN1 and consequently might affect patterns of protection in DNase I footprinting experiments. Accordingly, we purified IntN1 from a BL21(DE3) strain that has a mutation in ihfA as described in Materials and Methods. We were able to obtain a preparation that was estimated to be 90% pure, and this preparation was used for additional experiments examining IntN1 interactions with attachment site (see Fig. S1 in the supplemental material).
DNase I footprinting of IntN1.
DNase I footprinting was employed to identify the IntN1 binding site(s) on the NBU1 attachment sites. Positions of PCR primers containing 6-carboxyfluorescein phosphoramidate (6-FAM) were used to label the top or bottom strands of attL, attR, or attN1 as described in Materials and Methods and shown in Fig. 1B. Regions of protection from DNase I cleavage on the top strand of attL in the presence of IntN1 are shown in Fig. 2A. Nucleotides are numbered depending on location relative to the middle base of the overlap sequence which is designated 0. Bases to the left of the overlap sequence are assigned negative numbers, and bases to the right are given positive numbers (23). There is a region of protection located from positions −14 to +14 relative to the central base in the overlap sequence (Fig. 2A). Since the region includes the sites of cleavage and strand exchange, it is likely that there are two core-type sites located adjacent to the overlap sequence (Fig. 2B). Like other tyrosine recombinases, IntN1 likely binds to these sites while performing catalysis during excision. We have named these sites N′ and B. IntN1 protection corresponding to the B and N′ core-type sites was also detected on the bottom strand of attL from positions −16 to +17 (data not shown). Similar IntN1 protection in the attR core region was also detected from positions −19 to +17 on the top and bottom strands, corresponding to two core-type binding sites. The DNase I footprint of IntN1 on the bottom-strand attR core is shown in Fig. 3A and B. We have designated these core-type sites N and B′. Similar patterns of protection were also seen on the top strand of attR and the top and bottom strands of attN1 (data not shown).
Fig 2.

(A) DNase I footprint of IntN1 on the top strand of attL. Panel 1 shows a sequencing ladder generated using dideoxy sequencing reactions with the same 6-FAM-labeled primer used to make the footprinting substrate; green denotes adenine, blue denotes cytosine, black denotes guanine, and red denotes thymine. Panel 2 shows a DNase I digestion of top-strand-labeled attL in the absence of IntN1. Panels 3 through 5 show footprinting reactions with decreasing concentrations of IntN1. If IntN1 is diluted to 0.015 μM, a footprint is no longer detected (data not shown). The concentrations of IntN1 are indicated on the left sides of panels 3 through 5. Regions of protection from DNase I cleavage are denoted with bold black lines, and locations of enhanced cleavage are marked by asterisks. (B) IntN1 protection corresponding to the core-type sites and two arm-type sites on attL. Bolded bases denote regions protected from cleavage by DNase I in the presence of IntN1. Asterisks denote regions of enhanced DNase I cleavage in the presence of IntN1. Black boxes denote core-type sites B and N′, and gray boxes denote arm-type sites DR2a and DR2b. The arrows denote the sites of IntN1 cleavage during recombination. The overlap region is located between the N′ and B core-type sites, and the middle base of the overlap region is denoted by “0.”
Fig 3.

(A) IntN1 protection around the core of the bottom strand of attR. Panel 1 shows a sequencing ladder generated by dideoxy sequencing reactions. Green denotes adenine, blue denotes cytosine, black denotes guanine, and red denotes thymine. Panel 2 shows a DNase I footprinting reaction in the absence of IntN1. Panels 3 through 6 show footprinting reactions with decreasing dilutions of IntN1. IntN1 concentrations are shown at the left of panels 3 through 6. (B) IntN1 protection around the attR core. Bolded bases were protected from DNase I cleavage by IntN1. The black boxes denote core-type sites N and B′. The arrows indicate the cleavage sites.
In addition to the core-type sites, IntN1 arm-type sites were also identified through the DNase I footprinting experiments. Two regions of protection were detected on the top strand of attL. One, called DR2a, extends from positions +22 to +28 (Fig. 2A and B). There is also a region of enhanced DNase I cleavage located immediately upstream of DR2a (+19 to +21). An enhancement is also visible in DR2a at position +26. A second site, named DR2b, was detected from positions +33 to +40 (Fig. 2A and B). Protection corresponding to the DR2a and DR2b arm-type sites is also detectable on the bottom strand of attL from positions +17 to +28 and positions +33 to +40, and it was also detected on attN1 (data not shown). Two additional arm-type sites were also identified on attL. A site named DR3a was observed on the top and bottom strands from +141 to +150 (data not shown). In addition, weak protection from DNase I cleavage was detected on attN1 from positions +157 to +161 on the top and bottom strands of attN1 (data not shown). We have named this arm-type site DR3b.
Two arm-type sites were also detected on attR. IntN1 protection was observed from approximately positions −95 to −81 on the top strand of the excisive attachment site attR (data not shown). Another footprint was identified immediately downstream from positions −77 to −71 (data not shown). We believe that this region contains two arm-type sites, which we have named DR1a and DR1b (data not shown). DR1a and DR1b were also detected on the bottom strand of attR from positions −95 to −84 and positions −80 to −75 (data not shown). In total, we have identified four core-type sites and six arm-type sites through DNase I footprinting studies.
The DR1a, DR1b, DR3a, and DR3b arm-type sites are required for in vitro integration.
Presumably, specific IntN1 interactions with arm-type sites are required for the integration and excision of NBU1. In the lambda system, the P1 and P′3 arm-type sites are required for integration, the P2 arm-type site is required for excision, and intact P′1 and P′2 arm-type sites are necessary for both integration and excision (26–28). To determine which IntN1 arm-type sites are required for integration, site-directed mutations were constructed in each of the arm-type sites. In previous studies on the lambda system, multiple base pair mutations in some arm-type sites were necessary for detectable effects on recombination (26, 27). Cooperative interactions are involved in lambda Int binding to att DNA, and changing a single base pair in one of the arm-type sites was not necessarily sufficient to affect Int binding (17, 26, 27, 29). AatII sites were introduced in each IntN1 arm-type site to change the sequence of the binding sites as well as possibly disrupt any cooperative IntN1 interactions. The AatII mutations changed 6 bp in each arm-type site. The mutated arm-type sites were then tested in an in vitro integration assay.
The results of the in vitro integration assays are shown in Fig. 4. AatII mutations in the DR2a and DR2b arm-type sites did not affect the integration frequency (Fig. 4, lanes 4 and 5). Mutations in the DR1a and DR1b arm-type sites reduced integration to barely detectable levels, demonstrating that intact DR1a and DR1b arm-type sites are important for efficient integration (Fig. 4, lanes 2 and 3). The DR3a and DR3b arm-type sites are also required for in vitro integration, since mutations in either site abolished detectable recombination (Fig. 4, lanes 6 and 7).
Fig 4.
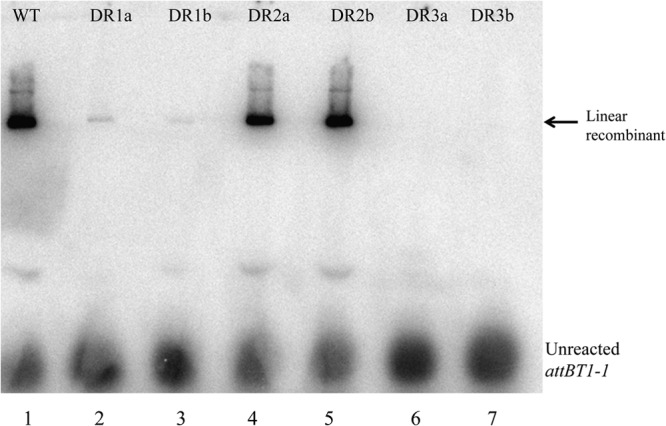
Results of in vitro integration reactions between a radiolabeled attB fragment and plasmid substrates containing attN1 sites with arm-type site mutations. Assays were performed as described in Materials and Methods. Lane 1, wild-type (WT) attN1 site; lanes 2 to 7, attN1 containing the indicated mutated arm-type site.
In a previous study, mutations were constructed in the sites that we identified here as DR1b, DR2b, and DR3b and the effects of the mutations were tested using an in vivo integration assay in E. coli (10). The results of the in vivo integration assays demonstrated that the mutated regions in what has now been identified as the DR1b and DR3b arm-type sites were required for efficient integration, while a mutation in the region containing the DR2b arm-type site had a minimal effect on integration frequency (10). At the time that these experiments were performed, it was not known whether these sites were IntN1 arm-type sites or host factor binding sites. The results described above agree with the experiments performed by Schmidt et al. (10). They show that the DR1b and DR3b sites are arm-type sites. In addition, the experiments detailed above also demonstrate that the DR1a and DR3a arm-type sites identified in this study are also required for NBU1 integration.
Orf2x interacts specifically with attL.
Orf2x is one of two NBU1-encoded proteins absolutely required for excision (Shoemaker, unpublished). The role of Orf2x in NBU1 excision had not been examined, although the presence of a potential helix-turn-helix motif suggested that it may be a DNA binding protein. We wanted to determine whether Orf2x interacted specifically with attL or attR. Orf2x was expressed in E. coli, and crude extract was used in gel shift assays with radiolabeled DNA fragments containing either attL or attR. Orf2x was able to shift the attL fragment, but no DNA binding was detected when Orf2x was incubated with attR (data not shown). An E. coli crude extract containing pET27b empty vector did not bind to the NBU1 attL or attR sites (data not shown). In addition, Orf2x did not shift a radiolabeled DNA substrate containing the attL site from CTnDOT, another mobile genetic element found in Bacteroides (data not shown). These findings demonstrated that Orf2x interacted specifically with the attL sequence of NBU1.
Orf2x was partially purified (using heparin-agarose and SP cation exchange chromatography) as described in Materials and Methods. We estimate that Orf2x was approximately 80% pure following the two purification steps (see Fig. S2A, lane 4, in the supplemental material). The partially purified Orf2x shifts attL (see Fig. S2B, lanes 2 and 4). A preparation of empty vector purified using the same protocol did not bind the labeled DNA (see Fig. S2B, lane 3). This preparation of Orf2x was used for further DNA binding and footprinting experiments.
DNase I footprinting of Orf2x on attL.
DNase I footprinting was employed to identify the binding site(s) of Orf2x on attL. Primers containing 6-carboxyfluorescein phosphoramidate (6-FAM) were used to label the top and bottom strands of attL as described in Materials and Methods. The results of DNase I footprinting of Orf2x on the bottom strand of attL are shown in Fig. 5A. Unfortunately, the region of attL where Orf2x binds does not contain many sites susceptible to DNase I cleavage. As a result, it is difficult to identify the exact sites bound by Orf2x. However, two regions of protection from DNase I cleavage showing a reproducible reduction in peak height were observed in the presence of Orf2x. One region was detected from positions +106 to +118 (Fig. 5A). Protection was especially pronounced from positions +106 to +108 and +116 to +118. Enhanced DNase I cleavage was also detected from positions +111 to +113. We have designated this region O1. Another region of protection was also detected on attL and spans from positions +126 to +142 (Fig. 5A). The region is dA+dT rich, and protection seen from positions +126 to +129 is subtle because DNase I does not cleave DNA efficiently in this region. This pattern was seen in four independent footprinting experiments (data not shown). Enhanced cleavage in the presence of DNase I is also visible at position +132 (Fig. 5A). We have called this region the dA+dT-rich element. The DNase I footprint of Orf2x is most visible with approximately 0.3 μM partially purified Orf2x, as seen in panel 3 of Fig. 5A. If Orf2x is diluted to approximately 0.08 μM, the two footprints disappear (Fig. 5A). Figure 5B illustrates the positions of the regions of Orf2x protection relative to the overlap sequence and direct repeats. These results suggest that there are either two Orf2x binding sites from positions +106 to +118 and positions +125 to +142 or one large site.
Fig 5.

(A) DNase I footprint of Orf2x on the bottom strand of attL. Fluorescently labeled attL DNA was PCR amplified and used in footprinting experiments with Orf2x. The top panel shows a sequencing ladder generated using dideoxy sequencing reactions with the same FAM-labeled primer used to make the footprinting substrate. Green denotes adenine, blue denotes cytosine, black denotes guanine, and red denotes thymine. Panel 2 shows a DNase I digestion performed in the absence of Orf2x. Panels 3 and 4 show footprinting reactions performed with decreasing concentrations of Orf2x. Orf2x concentrations are approximations and are indicated to the left of panels 2 through 4. Regions of protection from DNase I cleavage are denoted with bold black lines. Asterisks denote sites of enhanced DNase I cleavage in the presence of Orf2x. The identification of the binding sites is shown in the third panel. (B) The Orf2x binding sites as determined by DNase I footprinting reactions. Bolded bases were protected from DNase I cleavage in the presence of Orf2x. The orange boxes denote the O1 site and the dA+dT-rich element. Asterisks identify sites of enhanced DNase I cleavage in the presence of Orf2x. The system used for numbering the bases is described in Results.
Attempts to detect interactions with Orf2x and the top strand of attL were unsuccessful. Possible explanations for differences in Orf2x protection on the top and bottom strands of attL are presented in the Discussion.
A dA+dT-rich element is important for Orf2x binding.
As described above, it was not clear whether each region of protection on attL represented a distinct Orf2x binding site or whether attL contains only one Orf2x binding site. To determine how many Orf2x binding sites were present on attL, gel shift assays were performed with complementary oligonucleotides containing both regions of protection seen in DNase I footprinting experiments. The attL fragment used for gel shift analysis contained base pairs from +96 to +148. A single shift was observed when Orf2x was incubated with a substrate containing both regions (Fig. 6, lanes 2 and 3). This result suggests that there is only one Orf2x binding site, although it was also possible that Orf2x binding may be highly cooperative and the shift observed is two Orf2x proteins complexed with DNA.
Fig 6.

Gel shift assays with Orf2x and various attL substrates. The fragment used in lanes 1 through 3 contained the wild-type O1 site and dA+dT-rich element. The fragment utilized in gel shifts shown in lanes 4 through 6 contained an intact O1 site and the dA+dT-rich element mutated to the complementary sequence. The substrate used in lanes 7 through 9 contained an intact dA+dT-rich element and the O1 site changed to the complementary sequence. All three substrates are 52 bp in length and span the region from +96 to +148.
To further analyze Orf2x interactions with attL, gel shift assays were performed with substrates containing various deletions or mutations in each of the regions where Orf2x protection was detected in DNase I footprinting experiments. All substrates (excluding truncated substrates) were the same size as the wild-type substrate used in Fig. 6. Gel shift substrates containing the dA+dT-rich element alone were not shifted by Orf2x (data not shown). If the dA+dT-rich element (positions +126 to +142) was left intact but the O1 site sequence (positions +106 to +118) was changed to the complementary sequence (shown as “A/T intact, O1 comp” in Fig. 6), no detectable shift was seen (Fig. 6, lanes 8 and 9), demonstrating that the O1 site must be intact for Orf2x binding. Interestingly, Orf2x did not shift a substrate containing only the O1 site but lacking the dA+dT-rich element (data not shown). This result showed that O1 by itself was not sufficient for Orf2x binding.
To evaluate the contribution of the dA+dT-rich element, gel shifts were performed with substrates containing two different types of changes to the dA+dT-rich element. First, a substrate with the dA+dT-rich element changed to the complementary sequence with a wild-type O1 site (shown as “O1 intact, A/T comp” in Fig. 6) was tested. This mutation maintained the dA+dT richness of the dA+dT-rich element. Orf2x was shown to weakly shift the fragment (Fig. 6, lanes 5 and 6). The affinity of Orf2x for this substrate appeared to be reduced compared to that for the wild-type substrate (Fig. 6, lanes 5 and 6 compared to lanes 2 and 3). Next, the dA+dT base pairs in the dA+dT-rich element were changed to dG+dC base pairs to alter the dA/dT content of the region. Orf2x weakly shifted this substrate, and Orf2x appeared to have slightly less affinity for the “A/T to G/C” substrate than the “A/T Comp” substrate (data not shown). We interpret these experiments to show that Orf2x binds to the O1 site and that flanking DNA containing a dA+dT-rich sequence is required for efficient Orf2x binding.
DISCUSSION
In this report, we have identified the binding sites of IntN1 and Orf2x using DNase I footprinting. Like other tyrosine recombinases that display directionality, IntN1 interacts with two different classes of DNA binding sites. The CB and CAT domains bind core-type sites N and N′ on attN1 and B and B′ on attBT1-1 in the Bacteroides chromosome, while the N domain interacts with six arm-type sites (Fig. 7). Consensus sequences were derived by comparing the arm- and core-type sites to one another (Fig. 8A and B). The core-type sites do not share many base pairs in common; thus, the consensus sequence is weak. The only sequence conservation in all four core-type sites is a cytosine at position +4 of the N and B sites and position −4 of the N′ and B′ sites (Fig. 8A). The core-type binding sites of other well-characterized tyrosine recombinases such as lambda Int and IntDOT of CTnDOT show stronger core-type binding site consensus sequences (23, 30, 31). The arm-type sites have a much stronger consensus sequence (Fig. 8B), including six of nine bases that are found in all six sites and two bases that are found in five of six sites. The spacing between pairs of arm-type sites was also remarkably consistent. The pair of arm-type sites in each direct repeat was separated by 4 bp, and at least three of the base pairs were A/T base pairs (Fig. 7).
Fig 7.
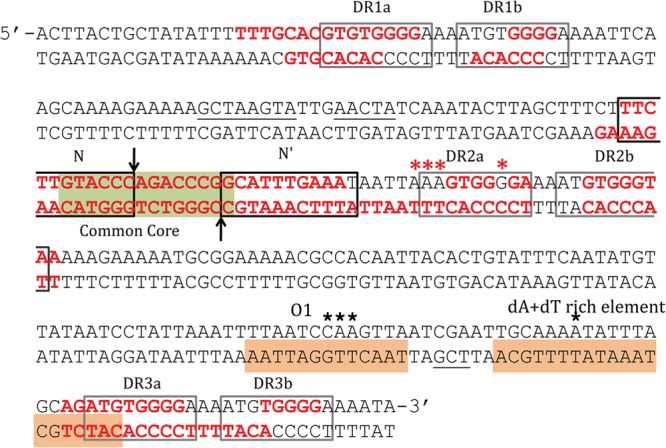
IntN1 and Orf2x protection on attN1 as determined by DNase I footprinting analysis. The black boxes indicate IntN1 core-type sites, and the gray boxes denote arm-type sites. The green box represents the common core region, and the vertical arrows indicate the sites of cleavage. IntN1 protection is shown by red bases, and enhancement of DNase I cleavage in the presence of IntN1 is shown by red asterisks. Orf2x protection is denoted by orange boxes, and black stars denote enhanced DNase I cleavage in the presence of Orf2x. Underlined bases represent bases protected from DNase I cleavage by IntN1 on only one strand.
Fig 8.
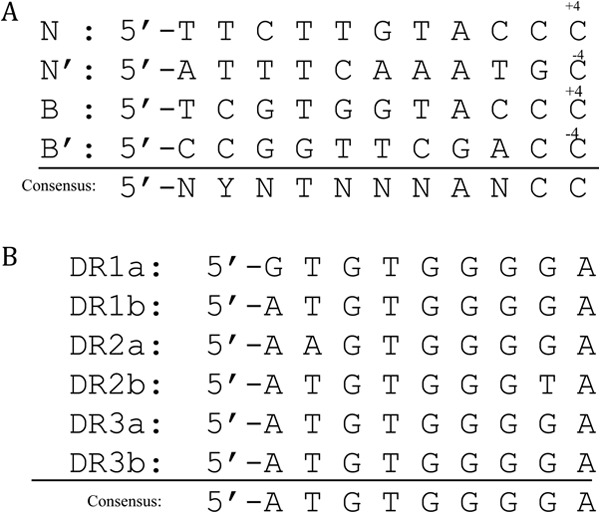
(A) Alignment of four core-type sites as determined by DNase I footprinting. The consensus sequence was derived by comparing each base between the four core-type sites. Y, T or C; N, any base. (B) Alignment of six arm-type sites as determined by DNase I footprinting. The consensus sequence is shown at the bottom.
Mutagenesis studies showed that the DR1a, DR1b, DR3a, and DR3b sites are required for efficient in vitro integration (Fig. 4). It is likely that IntN1 interactions with these four arm-type sites are required to promote integrative recombination. NBU1 resembles other systems, including lambda, CTnDOT, and mycobacteriophage L5, which also require multiple intact arm-type sites for integration (22, 26–28, 32). As described below, the DR2a and DR2b arm-type sites may be important for excision. Due to the complexity of the NBU1 excision reaction, there is currently no in vitro excision assay to examine the importance of the DR2a and DR2b sites or the other arm-type sites in NBU1 excision. Experiments are under way to develop an in vitro excision assay so that the roles of the arm-type sites can be further elucidated.
A unique feature of the arrangement of IntN1 binding sites is the close proximity of the N′ core-type site to the DR2a arm-type site (Fig. 7). In other site-specific recombination systems, including lambda, P22, Tn916, and CTnDOT, there are at least 40 bp between the central base in the overlap region and the nearest arm-type site (Fig. 9) (13, 23, 31, 33, 34). There are also host factor and/or excisionase binding sites between the core and the nearest arm-type site in the abovementioned systems. The DR2a arm-type site is separated from the N′ core-type site by only 5 bp, so there is insufficient room for an accessory factor to bind between the two binding sites (Fig. 9). It is possible that IntN1 monomers bound at DR2a or DR2b sites on attL are involved in intermolecular bridging, since direct interactions of an IntN1 bound to the DR2a arm-type site with the core would be hindered by the close proximity of the binding sites. IntN1 monomers bound at DR2a or DR2b may be able to simultaneously bind to N or B′ on attR. This possibility is supported by the results of mutagenesis studies that demonstrated that DR2a and DR2b are not necessary for integration (Fig. 4). In the lambda system, Int is capable of intermolecular interactions between attL and attR during excision (35, 36). It is also conceivable that DR2a and DR2b may be gratuitous arm-type sites and thus may play no role in the integration or excision reactions. Other systems have arm-type sites that are not required for either reaction (22, 37), and DR2a and DR2b may also be unnecessary.
Fig 9.

Alignment of integrase binding sites from other site-specific recombination systems, which are denoted with arrows. The “0” shown at the top of the figure represents the central base in the crossover region, with bases to the right indicated by positive numbers and bases to the left denoted with negative numbers. The arrows represent integrase binding sites. The core-type sites are inverted repeats that flank position 0. The numbering and spacing are approximate, and binding sites are not drawn to scale. The P6 and P7 arm-type sites from the mycobacteriophage L5 system are not included in the figure.
Many site-specific recombination systems display directionality by regulating the expression of RDFs that promote excision reactions. Excision systems can be simple or complex depending upon the number of RDFs involved. The lambda and HK022 systems are examples of simple systems that utilize one RDF for excision (14, 38). In contrast, complex systems like NBU1, CTnDOT, and Tn4555 use several element-encoded RDFs (11, 39, 40). Efficient NBU1 excision requires IntN1, Orf2x, and three additional NBU1-encoded proteins (Orf2, Orf3, and PrmN1) as well as a cis-acting DNA sequence (11). The functions of these proteins during NBU1 excision are not well understood. Orf2x has no close homologues, although it shares a helix-turn-helix motif and other similar characteristics with Xis of Tn4555 (11, 39). Exactly why NBU1 and Tn4555 excisions involve multiple proteins and (in the case of NBU1) a cis-acting DNA sequence is not known, but the additional complexity may be required for coordination with the coresident CTn to prevent transfer before the element is fully excised (11).
DNase I footprinting experiments identified an Orf2x binding site as well as a flanking dA+dT-rich element necessary for Orf2x binding (Fig. 5A and 7). Both regions are located near the end of attL proximal to the core (Fig. 5B and 7). Since only one shift was visible in gel shift assays, we believe that Orf2x binds only to the O1 site, while interactions with the flanking DNA stimulate Orf2x binding to O1. Even when the sequence of the dA+dT-rich element changed dramatically, Orf2x still bound weakly to a substrate containing an intact O1 site (Fig. 6). This result suggests that flanking DNA stimulates Orf2x binding to the O1 site, but the sequence of the flanking DNA is not as important. Interestingly, some IHF sites also contain dA+dT-rich elements. The H2 and H′ sites in the lambda attP, attL, and attR sites contain dA+dT-rich elements that are protected from nuclease digestion by IHF (41). The Orf2x dA+dT-rich element partially overlaps with the DR3a arm-type site. So, it is possible that Orf2x interactions with the dA+dT-rich element may preclude IntN1 from binding to DR3a, thus preventing an interaction necessary for integration. The requirement for an intact DR3a arm-type site for detectable in vitro integration (Fig. 4) supports this idea. In the lambda system, the P1 arm-type site is required for efficient integration (26–28). Lambda Xis inhibits Int binding to the P1 site, which promotes excisive recombination (42), and it is possible that Orf2x may similarly promote NBU1 excision. Interestingly, Orf2x alone does not inhibit NBU1 integration in vitro (data not shown). It is possible that one of the other NBU1 proteins involved in excision (Orf2, Orf3, or PrmN1) may be required to prevent the reintegration of NBU1 during excision.
Despite repeated attempts, we were unable to detect an Orf2x footprint on the top strand of attL when Orf2x was incubated alone with DNA (data not shown). We do not understand the basis of this result. DNase I cuts well in this region, so we do not believe the lack of a detectable footprint is due to inadequate cleavage. Most proteins produce DNase I footprints on both the top and bottom strands, although there are exceptions. The Xis of bacteriophage P22 produced a strong footprint on the top strand but not the bottom strand of the phage attR site (43). Interestingly, Orf2x protection at the O1 binding site was observed on the top strand of attL if IntN1 was also present (data not shown). Since IntN1 alone did not footprint in this region, we believe that the observed protection may be due to Orf2x binding to O1. It is possible that the presence of IntN1 may affect or even enhance Orf2x interactions with the O1 binding site. In the lambda system, DNase I footprinting experiments performed with Xis and Int demonstrated that DNA sequences protected from DNase I cleavage were the sum of the those protected by each protein individually (44). DNase I footprinting experiments have also shown that the integrase of mycobacteriophage L5 (Int-L5) cannot interact with the P1/P2 arm-type sites unless Xis-L5 is also present (37). The presence of IntN1 may be important for Orf2x interactions with the attL attachment site.
This is the first study to characterize the DNA binding sites of the two proteins required for the excision of NBU1. IntN1 interacts with six arm-type sites, and four arm-type sites (DR1a, DR1b, DR3a, and DR3b) are required for the in vitro integration of NBU1. We have also identified the binding site of Orf2x on the attL attachment site, O1, as well as a dA+dT-rich element that stimulates binding. The functions of the other NBU1 excision proteins, Orf2, Orf3, and PrmN1, are not known. Work is in progress to determine how these other proteins contribute to the complex NBU1 excision reaction.
Supplementary Material
ACKNOWLEDGMENTS
We thank Wilma Ross for technical advice and David McCurdy and Abigail Salyers for helpful suggestions.
Footnotes
Published ahead of print 4 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01011-13.
REFERENCES
- 1.Shoemaker NB, Salyers AA. 1988. Tetracycline-dependent appearance of plasmidlike forms in Bacteroides uniformis 0061 mediated by conjugal Bacteroides tetracycline resistance elements. J. Bacteriol. 170:1651–1657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stevens AM, Shoemaker NB, Salyers AA. 1990. The region of a Bacteroides conjugal chromosomal tetracycline resistance element which is responsible for production of plasmidlike forms from unlinked chromosomal DNA might also be involved in transfer of the element. J. Bacteriol. 172:4271–4279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whittle G, Shoemaker NB, Salyers AA. 2002. The role of Bacteroides conjugative transposons in the dissemination of antibiotic resistance genes. Cell. Mol. Life Sci. 59:2044–2054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferreira LQ, Avelar KE, Vieira JM, de Paula GR, Colombo AP, Domingues RM, Ferreira MC. 2007. Association between the cfxA gene and transposon Tn4555 in Bacteroides distasonis strains and other Bacteroides species. Curr. Microbiol. 54:348–353 [DOI] [PubMed] [Google Scholar]
- 5.Rajeev L, Salyers AA, Gardner JF. 2006. Characterization of the integrase of NBU1, a Bacteroides mobilizable transposon. Mol. Microbiol. 61:978–990 [DOI] [PubMed] [Google Scholar]
- 6.Shoemaker NB, Wang GR, Salyers AA. 1996. The Bacteroides mobilizable insertion element, NBU1, integrates into the 3′ end of a Leu-tRNA gene and has an integrase that is a member of the lambda integrase family. J. Bacteriol. 178:3594–3600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shoemaker NB, Wang GR, Stevens AM, Salyers AA. 1993. Excision, transfer, and integration of NBU1, a mobilizable site-selective insertion element. J. Bacteriol. 175:6578–6587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rajeev L, Segall A, Gardner J. 2007. The bacteroides NBU1 integrase performs a homology-independent strand exchange to form a Holliday junction intermediate. J. Biol. Chem. 282:31228–31237 [DOI] [PubMed] [Google Scholar]
- 9.Bauer CE, Gardner JF, Gumport RI. 1985. Extent of sequence homology required for bacteriophage lambda site-specific recombination. J. Mol. Biol. 181:187–197 [DOI] [PubMed] [Google Scholar]
- 10.Schmidt JW, Rajeev L, Salyers AA, Gardner JF. 2006. NBU1 integrase: evidence for an altered recombination mechanism. Mol. Microbiol. 60:152–164 [DOI] [PubMed] [Google Scholar]
- 11.Shoemaker NB, Wang GR, Salyers AA. 2000. Multiple gene products and sequences required for excision of the mobilizable integrated Bacteroides element NBU1. J. Bacteriol. 182:928–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stevens AM, Sanders JM, Shoemaker NB, Salyers AA. 1992. Genes involved in production of plasmidlike forms by a Bacteroides conjugal chromosomal element share amino acid homology with two-component regulatory systems. J. Bacteriol. 174:2935–2942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Azaro MA, Landy A. 2002. Lambda integrase and the lambda Int family, p 119–148 In Craig NL, Craigie R, Gellert M, Lambowitz AM. (ed), Mobile DNA II. ASM Press, Washington, DC [Google Scholar]
- 14.Bushman W, Yin S, Thio LL, Landy A. 1984. Determinants of directionality in lambda site-specific recombination. Cell 39:699–706 [DOI] [PubMed] [Google Scholar]
- 15.Cho EH, Gumport RI, Gardner JF. 2002. Interactions between integrase and excisionase in the phage lambda excisive nucleoprotein complex. J. Bacteriol. 184:5200–5203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swalla BM, Cho EH, Gumport RI, Gardner JF. 2003. The molecular basis of co-operative DNA binding between lambda integrase and excisionase. Mol. Microbiol. 50:89–99 [DOI] [PubMed] [Google Scholar]
- 17.Thompson JF, de Vargas LM, Skinner SE, Landy A. 1987. Protein-protein interactions in a higher-order structure direct lambda site-specific recombination. J. Mol. Biol. 195:481–493 [DOI] [PubMed] [Google Scholar]
- 18.Moitoso de Vargas L, Pargellis CA, Hasan NM, Bushman EW, Landy A. 1988. Autonomous DNA binding domains of lambda integrase recognize two different sequence families. Cell 54:923–929 [DOI] [PubMed] [Google Scholar]
- 19.Miller HI, Kirk M, Echols H. 1981. SOS induction and autoregulation of the himA gene for site-specific recombination in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 78:6754–6758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller HI. 1984. Primary structure of the himA gene of Escherichia coli: homology with the DNA-binding protein HU and association with the phenylalanyl-tRNA synthetase operon. Cold Spring Harbor Symp. Quant. Biol. 49:691–698 [DOI] [PubMed] [Google Scholar]
- 21.Bertani G. 1951. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 62:293–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wood MM, Dichiara JM, Yoneji S, Gardner JF. 2010. CTnDOT integrase interactions with attachment site DNA and control of directionality of the recombination reaction. J. Bacteriol. 192:3934–3943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dichiara JM, Mattis AN, Gardner JF. 2007. IntDOT interactions with core- and arm-type sites of the conjugative transposon CTnDOT. J. Bacteriol. 189:2692–2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng Q, Wesslund N, Shoemaker NB, Salyers AA, Gardner JF. 2002. Development of an in vitro integration assay for the Bacteroides conjugative transposon CTnDOT. J. Bacteriol. 184:4829–4837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson RC, Johnson L, Schmidt JW, Gardner JF. 2004. Major nucleoid proteins in the structure and function of the Escherichia coli chromosome, p 65–132 In Higgins NP. (ed), The bacterial chromosome. ASM Press, Washington, DC [Google Scholar]
- 26.Bauer CE, Hesse SD, Gumport RI, Gardner JF. 1986. Mutational analysis of integrase arm-type binding sites of bacteriophage lambda. Integration and excision involve distinct interactions of integrase with arm-type sites. J. Mol. Biol. 192:513–527 [DOI] [PubMed] [Google Scholar]
- 27.Numrych TE, Gumport RI, Gardner JF. 1990. A comparison of the effects of single-base and triple-base changes in the integrase arm-type binding sites on the site-specific recombination of bacteriophage lambda. Nucleic Acids Res. 18:3953–3959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hazelbaker D, Azaro MA, Landy A. 2008. A biotin interference assay highlights two different asymmetric interaction profiles for lambda integrase arm-type binding sites in integrative versus excisive recombination. J. Biol. Chem. 283:12402–12414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ross W, Landy A, Kikuchi Y, Nash H. 1979. Interaction of int protein with specific sites on lambda att DNA. Cell 18:297–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ross W, Landy A. 1983. Patterns of lambda Int recognition in the regions of strand exchange. Cell 33:261–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ross W, Landy A. 1982. Bacteriophage lambda int protein recognizes two classes of sequence in the phage att site: characterization of arm-type sites. Proc. Natl. Acad. Sci. U. S. A. 79:7724–7728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pena CE, Lee MH, Pedulla ML, Hatfull GF. 1997. Characterization of the mycobacteriophage L5 attachment site, attP. J. Mol. Biol. 266:76–92 [DOI] [PubMed] [Google Scholar]
- 33.Smith-Mungo L, Chan IT, Landy A. 1994. Structure of the P22 att site. Conservation and divergence in the lambda motif of recombinogenic complexes. J. Biol. Chem. 269:20798–20805 [PubMed] [Google Scholar]
- 34.Lu F, Churchward G. 1994. Conjugative transposition: Tn916 integrase contains two independent DNA binding domains that recognize different DNA sequences. EMBO J. 13:1541–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim S, Landy A. 1992. Lambda Int protein bridges between higher order complexes at two distant chromosomal loci attL and attR. Science 256:198–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim S, Moitoso de Vargas L, Nunes-Duby SE, Landy A. 1990. Mapping of a higher order protein-DNA complex: two kinds of long-range interactions in lambda attL. Cell 63:773–781 [DOI] [PubMed] [Google Scholar]
- 37.Lewis JA, Hatfull GF. 2003. Control of directionality in L5 integrase-mediated site-specific recombination. J. Mol. Biol. 326:805–821 [DOI] [PubMed] [Google Scholar]
- 38.Yagil E, Dolev S, Oberto J, Kislev N, Ramaiah N, Weisberg RA. 1989. Determinants of site-specific recombination in the lambdoid coliphage HK022. An evolutionary change in specificity. J. Mol. Biol. 207:695–717 [DOI] [PubMed] [Google Scholar]
- 39.Parker AC, Smith CJ. 2004. A multicomponent system is required for tetracycline-induced excision of Tn4555. J. Bacteriol. 186:438–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng Q, Sutanto Y, Shoemaker NB, Gardner JF, Salyers AA. 2001. Identification of genes required for excision of CTnDOT, a Bacteroides conjugative transposon. Mol. Microbiol. 41:625–632 [DOI] [PubMed] [Google Scholar]
- 41.Hales LM, Gumport RI, Gardner JF. 1994. Determining the DNA sequence elements required for binding integration host factor to two different target sites. J. Bacteriol. 176:2999–3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moitoso de Vargas L, Landy A. 1991. A switch in the formation of alternative DNA loops modulates lambda site-specific recombination. Proc. Natl. Acad. Sci. U. S. A. 88:588–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mattis AN, Gumport RI, Gardner JF. 2008. Purification and characterization of bacteriophage P22 Xis protein. J. Bacteriol. 190:5781–5796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yin S, Bushman W, Landy A. 1985. Interaction of the lambda site-specific recombination protein Xis with attachment site DNA. Proc. Natl. Acad. Sci. U. S. A. 82:1040–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.