

Abstract
Subgrouping of medulloblastoma by microarray expression profiling has dramatically changed our perspective of this malignant childhood brain tumour. Now, the availability of next-generation sequencing and complementary high-density genomic technologies has unmasked novel driver mutations in each medulloblastoma subgroup. The implications of these findings for the management of patients are readily apparent, pinpointing previously unappreciated diagnostic and therapeutic targets. Here, we summarize the ’explosion’ of data emerging from the application of modern genomics to medulloblastoma, and in particular the recurrent targets of mutation in medulloblastoma subgroups. These data are making their way into contemporary clinical trials as we seek to integrate conventional and molecularly targeted therapies.
Introduction
Central nervous system (CNS) tumours are the second most prevalent cancer in children after leukaemia, and remain the leading cause of cancer-related mortality in childhood1, 2. Medulloblastoma is the most common malignant childhood brain tumour. Overall survival rates for patients with medulloblastoma have reached 70-80% using treatment protocols that include a combination of surgery, cranio-spinal radiotherapy (in children ≥3 years old), and chemotherapy3-5. Current risk stratification tools have been in place for decades and are based solely on clinical features including age at diagnosis, extent of surgical resection, metastatic status, and in some cases histological features. Infants (≤3 years of age), patients with residual tumour (≥1.5cm2) following neurosurgery, and those exhibiting leptomeningeal dissemination at the time of diagnosis are all considered high-risk and all others deemed average-risk6. Although conventional therapies cure a large proportion of patients with medulloblastoma, the majority of survivors suffer from long-term side effects including developmental, neurological, neuroendocrine, and psychosocial deficits7-9. Molecular stratification of medulloblastoma patients has yet to be routinely implemented in the clinic and the use of rational, molecularly targeted therapy for the disease is still in its infancy10, 11. Through an improved understanding of the molecular and genetic basis of medulloblastoma, it is anticipated that patients will be stratified and treated according to the biological makeup of their disease in the future, leading to improved patient outcomes with reduced sequelae.
Studies of heritable forms of medulloblastoma provided the first insights into the processes that underlie the disease12. In the 1990s, Gorlin Syndrome was confirmed to be attributable to inherited mutations of the patched 1 (PTCH1) tumour suppressor gene (TSG) on chromosome 9q22.3213-15. This discovery paved the way for several follow-up studies, which revealed recurrent somatic mutations of PTCH1 in sporadic medulloblastomas16-18. The sonic hedgehog (SHH) pathway, a developmental signaling axis in which PTCH1 normally imposes an inhibitory effect, is aberrantly activated in about one third of all medulloblastomas19. Activation of SHH signaling in these tumours results from recurrent mutations and/or copy number aberrations targeting multiple specific levels of the pathway, as will be discussed below. Individuals with Turcot Syndrome are susceptible to the development of colorectal cancer and brain tumours, including medulloblastoma. Turcot Syndrome consists of two subtypes: type I cases exhibit an increased risk of medulloblastoma owing to inactivating germline mutations in adenomatous polyposis coli (APC), a TSG that negatively regulates β-catenin, the key effector of the WNT signaling pathway20. Deregulation of the WNT pathway secondary to somatic mutations of the β-catenin gene, CTNNB121,22, is now recognized to occur in ~10% of sporadic medulloblastomas19.
Extrapolation of insight gained from these medulloblastoma susceptibility syndromes to candidate gene approaches has revealed additional oncogenes and TSGs that are altered in familial and sporadic medulloblastoma, although mutations in the majority of these genes are exceedingly rare23-29. Recent technological advances in nucleic acid sequencing along with the availability of high-density microarrays of unprecedented resolution have opened new doors in the cancer genomics arena30, 31. Such ’next-generation’ technologies are now being applied to medulloblastoma and results from the first wave of medulloblastoma genome sequencing studies have now been published32-35. In this Review, we highlight the recent breakthroughs made in the field of medulloblastoma genomics and discuss how findings from large-scale copy number and next-generation sequencing studies will influence ongoing basic research and translate to improved treatment options for patients with medulloblastoma.
Molecular subgroups of medulloblastoma
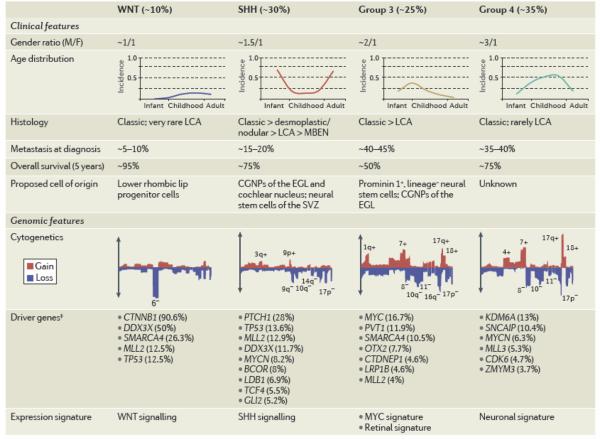
During the past few years, array-based transcriptional profiling of moderate-to-large cohorts of primary medulloblastomas has uncovered the existence of distinct molecular subgroups36-41. Despite the variable number of transcriptional ’clusters’ identified in each of these genomic studies, the current consensus is that medulloblastoma consists of four core subgroups: WNT, SHH, Group 3, and Group 442. Medulloblastoma subgroups exhibit highly disparate cytogenetics, mutational spectra, and gene expression signatures, in addition to divergent clinical phenotypes such as patient demographics, tumour cell histology, and patient outcome (Table 1)19, 43, 44. The establishment of the current four-subgroup structure of medulloblastoma has in a very short time revamped how medulloblastoma is viewed and studied, both in the laboratory and in the design of future clinical trials45, 46. What for many years was considered, and treated, as a single tumour entity47, is now regarded as four very distinct diseases, each requiring distinct therapeutic approaches.
Table 1. Clinical and genomic features of medulloblastoma subgroups.
Subgroup frequency, demographics, clinical features, and cytogenetic profiles were derived from a cohort of 827 subgrouped medulloblastomas described by Northcott et al99. Word cloud summaries of driver genes are based on the relative frequency of mutations or SCNAs affecting depicted genes within each subgroup as described in the recent medulloblastoma sequencing and copy number studies32-34, 99. Abbreviations: LCA, large cell and anaplastic; MBEN, medulloblastoma with extensive nodularity; Dx, diagnosis; OS, overall survival (5-year); CGNPs, cerebellar granule neuron precursors; EGL, external granule cell layer; SVZ, subventricular zone.
|
BCOR, BCL6 co-repressor; CDK6, cyclic-dependent kinase 6; CGNps, cerebellar greanule neuron precursors; CTDNEP1. CTD nuclear envelope phosphatase 1; CTNNB1, β-catenin; EGL, external granule cell layer; GLI2, GLI famnily zinc finger 2; KDM6A, lysine-specific demthylase 6A; LCA, large cell and anaplastic; LDB1, LIM domain binding 1; LRP1B, low density lipoprotein receptor-related protein 1B; MBEN, medulloblastoma with extensive nodularity; MLL, mixed lineage leukaemia; OTX2, orthodenticle homeobox 2; PTCH1, patched 1; SCNA, somatic copy number aberration; SHH, sonic hedgehog; SNCAIP. α-synuclein inteinteracting protein; SPTB, spectrin-β erythrocytic; SVZ, sinventricular zone; TCF4,transcription factor 4; TNXB, tenascin XB.
Subgruop frequency. demographics, clinical features and cytogenetic profiles were derived from a cohort of 827 medulloblastomas distributed into subgroups described by Northcott et al.99
WNT medulloblastoma
WNT subgroup medulloblastomas currently have the best prognosis of any subgroup; >95% of these patients will survive their disease (Table I)3, 5. WNT cases are equally distributed between males and females, despite a male-to-female preponderance of 1.5:1 in medulloblastoma overall. Cases of WNT medulloblastoma are the least common of the four subgroups, accounting for only about one of every ten diagnoses. These tumours typically occur in children over the age of three, exhibit classic histology, and are infrequently metastatic19.
Medulloblastomas harboring somatic mutations of CTNNB1 that promote stabilization and nuclear localization of the β-catenin protein belong to the WNT subgroup37, 48. In addition to harboring nearly ubiquitous CTNNB1 mutations, WNT subgroup medulloblastomas often carry heterozygous TP53 mutations49, 50. WNT tumours are readily identifiable by a WNT gene expression signature51 and the nuclear accumulation of β-catenin is routinely used as a biomarker for WNT pathway activation in both research and clinical settings. Cytogenetically, WNT medulloblastomas exhibit largely balanced genomes19, with the exception of monosomy 6, a hallmark chromosomal aberration found in almost all cases of WNT medulloblastoma that is very rarely seen in other subgroups.
Current evidence from mouse models of medulloblastoma, in combination with data on subgroup specific somatic events from cohorts of human medulloblastoma suggest that mutations are subgroup specific and therefore must be highly matched to their correct cell of origin (i.e. EGL for SHH medulloblastoma) in order for them to promote neoplastic transformation. A mouse model of WNT medulloblastoma (Blbp-Cre;Ctnnb1+/lox(Ex3);Trp53flx/flx) was recently generated in which a conditional stabilized allele of Ctnnb1 is targeted to progenitor cells of the lower rhombic lip (LRP, Table 2)52. Mice expressing the activated Ctnnb1 transgene in the context of Trp53 deletion develop classic medulloblastomas (penetrance of ~15%) after a relatively long latency. Extensive transcriptional characterization of tumours derived from these mice established their association with human WNT medulloblastoma counterparts. These tumours arise from the dorsal brainstem, in contrast to the external granule cell layer (EGL)-derived medulloblastomas arising in Ptch1+/− mice that model the human SHH medulloblastoma subgroup (discussed below). In a recent medulloblastoma genomics study conducted by Robinson et al (discussed in detail below), the authors supplemented the established WNT model by adding in a Pik3ca mutant allele (Pik3caE545K) identified in human WNT medulloblastoma, generating Blbp-Cre;Ctnnb1+/lox(Ex3);Trp53+/flx;Pik3caE545K mice32. These mice developed classic histology WNT medulloblastomas with 100% penetrance by 3 months of age, greatly increasing medulloblastoma incidence and decreasing the latency compared to the original WNT model. Although still relatively new reagents, it is anticipated that these mouse models will serve as an important vehicle for preclinical testing of compounds for future treatment of WNT subgroup patients - a subgroup of patients currently being considered for de-escalation of therapeutic intensity based on their favourable prognosis.
Table 2. Mouse models of medulloblastoma subgroups.
Abbreviations: LCA, large cell and anaplastic; CGNPs, cerebellar granule neuron progenitors; EGL, external granule cell layer; NSCs, neural stem cells.
| Genotype | Type of model | Phenotype | Refs |
|---|---|---|---|
| WNT subgroup | |||
|
Blbp–Cre;Ctnnb1+/lox[Ex3] Trp53flx/flx |
Transgenic: targeted expression and inactivation |
Classic histology medulloblastomas with 15% penetranceafteralong Latency(10–12 months). Tumours arise from LRPs |
52 |
|
Blbp–Cre;Ctnnb1+/lox[Ex3]. Trp53+/flx;Pik3caE545K |
Transgenic: targeted expression and inactivation |
Classic histology tumours with 100% penetrance by 12 weeks |
32 |
| SHH subgroup | |||
| Ptch1 +/− | Constitutive knockout | Sporadic medulioblastomas with a penetrance of 14–20% arising within 15–25 weeks. Tumours originate in CGNPs of the EGL |
60 |
| Ptchr+/−;Trp53+/− | Constitutive knockout | Medulioblastomas with a penetrance of 95–100% by 12 weeks |
61 |
| ND2–Swoal | Transgenic: targeted expression |
Mice hemizygousfor the transgene develop medulloblastoma with a 48% penetrance by 25 weeks. >90% of homozygous Smoal;Smoal mice develop highly metastatic medulloblastoma within 4–S weeks |
62,63 |
| Atoh1-Cre;Ptch1flx/flx | Transgenic: targeted inactivation |
100% of mice develop medulloblastoma within 3–12 weeks. Tumours arise in CGNPs of the ECL |
65 |
| GFAP–Cre;Ptch1flx/flx | Transgenic: targeted inactivation |
100% of mice develop medulloblastoma within 3–4 weeks. Tumours originate in NSCs from the subventricular zone |
65 |
|
Atoh1–SB11; T20nc;Ptch1+/− |
Transgenic: targeted mutagenesis and inactivation |
Highly metastatic medulioblastomas with a penetrance of 97% within 10 weeks |
163 |
| Group 3 subgroup | |||
|
Prom1+Lin− NSCs infected with MycT58A plus DNp53 (MP) retroviruses |
Orthotopic transplantation | 100% penetrant medulloblastomas that have features that are characteristic of the LCA subtype within 5–12 weeks |
75 |
|
Atoh1–GFP; Trp53−/− CGNPs infected with Myc-RFP retroviruses |
Orthotopic transplantation | 100% medulloblastoma incidence with features of the LCA subtype in 4 weeks |
75 |
| Group 4 subgroup? | |||
|
Glt1-tTA;TRE-MycN;Luc (known as GTML mice) |
Transgenic: targeted expression |
Mice deveLop either classic or LCA medulioblastomas by 30 weeks with a penetrance of 75%. Infrequent incidence of leptomeningeal metastases |
79,80 |
Atohl, atonal homologue 1:Blbp. brain lipid binding protein; CGNPs, cerebellar granule neuron precursors; Ctnnb1.β-catenin; EGL external granule cell layer; Ex3.exon 3; Flx. flox; GFAP.glial fibrillary acidic protein; GFR. green fluorescent protein; Glt1, glutamate transporter 1; LCA. large cell and anaplastic; Lin, lineage; LRPs, lower rhombic lip progenitors; Luc, luciferase; ND2, neurogenic differentiation 2; NSCs. neural stem cells; Pik3ca. PI3K catalytic-α polypeptide; Prom1, prominin l; Ptch1. patched 1; RFP. red fluorescent protein: SHH. sonic hedgehog; Smoal. activated mutant of smoothened; tTA. tetracycline transactivator.
SHH medulloblastoma
SHH-driven medulloblastomas represent an intermediate prognosis subgroup, with overall survival rates ranging from ~60-80% (Table 1)19, 39, 40, 53. These tumours exhibit a curious bimodal age distribution, accounting for the majority of both infant and adult medulloblastomas but only a minority of childhood cases. Desmoplastic (or nodular) histology is almost exclusively restricted to SHH medulloblastomas54, whereas classic and large-cell or anaplastic (LCA) cases also occur but are not confined to this subgroup.
Germline mutations affecting either PTCH1 or suppressor of fused homologue (SUFU), which encode negative regulators of the SHH signaling pathway, result in the development of SHH medulloblastoma13, 14, 24, 55-57. Somatic mutations of PTCH1 and occasionally additional pathway components, as well as somatic copy number aberrations (SCNAs) affecting the SHH target genes MYCN and GLI family zinc finger 2 (GLI2), are typical of this subgroup37-40. The SHH medulloblastoma genome contains significantly more regions of chromosomal gain and loss than WNT medulloblastoma, including frequent deletions of chromosomes 9q and 10q as well as other less common aberrations (Table 1)19.
Faithful mouse models recapitulating human SHH medulloblastoma have been available for more than a decade and have long been the workhorse in medulloblastoma basic research58, 59. Ptch1+/− mice60, 61 and transgenic mice overexpressing activated Smoothened (Nd2-Smoa1)62, 63 are amongst a multitude of mouse models mimicking defects in the canonical SHH pathway that develop medulloblastomas with variable frequencies (Table 2). Mouse SHH medulloblastomas can be initiated in cerebellar granule neuron precursors (CGNPs) of the EGL64-66 and those from cochlear nuclei67, or from neural stem cell populations residing in the subventricular zone65.
Antagonists of SHH signaling, primarily those inhibiting the pathway at the level of SMO, are currently an area of active interest in the pharmaceutical industry and in clinical trials for medulloblastoma10, 11. Despite showing a pronounced initial response to treatment with SMO inhibitors, both humans and mice acquired resistance to the treatment and relapsed within a short period, suggesting that such inhibitors may be ineffective when administered as monotherapy68-72. Furthermore, tumours harboring amplifications in key SHH pathway target genes (i.e. GLI1, GLI2, or MYCN)36, 39, 40, 73, might have an inherent primary resistance against these drugs. Nevertheless, young infants harboring germline PTCH1 mutations, such as those observed in Gorlin Syndrome medulloblastoma may be ideally suited for targeted therapy with SMO inhibitors, although concerns of complications with normal development in this age group have been raised74. In the future, appropriate patient selection will be critical in order to maximize the opportunity for effective treatment with SHH antagonists.
Group 3 medulloblastoma
Patients with Group 3 medulloblastoma currently have the worst outcome of the four subgroups (Table 1)19, 39, 40. These tumours are more common in males, restricted to paediatric patients, frequently of the LCA subtype, and nearly half are metastatic at the time of diagnosis19.
Focal, high-level amplification of the MYC proto-oncogene is highly enriched in Group 3 medulloblastoma and almost all cases exhibit aberrant MYC expression39, 40. The Group 3 genome exhibits high levels of genomic instability and often harbors gains of chromosomes 1q, 7, and 17q and deletions of 10q, 11, 16q, and 17p (Table 1)19. Aberrations on chromosome 17 are usually attributed to the presence of an isochromosome 17 – i(17q).
Earlier this year, two independent groups published MYC-driven mouse models of Group 3 medulloblastoma that exhibit similarly aggressive phenotypes as the human counterpart75, 76. Both of these models necessitate the loss of p53 activity to overcome MYC-induced apoptosis (Table 2). Since TP53 is not directly mutated in human Group 349, however, the search for more suitable candidates that cooperate with MYC in these tumours is currently underway. Nonetheless, the current models provide a highly desirable reagent for dissecting the biological basis of this poor outcome subgroup and for testing pharmacological agents that might help to selectively eradicate these tumours.
Group 4 medulloblastoma
The most common subgroup of medulloblastoma is Group 4, accounting for about two of every five cases (Table 1)19. Childhood Group 4 patients have an intermediate prognosis similar to the SHH subgroup but adults with Group 4 medulloblastoma may do significantly worse19, 77. There is a considerable gender bias in Group 4, with male cases being up to three times more common than female cases. These tumours are mainly of classic histology but can be of the LCA phenotype and metastases are found in about one third of patients.
MYCN and cyclin-dependent kinase 6 (CDK6) proto-oncogenes are recurrently amplified in Group 4 medulloblastomas, whereas they very rarely occur in Group 3 medulloblastoma39, 40. i(17q) is found in the majority of these cases and most female Group 4 medulloblastomas lose one copy of the X chromosome, suggesting the existence of one or more TSGs.
There are currently no confirmed preclinical mouse models of Group 478. An elegant transgenic mouse model that over-expresses both Mycn and luciferase using a bidirectional brain-specific promoter develops highly penetrant medulloblastomas, that exhibit properties of either SHH or non-SHH tumours which may depend on the timing of tumour initiation (Table 2)79, 80. It is not yet clear whether the non-SHH tumours arising in these mice are accurate representations of MYCN-driven Group 4 medulloblastoma, but they do nonetheless provide a valuable tool for studying the role of MYCN in the context of medulloblastoma pathogenesis.
Mutational analysis of medulloblastoma
Until recently, only a handful of genes were recognized as recurrently mutated either in the germline or somatically in patients with medulloblastoma. These included the TSGs PTCH1, SUFU, and TP53 and the oncogenes CTNNB1 and SMO81. Exceedingly rare mutations of APC, PMS2, CREB binding protein (CREBBP), and nibrin (NBS1) in individuals with hereditary syndromes that developed medulloblastoma had also been reported20, 24, 82-84. A significant breakthrough in our understanding of the genetic landscape of medulloblastoma came in 2010, when Parsons et al.85 published the first unbiased whole-exome sequencing study of medulloblastoma based on Sanger sequencing technology. Somatic mutations in histone methyltransferases (HMTs), mixed lineage leukaemia 2 (MLL2) and MLL3, were identified in 16% of cases, making them the first new candidates verified as recurrently mutated in medulloblastoma in nearly a decade. The bulk of the variants detected in these two histone modifiers were predicted to be inactivating, implying that they likely function as TSGs. Medulloblastoma subgroup status was unavailable for samples included in this study and thus the subgroup distribution of MLL2 and MLL3 mutations was not reported. Mutations affecting additional chromatin-associated genes SMARCA4, AT rich interactive domain 1A (ARID1A), and lysine-specific demethylase 6B (KDM6B) were also revealed in this study, reinforcing the theme of deregulation of chromatin modifiers (discussed below) as a common mode to medulloblastoma pathogenesis, as also implicated in prior copy number studies73.
Next-generation sequencing of medulloblastoma
This year, three independent next-generation sequencing studies of medulloblastoma were published in a single issue of Nature32-34, collectively screening whole genomes, whole exomes, and prioritized lists of candidate genes across a large cohort of 310 primary medulloblastoma specimens. These data represent the next important step in our understanding of medulloblastoma, in large part because each identified a common set of novel recurrent mutations, providing a firm footing for further research.
The median number of somatic, non-silent mutations single nucleotide variants (a nucleotide substitution resulting in the exchange of one nucleotide for another; SNVs) and indels (an insertion or deletion of nucleotides) per medulloblastoma genome appears to range from 10-12, which is significantly lower than mutation rates reported in solid tumours typical of adulthood, including adult brain tumours86-91. Likewise, a positive correlation between patient age and genome-wide mutation rate was also observed33, 34. Indeed, preliminary analysis of the mutational spectra of adult medulloblastoma suggests that adults harbour significantly more somatic SNVs and indels than subgroup-matched pediatric counterparts (Kool et al, in preparation). These observations might suggest that medulloblastoma, particularly when occurring in very young patients, requires deregulation of only a minimal number of driver genes. An alternative explanation could be that SCNAs or epigenetic deregulation play a more prominent role than point mutations. Indeed, SCNAs are common in non-WNT subgroups of medulloblastoma, as will be discussed below.
From the combined discovery cohorts of the three sequencing studies (n=189), each medulloblastoma subgroup was represented at a frequency comparable to what has been reported in the literature (Figure 1a)19. Within the individual studies, some subgroup bias was evident, with Group 3 samples accounting for 36% of the Broad cases and Group 4 contributing to 54% of the St. Jude cohort – both of which are higher than expected frequencies for a typical medulloblastoma subgroup distribution19. Since only a selection of candidate genes were sequenced in the verification cohorts from the DKFZ (n=2,734 genes in 65 samples) and St. Jude (n=136 genes in 56 samples) studies, the denominators used in the calculation of mutation frequency varied according to gene. Among the combined discovery cohorts that aimed to interrogate every coding exon in the genome, a total of 2,102 unique genes were identified as somatically mutated. Of these, only fifteen were reported as mutated in all three studies (Figure 1b). This limited list of commonly mutated genes includes titin (TTN), mucin 16 (MUC16), and ryanodine receptor 3 (RYR3), all of which are enormous genes (containing >300 exons in the case of TTN) that are routinely reported to be mutated in other sequencing studies89. The functional significance of these mutations is uncertain as they might be mutated by chance alone (i.e. passenger mutations) due to their very large size, with the mutations documented not having substantially contributed to the process of tumorigenesis. Indeed, when applying the MutSig algorithm (https://confluence.broadinstitute.org/display/CGATools/MutSig), which tests whether the observed mutations in a gene are simply a consequence of random background mutation processes by taking into account factors such as gene length, composition, and silent to non-silent mutation ratios, none of these common passenger genes were found to be mutated significantly above chance in the medulloblastoma sequencing studies33, 34. Subtraction of these genes leaves just twelve high-confidence candidates based on their detection in all three studies – suggesting that the list of prominent driver genes affected by somatic SNVs and indels in medulloblastoma is limited.
Figure 1. Meta-analysis of medulloblastoma next-generation sequencing studies.

(a) Pie chart showing the molecular subgroup distribution within the combined discovery cohort of the three medulloblastoma sequencing studies (n=189)32-34. (b) Venn diagram showing the number of mutated genes identified in the three next-generation sequencing studies of medulloblastoma (discovery cohorts, n=189) detailed in the text and the degree of overlap between them. Fifteen genes were commonly mutated in all three studies. (c) Frequency (%) and distribution of top candidate genes subject to recurrent non-synomonous mutation (SNV/indel) in medulloblastoma subgroups. Frequency is expressed as the percentage of affected cases within each subgroup and the denominator was dependent on the total number of cases screened for each individual gene (since not all genes were included in the targeted replication cohorts). Genes highlighted in bold were found to be mutated in each of the three sequencing studies. (d) Oncoprints showing the frequency and sample distribution of the most prevalent somatic mutations (SNVs/indels) and focal SCNAs within the medulloblastoma subgroups as determined in the four genomic studies described in the text32-34, 99.
The most common somatically mutated gene in medulloblastoma remains CTNNB1, sustaining activating SNVs in the third exon of 30 out of 32 (91%) cases of WNT medulloblastoma across the combined cohort from all three papers (Figure 1c, d). One CTNNB1 mutation was found in a Group 3 tumour32, but this was the sole exception and its significance is unknown.
Second to CTNNB1, the DEAD-box RNA helicase DDX3X is the next most frequently mutated gene in medulloblastoma (25/310; 8%; Figure 1c). Half of all WNT medulloblastomas (16/32) harbored mutations in DDX3X and 11% (7/66) of cases of SHH medulloblastoma were likewise targeted (Figure 1d). DDX3X is dynamically linked to multiple cellular processes including chromosome segregation, cell cycle regulation, transcription, and translation92-97. Through mapping of the mutations on to its crystal structure32-34, 98, it appears that they alter DDX3X-RNA binding and are likely to result in altered protein function as opposed to loss-of-function. Moreover, nearly all SNVs reported in DDX3X affect either of its two helicase domains suggesting these mutations impact their functionality (Figure 2). Functional studies investigating the impact of DDX3X mutations either in vitro or in vivo strongly suggest that DDX3X SNVs enhance cellular proliferation by potentiating the transactivation capacity of mutant β-catenin32, 34 and that DDX3X is required to maintain the lineage of LRPs32, the presumed WNT medulloblastoma cells-of-origin52.
Figure 2. Novel mutations and genomic targets in medulloblastoma subgroups.

Schematic representations of the most frequently targeted, novel candidate genes in medulloblastoma. Mutation positions are based on the three next-generation sequencing studies described in the text32-34. Genes are depicted within the subgroup for which they show the greatest enrichment. In Group 3, the zoom box adjacent to MYC depicts the general structure of the recurrent PVT1-MYC fusion genes reported by Northcott et al99. In Group 4, the zoom box adjacent to SNCAIP illustrates the general structure of the SNCAIP tandem duplications described by Northcott et al99. Abbreviations: DEAD, DEAD domain; LIM, LIM domain; ANK, ankyrin repeat; HLH, helix-loop-helix domain; NIF, NLI interacting factor-like phosphatase; TPR, tetratricopeptide repeat; JmjC, Jumonji C domain; InDel, insertion/deletion.
As expected, PTCH1 mutations were restricted to SHH (16/66; 24%; Figure 1c, d). Similarly, TP53 mutations, all of which mapped to the DNA binding domain, were distributed as previously reported49 and absent in all 76 cases of Group 3 medulloblastoma that were profiled. Mutations affecting MLL2 were found in all subgroups (18/310; 6% overall) but were enriched in both WNT and SHH medulloblastomas compared with Groups 3 and 4. Conversely, MLL3 mutations were less common than mutations of MLL2 (n=8/310 vs. 18/310, respectively) and were found only in Groups 3 and 4. SMARCA4, which had been previously implicated in medulloblastoma85, was frequently targeted in both WNT (8/32; 25%) and Group 3 (8/76; 11%) medulloblastomas, making it among the top five most commonly mutated genes in medulloblastoma (Figure 1c, d).
Histone modifiers in medulloblastoma
Deregulation of factors involved in the covalent post-translational modification of histones, in particular, HMTs, histone demethylases (HDMs), histone acetyltransferases (HATs), and histone deacetylases (HDACs), as well as various other chromatin-associated genes, have been repeatedly identified to occur in medulloblastoma32-34, 73, 85. Indeed, of the recurrently mutated genes identified in the next-generation sequencing studies (n=335), 24 are predicted to function in processes related to chromatin based on Gene Ontology analysis (GO term: chromatin modification), affecting 33% (62/189) of medulloblastomas (Figure 3, Table 3). Additional chromatin-associated genes appear to be preferentially altered by SCNA73, 99, although not to the same extent as those sustaining SNVs and indels.
Figure 3. Convergent deregulation of the histone code in medulloblastoma.

Cartoon summarizing a selection of genes recurrently mutated and/or undergoing SCNA in medulloblastoma that are functionally associated with chromatin regulation. Both inactive heterochromatin and active euchromatin and the typical histone marks associated with these chromatin states are illustrated.
Table 3. Chromatin modifiers recurrently mutated in medulloblastoma.
Abbreviations: H3K4, histone 3 lysine 4; H3K9, histone 3 lysine 9; H3K27, histone 3 lysine 27; H3K36, histone 3 lysine 36; HDAC, histone deacetylase; HAT, histone acetyltransferase.
| Gene | Somatic mutation (%} | Subgroup enrichment | Function |
|---|---|---|---|
| MLLZ | 5.8 | WNT and SHH | H3K4 methyltransferase |
| SMARCA4 | 5.8 | WNT and Group 3 | Chromatin remodeller |
| KDM6A | 5.2 | Group 4 | H3K27me2 and H3K27me3 demetbylase |
| MLL3 | 2.6 | Group 3 and Group 4 | H3K4 methyltransferase |
| BCOR | 2.0 | SHH | Transcriptional repressor |
| CHD7 | 1.9 | Group 3 and Group 4 | Chromatin remodeller |
| LDB1 | 1.6 | SHH | Transcriptional regulator |
| CREBBP | 1.3 | WNT and SHH | HAT |
| ZMYM3 | 1.3 | Group 4 | Associated with HOAC complex |
| ARID1B | 1.2 | WNT and SHH | Chromatin remodeller |
| EYA4 | 1.2 | None | Histone phosphatase |
| EP300 | 1.0 | SHH | HAT |
| TRRAP | 1.0 | None | Associated with HAT complex |
| KDM4C | 1.0 | Group 3 and Group 4 | H3K9me3 and H3K3Gme3 demethylsse |
| CTR9 | <1.0 | NA | Associated with RNA Pol II activity |
| TLK2 | <1.0 | NA | Modulates chromatin assembly |
| SETD2 | <1.0 | NA | H3K36 methyltransferase |
| TAF1 | <1.0 | NA | Component of TFIID-RNA Pol II complex |
| KDM1A | <1.0 | NA | H3K4mel,H3K4me2 and H3K9 demethylase |
| KDM5A | <1.0 | NA | H3K4me2 and H3K4me3 demethylase |
| BRCA2 | <1.0 | NA | Regulates DNA double-strand break repair |
| CTCF | <1.0 | NA | Chromatin binding and remodelling factor |
| REST | <1.0 | NA | Recruits histone demethylases and HDACs |
| HDAC9 | <1.0 | NA | Histone lysine deacetylase |
ARID1B, AT-rich interactive domain 1B; BCOR, BCL6 co-repressor; CHD7. chromodomain helicase DNA binding protein 7; CREBBP. CREB binding protein; CTCF. CCCTC-binding factor; EP300, E1A binding protein p300; EYA4. eyes absent homologue 4; H3K4, histone H3 lysine 4; H3K9, histone H3 lysine 9; H3K27. histone H3 lysine 27; H3K36, histone H3 lysine 36; HAT, histone acetyltransferase; HDAC. histone deacetylase; KDM, lysine-specific demethylase: LDB1, LIM domain binding 1; me1. monomethylated; me2, dimethylated; me3. trimethylated; MLL mixed lineage leukaemia: NA, not applicable; Pol. polymerase; REST. RE1-silencing transcription factor; SETD2. SET domain containing 2; SHH, sonic hedgehog; TFIID, transcription factor IID; TLK2. tousled-like kinase 2; TRRAP. transformation/transcription domain-associated protein.
In addition to the previously described mutations affecting MLL2, MLL3, and SMARCA4, several new candidates sharing similar functionality have emerged. KDM6A (also known as UTX), a histone H3 lys27 demethylase (i.e H3K27me2 or H3K27me3)100, 101, is the most frequently mutated gene in Group 4 medulloblastoma (13/108; 12%; Figure 1c, d). KDM6A belongs to the Jumonji C family of HDMs that also includes KDM6B102, also reported as mutated in medulloblastoma85. Ten of sixteen mutations affecting KDM6A were nonsense mutations implicating it as a novel medulloblastoma TSG (Figure 2). Likewise, the KDM6A locus on chromosome Xp11.3 was reported as homozygously deleted in multiple Group 4 cases profiled in the recent Medulloblastoma Advanced Genomics International Consortium (MAGIC) copy number study (discussed below)99, further substantiating its potential significance in Group 4 biology.
Additional chromatin remodeling genes, ZMYM3 and chromodomain helicase DNA binding protein 7 (CHD7)103-106, are also recurrently mutated in Group 4 medulloblastoma (Figure 1c). Robinson et al.32 proposed an elegant connection between KDM6A, ZMYM3, and CHD7 mutations and enhancer of zeste homologue 2 (EZH2) activity. EZH2 is an H3K27 methyltransferase that functions to maintain the undifferentiated state of stem cells through repression of lineage-specific gene expression107, 108, essentially opposing the H3K27 demethylase activity of KDM6A, which promotes differentiation. EZH2 is selectively over-expressed in Groups 3 and 4 medulloblastoma, particularly those cases exhibiting chromosome 7 gain32, 109, and EZH2 inhibition has been shown to suppress medulloblastoma cell growth and promote apoptosis109. Robinson et al. demonstrated a mutually exclusive correlation between the copy number-driven expression of EZH2 and inactivating mutations affecting KDM6A, CHD7 and/or ZMYM3 as alternative yet complementary mechanisms of maintaining constitutive H3K27me3 in Groups 3 and 4 medulloblastoma. Although these observations provide an attractive model for the disruption of physiological H3K27me3 in these subgroups, functional experiments will be required to validate this relationship.
A number of genes associated with chromatin modification also appear to be specifically mutated at relatively modest frequencies in the SHH medulloblastoma subgroup (Figure 1c, d). Mutations targeting components of the nuclear receptor co-repressor (N-CoR) complex110, BCL6 corepressor (BCOR) and LIM domain binding 1 (LDB1), were mutually exclusive in SHH medulloblastoma and collectively accounted for 14% (8/58) of cases (Figure 1d). All BCOR mutations were truncating, as were 2 of 4 mutations affecting LDB1, suggesting that these genes undergo loss-of-function in SHH medulloblastoma (Figure 2). The N-CoR complex is associated with HDAC activity and is presumed to mediate transcriptional repression by influencing the deacetylation and condensation of chromatin110. Recurrent mutations affecting this complex in SHH medulloblastoma, taken together with the apparent deregulation of the H3K27 methylation state described in Groups 3 and 4, strengthens the argument that disruption of the histone code appears to occur across subgroups of medulloblastoma and continues to evolve as one of the most prominent themes underlying medulloblastoma pathogenesis (Figure 3).
Correlation between SNVs and SCNAs
Efforts to integrate different sources of genomic data in order to extract biologically meaningful information remain a centrally important challenge in bioinformatics111 and relatively few studies have accomplished such a feat on a global scale. But there are examples of genes that are targeted by multiple mechanisms (SNVs, indels, SCNAs, etc.). For instance, PTCH1 on chromosome 9q22.32 is both the most frequently mutated gene and the most frequent target of focal deletion in SHH medulloblastomas (Figure 1c, d)99. Additionally, BCOR, DDX3X, and KDM6A occur in a region on chromosome X (Xp11.4-p11.3) that is the target of both recurrent focal and broad (i.e. whole-chromosome and chromosome arm) deletions in medulloblastoma99.
A novel candidate gene, CTD nuclear envelope phosphatase 1 (CTDNEP1; also known as DULLARD), was identified as a recurrent target of mutation in Groups 3 and 4 medulloblastomas (four cases from these subgroups; two cases without subgroup information available) and all but one of these resulted in a premature stop codon (Figure 2). CTDNEP1 encodes a nuclear envelope serine/threonine phosphatase that is thought to function as an antagonist of bone morphogenetic protein (BMP) signaling that regulates neural induction in Xenopus laevis112. In mammals, it is involved in lipin pathway activation, which is responsible for the formation of diacylglycerol113, 114. CTDNEP1 occurs on chromosome 17p13.1, a historical hotspot of deletion and loss-of-heterozygosity (LOH) in medulloblastoma115-117, especially in Groups 3 and 4. Indeed, the majority (5 of 6) of CTDNEP1 mutations were homozygous as a result of corresponding 17p deletion in the same tumours. Although CTDNEP1 is unlikely to be the sole TSG that is important in 17p LOH in medulloblastoma, this observation convincingly qualifies CTDNEP1 as a candidate TSG, and as such warrants further functional characterization.
Inspection of key driver mutations identified in the sequencing studies and their respective sample distribution within the individual subgroups leads to some intriguing observations. Specifically, 100% of WNT medulloblastomas in the combined discovery cohorts (n=18/18) have mutations in just a handful of genes. This suggests that WNT medulloblastoma is the most homogenous of the four subgroups and may represent the best candidate for future targeted therapies, since most patients will share similar mutational profiles. Likewise, the majority (28/42; 67%) SHH medulloblastomas are associated with recurrent somatic mutations – this not considering the potential contribution of common germline mutations in known TSGs: PTCH1, SUFU and TP53. By contrast, only 31% (16/52) of Group 3 and 20% (14/69) of Group 4 medulloblastomas are associated with the more common SNVs and indels reported in these subgroups (Figure 4). Overlaying gains and losses of significantly altered chromosomes in Groups 3 and 4 medulloblastoma reveals that cases devoid of SNVs and indels exhibit more pronounced chromosomal instability (CIN) – that is, broad SCNAs – than those harboring mutations (Figure 4). Collectively, these observations suggest that up to two-thirds of all Group 3 and 4 medulloblastomas are more likely to be driven by SCNAs (or alternative mechanisms) than SNVs and indels. Although this requires validation, the large proportion of Groups 3 and 4 medulloblastomas that seemingly lack any obvious individual driver mutation(s) is an unexpected finding, warranting further investigation to determine what is driving tumorigenesis in this large subset of patients. Likewise, it will be important to establish whether the respective genotypes of these SNV-impoverished patients confer a better or worse outcome.
Figure 4. Correlation of cytogenetic alterations and somatic mutations within Group 3 and Group 4 medulloblastoma.

Distribution of prevalent broad SCNAs and recurrent mutations (SNVs/indels) in Group 3 and 4 medulloblastomas. The majority of both Group 3 and Group 4 medulloblastomas are devoid of somatic mutations in any of the most recurrently mutated genes within these subgroups.
Structural variation in medulloblastoma
Although considerable attention has been devoted to the newly identified, recurrent somatic SNVs and indels in medulloblastoma, an equally compelling case can be made for novel findings attributable to structural variation (SV). In Groups 3 & 4, focal SCNAs appear to account for as many cases as do SNVs, underscoring the importance of considering all possible mechanisms of mutation when tallying potential driver events. Additional forms of SV that affect the integrity of the genome, including chromothripsis and tetraploidy have both been described to occur in specific medulloblastoma subgroups (see below)33, 35, 99 and provide new explanations for some of the distinct genomic architecture previously underappreciated in this genome.
Somatic copy number aberrations (SCNAs)
Array-based genome-wide copy number analyses of medulloblastoma have been ongoing for the better part of a decade.81 The majority of studies have identified prevalent cytogenetic anomalies in medulloblastoma, including i(17q) and isolated chromosome 17 alterations, monosomy 6, deletion of chromosomes 9q, 10q, 16q, and gain of chromosomes 1q, 7, and 18 – many of which are now recognized as enriched in a particular subgroup(s)19. Focal, oncogenic amplifications of MYCN, MYC, GLI2, CDK6, and orthodenticle homeobox 2 (OTX2) have been the most widely noted39, 40, 73, 118-121. Aside from these characteristic aberrations, most copy number studies of medulloblastoma have investigated an insufficient number of samples to adequately account for the inherent heterogeneity of medulloblastoma, especially according to molecular subgroup.
However, a large international study led by MAGIC has now described SCNAs across an unprecedented collection of 1,087 primary medulloblastoma samples99. WNT medulloblastomas (n=76) were confirmed to exhibit virtually no significant regions of focal SCNA. By contrast, SHH medulloblasomas are characterized by frequent focal, high amplitude SCNAs (i.e. high-level amplifications and homozygous deletions). In addition to SCNAs targeting prominent SHH pathway genes, a multitude of new genes were found to be recurrently affected by SCNA in SHH medulloblastomas, especially those involved in either p53 or receptor tyrosine kinase (RTK)-PI3K signaling99. High-level amplification of protein phosphatase 1D (PPM1D) on chromosome 17q23.2, a gene previously reported to be amplified in medulloblastoma120, was found to occur exclusively in SHH medulloblastomas. PPM1D encodes WIP1, a p53-induced phosphatase that inhibits wild type p53 activity122. WIP1 is aberrantly expressed in medulloblastomas123 and promotes the development of SHH medulloblastoma in mice124. Paradoxically, broad gains of chromosome 17q are extremely rare in cases of SHH medulloblastoma but are common to Groups 3 and 4, suggesting that PPM1D amplification is specific to SHH and distinct from the target(s) of 17q gain in other subgroups.
Genes associated with RTK-PI3K signaling were also selectively altered in SHH medulloblastoma, including amplifications of insulin-like growth factor 1 receptor (IGF1R), insulin receptor substrate 2 (IRS2), PIK3C2G, PIK3C2B, and yes-associated protein 1 (YAP1) as well as focal deletions of PTEN on chromosome 10q23.31. PTEN was the most frequent target of homozygous deletion in the MAGIC cohort, occurred exclusively in pediatric SHH cases, and is a prominent candidate target for the characteristic chromosome 10q deletions observed in SHH. The apparently specific deregulation of PI3K signaling in SHH medulloblastoma is poignant because combination therapy consisting of both SHH (i.e. SMO) and PI3K pathway inhibitors has shown preliminary efficacy in preclinical SHH medulloblastoma mouse models, delaying, preventing, or overcoming the resistance encountered in response to treatment with SMO inhibitors alone70, 71.
Despite some clear underlying similarities, Group 3 and Group 4 medulloblastomas exhibit distinct patterns of broad and focal SCNAs that permit their discrimination19. Group 3 medulloblastomas more commonly sustain chromosome 1q gains and deletions on 5q, whereas chromosome 4 gains and deletion of X (females) are more frequently seen in Group 438-40. With respect to driver genes altered by SCNAs, MYC amplifications predominate in Group 3 (30/178; 17% of Group 3 cases in the MAGIC study; Figure 1d) 99. Fusion transcripts involving MYC and PVT1 (i.e. PVT1-MYC) have also been identified in at least 60% of MYC-amplified Group 3 medulloblastomas (Figure 2)99. PVT1 is a non-coding gene mapping adjacent to MYC on chr8q24.21 that also encodes multiple miRNAs (miR-1204-1207); some reports suggest that these miRNAs enhance the oncogenic properties of MYC125-127. PVT1-MYC fusions are the first recurrent gene fusions reported in medulloblastoma, adding further complexity to the role of MYC in driving Group 3 medulloblastoma tumorigenesis.
Tandem duplication
A novel, and somewhat subtle structural aberration that became apparent in the Group 4 medulloblastoma samples (n=317) in the MAGIC cohort concerns focal, single copy gain of α-synuclein interacting protein (SNCAIP) on chr5q23.299. SNCAIP encodes synphilin 1, a binding partner of α-synuclein that constitutes the major component of the characteristic protein aggregates (i.e. Lewy bodies) that occur in Parkinson’s disease128-130. SNCAIP gains were confined to Group 4 medulloblastoma and found in ~25% of Group 4α, a subtype of Group 4 medulloblastoma that is characterized by a mostly balanced genome39, 99. SNCAIP gains are the result of a highly stereotypical tandem duplication juxtaposing a slightly truncated SNCAIP gene adjacent to the germline allele (Figure 2). Results from multiple published expression datasets suggest that SNCAIP is a bona fide Group 4 signature gene that may represent a lineage marker for the yet to be identified Group 4 cell-of-origin99. Recurrent tandem duplication of SNCAIP in Group 4 places this candidate among the relatively few genes altered by this mechanism in cancer, including the highly prevalent BRAF duplications observed in another paediatric brain tumour, pilocytic astrocytoma131, 132.
Chromothripsis
Individuals with Li-Fraumeni syndrome (LFS) harbor germline TP53 mutations133, 134 and are predisposed to the development of a variety of different cancers, including SHH medulloblastoma12. To investigate the somatic mutations cooperating with germline inactivation of TP53 in medulloblastoma, Rausch et al.35 performed whole genome sequencing (WGS) on a female patient with LFS who had developed SHH medulloblastoma. Strikingly, the genome of this case was characterized by a very complex series of somatic rearrangements, which included fusions of multiple highly-amplified regions clustered on individual chromosomes (intrachromosomal) as well as those from different chromosomes (interchromosomal) that were organized as double-minute chromosomes. The pattern of rearrangements observed in this index case was inferred as prototypical chromothripsis as originally defined by Stephens et al.135. Further genomic analyses verified an intricate relationship between TP53 mutation and chromothripsis in SHH medulloblastomas35. Moreover, multiple oncogenes relevant to the SHH subgroup, including MYCN, GLI2, BOC, and IGF1R, were shown in this study to be selectively amplified in the context of chromothripsis, providing a mechanism for their deregulation in SHH. Demographically, LFS-SHH medulloblastomas show a clear enrichment in childhood and early adolescence (i.e. ~8-14 years), comprising a subtype of SHH medulloblastoma that is both genetically and clinically distinct from the more common infant and adult subtypes33.
Amplification patterns associated with chromothripsis were also reported in the MAGIC study99. Interestingly, a considerable number of MYC-amplified, PVT1-MYC fusion-positive cases of Group 3 medulloblastoma were inferred by single nucleotide polymorphism (SNP) array and verified by WGS as probable chromothripsis candidates. As TP53 mutations are virtually absent in Group 3 medulloblastomas, alternative, p53-independent mechanisms could be responsible for the chromothripsis observed in this subgroup.
Since chromothripsis seems to be tightly linked to specific patient subsets, it may prove beneficial to identify these patients early in the course of treatment such that they can be stratified accordingly and their therapeutic regimens adjusted. Indeed, in LFS patients with SHH medulloblastomas, it may be advantageous to avoid the administration of excessive DNA damaging agents (such as alkylating drugs), and radiotherapy, given that their underlying genotypes confer a high risk of secondary malignancies.
Tetraploidy
Aneuploidy has been recognized as a hallmark feature of cancer cells for more than a century136. Similarly, polyploidy, ranging from hypodiploid cells consisting of considerably fewer than 46 chromosomes, up to hypertetraploid cells that can harbour up to 200 chromosomes, has long been observed in tumour cell karyotypes137, 138. Early karyotyping studies of primary medulloblastoma samples and patient-derived medulloblastoma cell lines found that aneuploidy and polyploidy, including tetraploidy, commonly occurred139-144. Jones et al identified an unexpectedly high number of tetraploid medulloblastomas33 through inspection of mutant allele frequencies, with a sizable proportion observed at 25% as opposed to the expected 50% allele frequency for clonal heterozygous mutations. Tetraploidy occurred mostly in Group 3 and Group 4 medulloblastomas (~40-50%). Of particular interest, all of the tetraploid SHH medulloblastomas exhibited TP53 mutations and chromothripsis. Although the biological and clinical significance of tetraploidy in medulloblastoma requires further investigation, a number of recent studies have suggested that tetraploidization is associated with CIN and can drive tumorigenesis in mice145, 146. Tetraploid cells are also known to be more resistant to chemo- and radiotherapy, which might partially account for the poor response of these patient subgroups, particularly Group 3, to standard therapies147. Thus, targeting the maintenance of a tetraploid state through successive cell divisions using novel classes of drugs, such as mitotic checkpoint kinase or kinesin inhibitors, may serve as an attractive therapeutic option for a considerable subset of medulloblastomas148, 149.
Medulloblastoma in the ’post-genomics’ era
Medulloblastoma predisposition
To date, most if not all next-generation genomic approaches to studying medulloblastoma have focused exclusively on alterations present in the tumour that were acquired somatically during tumourigenesis. As a result, the germline of individuals who go on to develop medulloblastoma is largely unexplored, leaving a lot of open questions regarding the potential contribution of germline variants to medulloblastoma predisposition and development. Is there more to the medulloblastoma germline than just mutations in well known hereditary cancer genes, PTCH1, SUFU, and TP53? Historical estimates are that <5% of medulloblastomas can be attributed to underlying predisposition150, 151. Could this be a gross underestimate of the true frequency of causative germline mutations in these patients? Concerted efforts focused on answering these questions will necessitate the next-generation sequencing of large numbers of germline samples obtained from medulloblastoma patients and normal healthy controls, followed by cataloguing of pathogenic variants present in affected individuals. Although such a study will undoubtedly pose significant challenges, particularly when attempting to discriminate pathogenic variation from so-called ’private’ SNPs, it is anticipated that such a comprehensive investigation will yield novel candidates that predispose to medulloblastoma and potentially provide an explanation for some of the cases that currently cannot be justified by somatic alterations.
Beyond the coding genome
Given that many of the ongoing sequencing projects are based on WGS, the ability to look outside the roughly 2% of the genome that codes for proteins and functional RNAs is now feasible. Little investigation thus far has been focused on the potential consequence(s) of somatic intragenic or intergenic mutations or SCNAs. Although a daunting task when considering the large number of SNPs that exist in an individual152, 153, recurrent mutations that affect promoters, enhancers, and other regulatory elements may have profound effects on neighbouring genes and will undoubtedly be a worthwhile area of further investigation. In medulloblastoma, it will be interesting to identify non-coding DNA mutations that are enriched or restricted to a particular subgroup and subsequently link these alterations with driver genes.
Integrative genomics
Now that detailed maps of the most prominent SCNAs, SNVs and indels that contribute to medulloblastoma have been outlined, epigenetic alterations, including the DNA methylome and genome-wide maps of histone modifications, will become a priority154, 155. Moreover, the capacity to identify recurrent gene fusions, alternative splicing events, and previously unreported gene products are now possible with RNA sequencing (RNASeq) technology156, and large-scale RNASeq projects of medulloblastoma are currently underway.
Global integration of medulloblastoma genomic, epigenomic, and transcriptional data is likely to result in a much greater depth of knowledge. A seminal breast cancer study consisting of nearly 2,000 biopsies recently combined SCNA and gene expression data generated from these samples, ultimately revealing novel subgroups of the disease with distinct clinical outcomes157. Likewise, a recent study of colorectal cancer systematically analyzed exome sequence, SCNAs, promoter methylation, and the transcriptome (mRNA and miRNA) in a large series of 276 cases, unveiling distinct genomic patterns in hypermutated versus non-hypermutated disease90. Such integrative approaches have not yet been applied to medulloblastoma but are undoubtedly on the horizon. The simple comparison of cytogenetic events with somatic SNVs and indels (as in this Review) suggests that future, integrative efforts will yield further insight into medulloblastoma, especially into the clinically-challenging Group 3 and Group 4 medulloblatomas, the pathogenesis of which cannot be entirely explained by the SCNA and mutational data currently available.
Remaining challenges in medulloblastoma genomics
The next challenge faced by the medulloblastoma community will be to discriminate true driver events from passengers that provide no selective advantage to the tumours158, 159. The functional significance of high priority candidates such as DDX3X has already been assessed, but individual verification of all genes undergoing recurrent focal SCNA or mutation in medulloblastoma is unrealistic. Methodical candidate evaluation will require broader and more sophisticated approaches such as the use of in vivo RNAi160, 161 and modern mutagenesis screens such as the sleeping beauty (SB) transposon system162, the latter having already demonstrated utility in medulloblastoma studies163. Studies comparing the genomics of primary tumours with their matched metastases or relapses should also be undertaken. Wu et al.163 identified dramatic differences in the genes targeted in primary medulloblastomas compared with matched metastases in mice; a trend that was supported in paired primary and metastatic human medulloblastoma biopsies. Genomic studies further assessing the similarity of primary medulloblastomas with their matched metastases or relapses will be critical to elucidate whether the mutations required for the primary tumour are maintained in resistant disease and likewise, to discover the alterations that are prevalent in metastases and relapses but are absent or subclonal in the primary tumour. This information is particularly important because metastases and relapsed disease account for most of the morbidity associated with medulloblastoma164-166.
Most if not all neuro-oncologists and neuro-psychologists would attest that current treatment options for medulloblastoma remain inadequate6, 167. The search for new therapeutic options in the form of small molecules targeting the pathways and processes recurrently altered in medulloblastoma subgroups, or the specific cell types responsible for tumour maintenance, will clearly become the focus of many labs. High-throughput drug screens for agents with activity against medulloblastoma are now in progress. In order to effectively test large libraries of compounds for the treatment of medulloblastoma, not only will representative preclinical models of each medulloblastoma subgroup be required, but also multiple different models of each subgroup mimicking the prevalent mutations and SCNAs observed in the human disease will be necessary. Once established, these transgenic mice, xenografts, primary cells, and immortalized cell lines will be challenged with literally thousands of small molecules, ideally those already FDA approved, in order to pinpoint new therapies that are less toxic and more effective for specific patient subgroups or even individual patients. Finally, early testing of targeted therapy for medulloblastoma suggests that combination therapy will be required in order to cure the disease and prevent acquired resistance10. This implies that not only will thousands of compounds need to be tested, but also seemingly infinite combinations of small molecules may be necessary in order to uncover the best ’recipe’ for individualized disease eradication. Focused screens intended to target specific genes and gene families, such as agents that inhibit the activity of chromatin modifiers, or specific karyotypes such as those selective for tetraploid cells, are also equally likely given the findings from the recent genomic studies.
Conclusions
An immense amount of genomic data has identified multiple candidate genes that contribute to the pathogenesis of different subgroups of medulloblastoma. The RNA helicase DDX3X, chromatin regulators KDM6A and ZMYM3, N-CoR complex genes BCOR and LDB1, and the Parkinson’s disease gene SNCAIP are amongst a host of novel, somatically targeted genes that show clear enrichment in a particular subgroup. Other candidates, including MLL2 and SMARCA4, sustain mutations in multiple subgroups suggesting they may have a broader role in medulloblastoma biology. Indeed, the next phase of research will be aimed at functional validation, and will lead to an improved understanding of how mutations and SCNAs contribute to the initiation, maintenance, and progression of the disease. Likewise, tetraploidy and chromothripsis provide new insight into the broader mechanisms of SV in medulloblastoma and raise the possibility of therapeutically targeting these processes, as opposed to targeting a single gene or pathway. Finally, the interplay between mutations and SV, as well as the importance of the medulloblastoma epigenome and the complexity of the transcriptome, will all be better understood through integrative genomic studies. As can be concluded from the studies presented in this Review, the first phase of next-generation ’medulloblastomics’ has reached a milestone, but this is likely just the end of the beginning.
Box 1. Cellular origins of medulloblastoma subgroups.
Identification and characterization of the normal cells that undergo neoplastic transformation and give rise to cancer is a topic of keen interest to the medical research community. In parallel with deciphering the repertoire of somatic genomic alterations present in a given cancer, assigning an identity to the cell(s)-of-origin is an essential step in gaining a more comprehensive understanding of tumorigenesis168. By knowing which cells are susceptible to transformation, experimental approaches intended to functionally characterize candidate genes can be undertaken in the correct cellular context, and strategies for specifically targeting these cells-of-origin can be designed and tested.
Considerable insight into the cellular origins of medulloblastoma has been made during the past few years, partially owing to the recent genomic characterization of medulloblastoma subgroups. Elegant mapping of transcriptional profiles from the human medulloblastoma subgroups to transcriptional data generated from normal cells of the developing mouse cerebellum or data from genetically engineered mouse medulloblastomas has implicated cellular origins for three out of the four subgroups. WNT medulloblastomas arise in lower rhombic lip progenitors (LRPs) of the dorsal brainstem and can be initiated in mice by transgenic expression of an activated mutant Ctnnb1 allele compounded with Trp53 deletion52. SHH medulloblastomas have long been known to arise in CGNPs of the cerebellar EGL64, 66, 169, 170 and more recently CGNPs derived from cochlear nuclei of the brainstem67. Neural stem cells (NSCs) residing in the subventricular zone (SVZ) are also capable of giving rise to SHH-dependent medulloblastoma65. Nearly all mouse models of SHH medulloblastoma rely on perturbations of the canonical SHH pathway in these respective cell types, with inactivation of Ptch1 and activation of SMO (i.e. SMOA1) being the most commonly used initiating events. When combined with Trp53 inactivation, MYC-driven Group 3 medulloblastomas can be generated in at least three possible cell types: 1) Atoh1-positive CGNPs from the EGL76; 2) Atoh1-negative CGNPs from the EGL76; or 3) Prominin1-positive, lineage-negative neural stem cells75. The specific cellular origin(s) of Group 4 medulloblastoma have thus far remained elusive. However, targeted expression of MYCN, a bona fide SHH and Group 4 driver gene, to different neural stem cell populations from the developing mouse brain generates either SHH-dependent or SHH-independent medulloblastomas suggesting the latter may represent a faithful model of Group 479, 80
At a glance.
Medulloblastoma is the most common malignant pediatric brain tumour and is a leading cause of cancer-related morbidity and mortality in children.
Integrative genomic studies have recently identified at least four distinct molecular subgroups of medulloblastoma – WNT, SHH, Group 3 and Group 4 – exhibiting highly discriminate transcriptional, cytogenetic, and mutational spectra in addition to divergent patient demographics and clinical behavior.
Recent next-generation sequencing of medulloblastoma samples and their matched germline material has led to the identification of a multitude of previously unknown candidate genes somatically mutated in this cancer, including many that are mutated in a subgroup-specific manner.
Different mechanisms of structural variation including somatic copy number aberrations, chromothripsis, and tetraploidy contribute to a considerable proportion of medulloblastomas.
Despite the extensive amount of copy number and sequence data that has become available for large cohorts of medulloblastoma, the majority of Group 3 and Group 4 tumours cannot be attributed to a specific driver mutation(s), suggesting that additional mechanisms, such as epigenetic deregulation, may also play a prominent role.
Research on medulloblastoma during the next several years will be focused on the functional validation of candidate genes and molecular processes reported to be deregulated in the recent human genomic studies. The generation of faithful models recapitulating these mutational events will become a priority, and such reagents will serve as a valuable resource for the development of rational, molecularly targeted therapies.
Acknowledgements
This work was principally supported by the PedBrain Tumor Project contributing to the International Cancer Genome Consortium (ICGC), funded by German Cancer Aid (109252) and the German Federal Ministry of Education and Research (BMBF, NGFNplus #01GS0883). We also acknowledge the Pediatric Brain Tumor Foundation (PBTFUS), the National Institutes of Health (R01CA148699 and R01CA159859) and the Dutch Cancer Foundations KWF (2010-4713) and KIKA. PAN is a Roman Herzog Post-Doctoral Fellow supported by the Hertie Foundation and the DKFZ. We are deeply indebted to Christian Smith of Creative Science Studios (http://www.creativesciencestudios.com/) for assistance with artwork. We also acknowledge Charles Imbusch (DKFZ) for bioinformatic support pertaining to the meta-analysis of next-generation sequencing studies. Lastly, special acknowledgments are warranted for Trevor J. Pugh (Broad Institute), Natalie Jäger (DKFZ), and David J. H. Shih (SickKids) who played lead bioinformatic roles in the respective medulloblastoma genomic studies highlighted in this Review.
Glossary
- Leptomeningeal dissemination
Metastasis of tumour cells to the leptomeninges (arachnoid mater and pia mater) that wrap the brain and spinal cord. Metastatic medulloblastoma cells typically disseminate to the leptomeninges via the cerebrospinal fluid (CSF).
- Gorlin Syndrome (nevoid basal cell carcinoma syndrome; NBCCS)
An autosomal dominant condition in which affected individuals harbor germline mutations in the PTCH1 tumour suppressor gene and whose cells therefore display aberrant activation of the SHH signaling pathway. Individuals with Gorlin’s exhibit a variety of developmental defects including jaw cysts, palmar pits, and skeletal abnormalities and are predisposed to the development of extensive basal cell carcinomas and medulloblastoma, among other cancers.
- Turcot Syndrome
Typically an autosomal dominant condition characterized by multiple adenomatous colon polyps and predisposition to colorectal cancer and brain tumours. The genetic basis is linked to mutations in APC (type I) or in the mismatch repair genes MLH1 or PMS2, with APC mutations predisposing to medulloblastoma and mismatch repair mutations to glioblastoma multiforme, respectively.
- Next-generation sequencing (NGS)
Sequencing technologies emerging since 2005 that have massively increased the output of the nucleic acid sequencing process. They produce millions to hundreds-of-millions of typically short sequence reads (50–400bp) from amplified DNA clones.
- Lower rhombic lip (LRL)
A division of the rhombic lip, a specialized germinal matrix situated at the interface between the neural tube and the roofplate of the fourth ventricle of the developing cerebellum. Precursor cells from the LRL are presumed cells-of-origin for WNT medulloblastoma.
- Dorsal brainstem
The hindmost parts of the structure of the brainstem.
- External granule cell layer (EGL)
A germinal zone comprised of CGNPs that line the surface of the developing cerebellum.
- Desmoplastic (or nodular) histology
Histological variant of medulloblastoma characterized by the presence of a varying number of nodules that consist of differentiated neurocytic cells and internodular desmoplasia (fibrous or connective tissue), best demonstrated by reticulin staining. Mostly restricted to the SHH medulloblastoma subgroup.
- Classic histology
The most common histological subtype of medulloblastoma, displaying prototypical sheets of repetitive small cells with a high nuclear:cytoplasmic ratio and round nuclei. Found in all medulloblastoma subgroups.
- Large-cell or anaplastic (LCA) histology
An admixture of two usually co-occuring histological variants of medulloblastoma associated with a more aggressive clinical phenotype and the Group 3 subgroup. Collectively, LCA medulloblastomas appear as groups of large cells with round nuclei (i.e. large cell) and cells exhibiting marked cytological pleomorphisms (anaplasia), especially nuclear pleomorphism.
- Cerebellar granule neuron precursors (CGNPs)
As a precursor to the most abundant neuron in the brain, CGNPs arise in the hindbrain during late embryonic development and migrate postnatally to the external granule cell layer of the cerebellum. Here they undergo a period of massive proliferation before eventually migrating inward to the internal granule layer and terminally differentiating.
- Cochlear nuclei
Structures in the brainstem which derive from the auditory lower rhombic lip, receiving inputs on sound from the cochlear nerve. CGNPs residing in the cochlear nuclei have been implicated as a possible cell-of-origin for SHH medulloblastoma.
- Isochromosome
An abnormal chromosome produced during mitosis or meiosis that is characterized by the presence of two genetically and morphologically identical chromosome arms fused at the centromere. Strictly, the common alteration of chromosome 17 in medulloblastoma is more often an isodicentric (17q), as the breakpoint is typically in the p-arm proximal to the centromere rather than at the centromere itself.
- Single nucleotide variants (SNVs)
A nucleotide substitution resulting in the exchange of one nucleotide for another. In cancer, SNVs refer to single nucleotide changes present in the tumour DNA which are not present in the patient’s normal genome.
- Indels
A mutation involving the insertion or deletion of nucleotides, which often lead to a loss-of-function by causing a shift in the reading frame of a gene.
- Chromothripsis
A process of erroneous DNA repair following a single catastrophic breakage event that results in massive genomic rearrangement
- Tetraploidy
A whole-genome duplication event such that each chromosome is present in four rather than two copies.
- Lewy bodies
Abnormal protein aggregates found in the brain of individuals with Parkinson’s disease, Lewy Body Dementia, and other disorders.
- Tandem duplication
A structural variant caused by a duplication event that results in two genomic segments of at least 1kb in size that share >90% sequence identity positioned in a contiguous manner within the genome.
- Double-minute chromosomes
Typically acentric, extra-chromosomally amplified chromatin, usually containing a particular chromosomal segment or gene. Double minutes are common in cancer and often result in oncogene amplification through replication and asymmetric distribution after cell division.
- Single nucleotide polymorphism (SNP)
A single nucleotide DNA sequence variant that differs between individuals or paired chromosomes from the same individual. In contrast to SNVs, these are present in the normal genome of an individual.
- Aneuploidy
A cellular state characterized by an abnormal number of chromosomes.
Biographies
Paul A Northcott, PhD Paul Northcott earned his PhD at the University of Toronto while working under the mentorship of Michael Taylor, MD, PhD at the Arthur & Sonia Labatt Brain Tumour Research Centre affiliated with the Hospital for Sick Children (SickKids). Paul continued at SickKids as a Research Fellow before accepting a new position within Dr. Stefan Pfister’s group at the German Cancer Research Centre (DKFZ) in early 2012. The focus of Paul’s research over the past 8 years has been the genomic characterization of medulloblastoma, with a particular interest in dissecting the genomic landscape of medulloblastoma subgroups. Paul is currently using next-generation sequencing approaches to comprehensively explore the mutational spectra of medulloblastoma subgroups and gain insight into medulloblastoma predisposition through large-scale genomic studies.
David TW Jones, PhD David Jones has played a major role in the ICGC PedBrain Tumor sequencing project since moving to Heidelberg from the group of V. Peter Collins at the University of Cambridge two years ago. In 2008, he was the first to describe a highly recurrent BRAF fusion gene occurring in two-thirds of the most common childhood brain tumor (pilocytic astrocytoma). His primary research focus is the application of cutting-edge genomics techniques to identify new diagnostic, prognostic and therapeutic targets in the field of neurooncology.
Marcel Kool, PhD Marcel Kool’s expertise is the genomics of pediatric brain tumors. His aim is to characterize each brain tumor entity in full detail at the genomic level in order to identify clinical relevant subgroups, to find diagnostic and/or prognostic/predictive biomarkers for each of these tumors and their subgroups for use in clinical settings, to find the oncogenic driving events in these tumors, and to find the best therapeutic targets.
Giles W Robinson, MD Giles Robinson is a Research Associate in Neuro-Oncology at St Jude Children’s Research Hospital. He obtained his MD from Warren Albert Medical School of Brown University and completed his pediatric residency training in Children’s Hospital Colorado. He came to St Jude Children’s Hospital in 2007 where he completed his pediatric hematology-oncology and neuro-oncology fellowship training prior to joining the division as faculty. He treats children with all types of brain tumors and has a particular research focus in medulloblastoma. He has conducted extensive genomic analyses of this tumor, with the ultimate goal of enhancing the knowledge of tumor biology and translating that knowledge into novel curative therapies.
Richard J Gilbertson, MD, PhD Richard Gilbertson trained as a pediatric oncologist in England, where he also completed his Ph.D., studying the biology of pediatric medulloblastoma. He moved to St. Jude Children’s Research Hospital, Memphis, in 2000 where he is Director of the Comprehensive Cancer Center, Executive Vice President, and Director of the Division of Brain Tumor Research. He holds the Lillian R. Cannon Comprehensive Cancer Center Director Endowed Chair. His laboratory research is focused on understanding the link between normal development and the cellular and molecular origins of cancer, particularly pediatric brain tumors. He is involved in a series of clinical trials of new treatments of cancer.
Yoon-Jae Cho, MD, PhD Yoon-Jae Cho obtained his MD from the Oregon Health & Science University and then completed training in Pediatric Neurology and Neuro-oncology at Children’s Hospital Boston and Dana-Farber Cancer Institute, respectively. He performed post-doctoral studies with Scott Pomeroy, MD, PhD, characterizing the genomic landscape of medulloblastoma. He is now Assistant Professor of Neurology and Neurosurgery at Stanford University where his laboratory is focused on translating genomic studies of pediatric brain tumors into novel diagnostic, prognostic and therapeutic applications.
Scott L Pomeroy, MD, PhD The lab of Scott Pomeroy is focused on understanding the molecular and cellular basis of medulloblastomas and other embryonal brain tumors. They have used integrative genomics, including transcriptome, DNA copy number and exome sequencing, to analyze medulloblastoma samples from more than 200 children, including a cohort that participated in clinical trials conducted by the Children’s Oncology Group. It was discovered that medulloblastomas are quite heterogeneous, consisting of multiple molecular subtypes that each have unique gene expression and DNA copy number changes and somatic mutations reflecting the mechanisms that regulate tumor growth. In collaboration with Jill Mesirov and Pablo Tamayo at the Broad Institute and our colleagues at the University of Toronto and DKFZ in Heidelberg, outcome prediction models based on genomic data are being developed for risk stratification and molecular subtyping to be used in the next generation of clinical trials. Moreover, molecular mechanisms of identified from genomic data will be used in the development and implementation of targeted therapies to further reduce and ultimately replace conventional therapies based on radiation and chemotherapy. Dr. Pomeroy currently is the Chair of the Department of Neurology and Neurologist-in-Chief of Boston Children’s Hospital, the Bronson Crothers Professor of Neurology at Harvard Medical School, and the Director of the Eunice K. Shriver National Institutes of Child Health and Human Development funded Intellectual and Developmental Disabilities Research Center of Boston Children’s Hospital and Harvard Medical School.
Andrey Korshunov, MD Andrey Korshunov is professor of neuropathology and senior research associate of Clinical Neuropathology Unit at the German Cancer Research Center and Department of Neuropathology at the Heidelberg University. Previously, he was a chairperson in the Department of Neuropathology at the Neurosurgical Burdenko Institute (Moscow; Russia), where Dr. Korshunov received his “doctor of sciences” degree. He is actively involved in basic research focusing on genetics and molecular pathology of pediatric tumors of the nervous system.
Peter Lichter, PhD Peter Lichter pioneered the development of technologies to delineate virtually any chromosomal region by fluorescence in situ hybridization (FISH) and to detect DNA copy number alterations via high resolution comparative genomic hybridization (arrayCGH). Applying these as well as next generation DNA sequencing approaches, he made major contributions to decipher the higher order genome organization and to elucidate pathomechanisms of tumor etiology and progression, including the description of novel prognostic or predictive gene signatures.
Michael D Taylor, MD, PhD Michael D. Taylor is a pediatric neurosurgeon and senior scientist at The Hospital for Sick Children in Toronto, Canada. His clinical practice is focused on the treatment of children with malignant brain tumors. The Taylor laboratory uses tools from human cancer genetics and genomics in combination with mouse modeling and functional genomics to better understand the molecular underpinnings of childhood medulloblastoma.
Stefan M Pfister, MD Stefan Pfister was appointed Head of the Division Pediatric Neurooncology at the German Cancer Research Center (DKFZ), Heidelberg, Germany, in 2012. He is a paediatrician by training, and received his M.D. from Tübingen University, and his clinical education at Mannheim and Heidelberg University Hospitals. As a physician-scientist, he completed postdoctoral fellowships with Christopher Rudd at the Dana-Faber Cancer Institute/Harvard Medical School, Boston, USA, and with Peter Lichter at the German Cancer Research Center, Division of Molecular Genetics, Heidelberg, Germany. His research is focused on the genetic characterization of childhood brain tumors through the application of next-generation profiling methods and subsequently translating novel findings into a clinical context.
Footnotes
Competing interests statement The authors declare no competing financial interests.
References
- 1.Pui CH, Gajjar AJ, Kane JR, Qaddoumi IA, Pappo AS. Challenging issues in pediatric oncology. Nature reviews. Clinical oncology. 2011;8:540–9. doi: 10.1038/nrclinonc.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA: a cancer journal for clinicians. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 3.Gajjar A, et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. The lancet oncology. 2006;7:813–20. doi: 10.1016/S1470-2045(06)70867-1. [DOI] [PubMed] [Google Scholar]
- 4.Rutkowski S, et al. Survival and prognostic factors of early childhood medulloblastoma: an international meta-analysis. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28:4961–8. doi: 10.1200/JCO.2010.30.2299. [DOI] [PubMed] [Google Scholar]
- 5.Ellison DW, et al. beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children’s Cancer Study Group Brain Tumour Committee. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2005;23:7951–7. doi: 10.1200/JCO.2005.01.5479. [DOI] [PubMed] [Google Scholar]
- 6.Packer RJ, Vezina G. Management of and prognosis with medulloblastoma: therapy at a crossroads. Archives of neurology. 2008;65:1419–24. doi: 10.1001/archneur.65.11.1419. [DOI] [PubMed] [Google Scholar]
- 7.Mabbott DJ, Penkman L, Witol A, Strother D, Bouffet E. Core neurocognitive functions in children treated for posterior fossa tumors. Neuropsychology. 2008;22:159–68. doi: 10.1037/0894-4105.22.2.159. [DOI] [PubMed] [Google Scholar]
- 8.Mabbott DJ, et al. Serial evaluation of academic and behavioral outcome after treatment with cranial radiation in childhood. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005;23:2256–63. doi: 10.1200/JCO.2005.01.158. [DOI] [PubMed] [Google Scholar]
- 9.Spiegler BJ, Bouffet E, Greenberg ML, Rutka JT, Mabbott DJ. Change in neurocognitive functioning after treatment with cranial radiation in childhood. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2004;22:706–13. doi: 10.1200/JCO.2004.05.186. [DOI] [PubMed] [Google Scholar]
- 10.Low JA, de Sauvage FJ. Clinical experience with Hedgehog pathway inhibitors. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2010;28:5321–6. doi: 10.1200/JCO.2010.27.9943. [DOI] [PubMed] [Google Scholar]
- 11.Ng JM, Curran T. The Hedgehog’s tale: developing strategies for targeting cancer. Nature reviews. Cancer. 2011;11:493–501. doi: 10.1038/nrc3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor MD, Mainprize TG, Rutka JT. Molecular insight into medulloblastoma and central nervous system primitive neuroectodermal tumor biology from hereditary syndromes: a review. Neurosurgery. 2000;47:888–901. doi: 10.1097/00006123-200010000-00020. [DOI] [PubMed] [Google Scholar]
- 13.Hahn H, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841–51. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- 14.Johnson RL, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668–71. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- 15.Gailani MR, et al. Developmental defects in Gorlin syndrome related to a putative tumor suppressor gene on chromosome 9. Cell. 1992;69:111–7. doi: 10.1016/0092-8674(92)90122-s. [DOI] [PubMed] [Google Scholar]
- 16.Raffel C, et al. Sporadic medulloblastomas contain PTCH mutations. Cancer research. 1997;57:842–5. [PubMed] [Google Scholar]
- 17.Pietsch T, et al. Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of Drosophila patched. Cancer research. 1997;57:2085–8. [PubMed] [Google Scholar]
- 18.Wolter M, Reifenberger J, Sommer C, Ruzicka T, Reifenberger G. Mutations in the human homologue of the Drosophila segment polarity gene patched (PTCH) in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer research. 1997;57:2581–5. [PubMed] [Google Scholar]
- 19.Kool M, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta neuropathologica. 2012;123:473–84. doi: 10.1007/s00401-012-0958-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamilton SR, et al. The molecular basis of Turcot’s syndrome. The New England journal of medicine. 1995;332:839–47. doi: 10.1056/NEJM199503303321302. [DOI] [PubMed] [Google Scholar]
- 21.Zurawel RH, Chiappa SA, Allen C, Raffel C. Sporadic medulloblastomas contain oncogenic beta-catenin mutations. Cancer research. 1998;58:896–9. [PubMed] [Google Scholar]
- 22.Eberhart CG, Tihan T, Burger PC. Nuclear localization and mutation of beta-catenin in medulloblastomas. Journal of neuropathology and experimental neurology. 2000;59:333–7. doi: 10.1093/jnen/59.4.333. [DOI] [PubMed] [Google Scholar]
- 23.Pastorino L, et al. Identification of a SUFU germline mutation in a family with Gorlin syndrome. American journal of medical genetics. Part A. 2009;149A:1539–43. doi: 10.1002/ajmg.a.32944. [DOI] [PubMed] [Google Scholar]
- 24.Taylor MD, et al. Mutations in SUFU predispose to medulloblastoma. Nature genetics. 2002;31:306–10. doi: 10.1038/ng916. [DOI] [PubMed] [Google Scholar]
- 25.Dahmen RP, et al. Deletions of AXIN1, a component of the WNT/wingless pathway, in sporadic medulloblastomas. Cancer research. 2001;61:7039–43. [PubMed] [Google Scholar]
- 26.Baeza N, Masuoka J, Kleihues P, Ohgaki H. AXIN1 mutations but not deletions in cerebellar medulloblastomas. Oncogene. 2003;22:632–6. doi: 10.1038/sj.onc.1206156. [DOI] [PubMed] [Google Scholar]
- 27.Smyth I, et al. Isolation and characterization of human patched 2 (PTCH2), a putative tumour suppressor gene inbasal cell carcinoma and medulloblastoma on chromosome 1p32. Human molecular genetics. 1999;8:291–7. doi: 10.1093/hmg/8.2.291. [DOI] [PubMed] [Google Scholar]
- 28.Koch A, et al. Mutations of the Wnt antagonist AXIN2 (Conductin) result in TCF-dependent transcription in medulloblastomas. International journal of cancer. Journal international du cancer. 2007;121:284–91. doi: 10.1002/ijc.22675. [DOI] [PubMed] [Google Scholar]
- 29.Lam CW, et al. A frequent activated smoothened mutation in sporadic basal cell carcinomas. Oncogene. 1999;18:833–6. doi: 10.1038/sj.onc.1202360. [DOI] [PubMed] [Google Scholar]
- 30.Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nature reviews. Genetics. 2010;11:685–96. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- 31.Stratton MR. Exploring the genomes of cancer cells: progress and promise. Science. 2011;331:1553–8. doi: 10.1126/science.1204040. [DOI] [PubMed] [Google Scholar]
- 32.Robinson G, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012 doi: 10.1038/nature11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones DT, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012 doi: 10.1038/nature11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pugh TJ, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012 doi: 10.1038/nature11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rausch T, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148:59–71. doi: 10.1016/j.cell.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Northcott PA, et al. The miR-17/92 polycistron is up-regulated in sonic hedgehog-driven medulloblastomas and induced by N-myc in sonic hedgehog-treated cerebellar neural precursors. Cancer research. 2009;69:3249–55. doi: 10.1158/0008-5472.CAN-08-4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thompson MC, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24:1924–31. doi: 10.1200/JCO.2005.04.4974. [DOI] [PubMed] [Google Scholar]
- 38.Kool M, et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PloS one. 2008;3:e3088. doi: 10.1371/journal.pone.0003088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cho YJ, et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:1424–30. doi: 10.1200/JCO.2010.28.5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Northcott PA, et al. Medulloblastoma comprises four distinct molecular variants. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:1408–14. doi: 10.1200/JCO.2009.27.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Remke M, et al. FSTL5 is a marker of poor prognosis in non-WNT/non-SHH medulloblastoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29:3852–61. doi: 10.1200/JCO.2011.36.2798. [DOI] [PubMed] [Google Scholar]
- 42.Taylor MD, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta neuropathologica. 2012;123:465–72. doi: 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Northcott PA, Korshunov A, Pfister SM, Taylor MD. The clinical implications of medulloblastoma subgroups. Nature reviews. Neurology. 2012 doi: 10.1038/nrneurol.2012.78. [DOI] [PubMed] [Google Scholar]
- 44.Northcott PA, Dubuc AM, Pfister S, Taylor MD. Molecular subgroups of medulloblastoma. Expert review of neurotherapeutics. 2012;12:871–84. doi: 10.1586/ern.12.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Packer RJ. Risk stratification of medulloblastoma: a paradigm for future childhood brain tumor management strategies. Current neurology and neuroscience reports. 2011;11:124–6. doi: 10.1007/s11910-010-0168-5. [DOI] [PubMed] [Google Scholar]
- 46.Leary SE, Olson JM. The molecular classification of medulloblastoma: driving the next generation clinical trials. Current opinion in pediatrics. 2012;24:33–9. doi: 10.1097/MOP.0b013e32834ec106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pomeroy SL, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415:436–42. doi: 10.1038/415436a. [DOI] [PubMed] [Google Scholar]
- 48.Clifford SC, et al. Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub-group of medulloblastomas associated with a favorable prognosis. Cell cycle. 2006;5:2666–70. doi: 10.4161/cc.5.22.3446. [DOI] [PubMed] [Google Scholar]
- 49.Pfaff E, et al. TP53 mutation is frequently associated with CTNNB1 mutation or MYCN amplification and is compatible with long-term survival in medulloblastoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28:5188–96. doi: 10.1200/JCO.2010.31.1670. [DOI] [PubMed] [Google Scholar]
- 50.Lindsey JC, et al. TP53 mutations in favorable-risk Wnt/Wingless-subtype medulloblastomas. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:e344–6. doi: 10.1200/JCO.2010.33.8590. author reply e347-8. [DOI] [PubMed] [Google Scholar]
- 51.Northcott PA, et al. Rapid, reliable, and reproducible molecular sub-grouping of clinical medulloblastoma samples. Acta neuropathologica. 2012;123:615–26. doi: 10.1007/s00401-011-0899-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gibson P, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010;468:1095–9. doi: 10.1038/nature09587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Northcott PA, et al. Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta neuropathologica. 2011;122:231–40. doi: 10.1007/s00401-011-0846-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ellison DW, et al. Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta neuropathologica. 2011;121:381–96. doi: 10.1007/s00401-011-0800-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brugieres L, et al. High Frequency of Germline SUFU Mutations in Children With Desmoplastic/Nodular Medulloblastoma Younger Than 3 Years of Age. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30:2087–93. doi: 10.1200/JCO.2011.38.7258. [DOI] [PubMed] [Google Scholar]
- 56.Brugieres L, et al. Incomplete penetrance of the predisposition to medulloblastoma associated with germ-line SUFU mutations. Journal of medical genetics. 2010;47:142–4. doi: 10.1136/jmg.2009.067751. [DOI] [PubMed] [Google Scholar]
- 57.Garre ML, et al. Medulloblastoma variants: age-dependent occurrence and relation to Gorlin syndrome--a new clinical perspective. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:2463–71. doi: 10.1158/1078-0432.CCR-08-2023. [DOI] [PubMed] [Google Scholar]
- 58.Huse JT, Holland EC. Genetically engineered mouse models of brain cancer and the promise of preclinical testing. Brain pathology. 2009;19:132–43. doi: 10.1111/j.1750-3639.2008.00234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu X, Northcott PA, Croul S, Taylor MD. Mouse models of medulloblastoma. Chinese journal of cancer. 2011;30:442–9. doi: 10.5732/cjc.011.10040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–13. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 61.Wetmore C, Eberhart DE, Curran T. The normal patched allele is expressed in medulloblastomas from mice with heterozygous germ-line mutation of patched. Cancer research. 2000;60:2239–46. [PubMed] [Google Scholar]
- 62.Hatton BA, et al. The Smo/Smo model: hedgehog-induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer research. 2008;68:1768–76. doi: 10.1158/0008-5472.CAN-07-5092. [DOI] [PubMed] [Google Scholar]
- 63.Hallahan AR, et al. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer research. 2004;64:7794–800. doi: 10.1158/0008-5472.CAN-04-1813. [DOI] [PubMed] [Google Scholar]
- 64.Oliver TG, et al. Loss of patched and disruption of granule cell development in a pre-neoplastic stage of medulloblastoma. Development. 2005;132:2425–39. doi: 10.1242/dev.01793. [DOI] [PubMed] [Google Scholar]
- 65.Yang ZJ, et al. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer cell. 2008;14:135–45. doi: 10.1016/j.ccr.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schuller U, et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer cell. 2008;14:123–34. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Grammel D, et al. Sonic hedgehog-associated medulloblastoma arising from the cochlear nuclei of the brainstem. Acta neuropathologica. 2012;123:601–14. doi: 10.1007/s00401-012-0961-0. [DOI] [PubMed] [Google Scholar]
- 68.Rudin CM, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. The New England journal of medicine. 2009;361:1173–8. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yauch RL, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science. 2009;326:572–4. doi: 10.1126/science.1179386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Buonamici S, et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Science translational medicine. 2010;2:51ra70. doi: 10.1126/scitranslmed.3001599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dijkgraaf GJ, et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer research. 2011;71:435–44. doi: 10.1158/0008-5472.CAN-10-2876. [DOI] [PubMed] [Google Scholar]
- 72.Metcalfe C, de Sauvage FJ. Hedgehog fights back: mechanisms of acquired resistance against Smoothened antagonists. Cancer research. 2011;71:5057–61. doi: 10.1158/0008-5472.CAN-11-0923. [DOI] [PubMed] [Google Scholar]
- 73.Northcott PA, et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nature genetics. 2009;41:465–72. doi: 10.1038/ng.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kimura H, Ng JM, Curran T. Transient inhibition of the Hedgehog pathway in young mice causes permanent defects in bone structure. Cancer cell. 2008;13:249–60. doi: 10.1016/j.ccr.2008.01.027. [DOI] [PubMed] [Google Scholar]
- 75.Pei Y, et al. An animal model of MYC-driven medulloblastoma. Cancer cell. 2012;21:155–67. doi: 10.1016/j.ccr.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kawauchi D, et al. A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer cell. 2012;21:168–80. doi: 10.1016/j.ccr.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Remke M, et al. Adult medulloblastoma comprises three major molecular variants. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:2717–23. doi: 10.1200/JCO.2011.34.9373. [DOI] [PubMed] [Google Scholar]
- 78.Eberhart CG. Three down and one to go: modeling medulloblastoma subgroups. Cancer cell. 2012;21:137–8. doi: 10.1016/j.ccr.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Swartling FJ, et al. Distinct Neural Stem Cell Populations Give Rise to Disparate Brain Tumors in Response to N-MYC. Cancer cell. 2012;21:601–13. doi: 10.1016/j.ccr.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Swartling FJ, et al. Pleiotropic role for MYCN in medulloblastoma. Genes & development. 2010;24:1059–72. doi: 10.1101/gad.1907510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Northcott PA, Rutka JT, Taylor MD. Genomics of medulloblastoma: from Giemsa-banding to next-generation sequencing in 20 years. Neurosurgical focus. 2010;28:E6. doi: 10.3171/2009.10.FOCUS09218. [DOI] [PubMed] [Google Scholar]
- 82.Ciara E, et al. Heterozygous germ-line mutations in the NBN gene predispose to medulloblastoma in pediatric patients. Acta neuropathologica. 2010;119:325–34. doi: 10.1007/s00401-009-0608-y. [DOI] [PubMed] [Google Scholar]
- 83.Distel L, Neubauer S, Varon R, Holter W, Grabenbauer G. Fatal toxicity following radio- and chemotherapy of medulloblastoma in a child with unrecognized Nijmegen breakage syndrome. Medical and pediatric oncology. 2003;41:44–8. doi: 10.1002/mpo.10275. [DOI] [PubMed] [Google Scholar]
- 84.Taylor MD, et al. Medulloblastoma in a child with Rubenstein-Taybi Syndrome: case report and review of the literature. Pediatric neurosurgery. 2001;35:235–8. doi: 10.1159/000050428. [DOI] [PubMed] [Google Scholar]
- 85.Parsons DW, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331:435–9. doi: 10.1126/science.1198056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Parsons DW, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hodis E, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–63. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Banerji S, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486:405–9. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barbieri CE, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nature genetics. 2012;44:685–9. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pek JW, Kai T. DEAD-box RNA helicase Belle/DDX3 and the RNA interference pathway promote mitotic chromosome segregation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:12007–12. doi: 10.1073/pnas.1106245108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lai MC, Chang WC, Shieh SY, Tarn WY. DDX3 regulates cell growth through translational control of cyclin E1. Molecular and cellular biology. 2010;30:5444–53. doi: 10.1128/MCB.00560-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schroder M. Human DEAD-box protein 3 has multiple functions in gene regulation and cell cycle control and is a prime target for viral manipulation. Biochemical pharmacology. 2010;79:297–306. doi: 10.1016/j.bcp.2009.08.032. [DOI] [PubMed] [Google Scholar]
- 95.Lee CS, et al. Human DDX3 functions in translation and interacts with the translation initiation factor eIF3. Nucleic acids research. 2008;36:4708–18. doi: 10.1093/nar/gkn454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lai MC, Lee YH, Tarn WY. The DEAD-box RNA helicase DDX3 associates with export messenger ribonucleoproteins as well as tip-associated protein and participates in translational control. Molecular biology of the cell. 2008;19:3847–58. doi: 10.1091/mbc.E07-12-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rosner A, Rinkevich B. The DDX3 subfamily of the DEAD box helicases: divergent roles as unveiled by studying different organisms and in vitro assays. Current medicinal chemistry. 2007;14:2517–25. doi: 10.2174/092986707782023677. [DOI] [PubMed] [Google Scholar]
- 98.Hogbom M, et al. Crystal structure of conserved domains 1 and 2 of the human DEAD-box helicase DDX3X in complex with the mononucleotide AMP. Journal of molecular biology. 2007;372:150–9. doi: 10.1016/j.jmb.2007.06.050. [DOI] [PubMed] [Google Scholar]
- 99.Northcott PA, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012 doi: 10.1038/nature11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sengoku T, Yokoyama S. Structural basis for histone H3 Lys 27 demethylation by UTX/KDM6A. Genes & development. 2011;25:2266–77. doi: 10.1101/gad.172296.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kim E, Song JJ. Diverse ways to be specific: a novel Zn-binding domain confers substrate specificity to UTX/KDM6A histone H3 Lys 27 demethylase. Genes & development. 2011;25:2223–6. doi: 10.1101/gad.179473.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kooistra SM, Helin K. Molecular mechanisms and potential functions of histone demethylases. Nature reviews. Molecular cell biology. 2012;13:297–311. doi: 10.1038/nrm3327. [DOI] [PubMed] [Google Scholar]
- 103.Lee MG, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005;437:432–5. doi: 10.1038/nature04021. [DOI] [PubMed] [Google Scholar]
- 104.Schnetz MP, et al. CHD7 targets active gene enhancer elements to modulate ES cell-specific gene expression. PLoSgenetics. 2010;6:e1001023. doi: 10.1371/journal.pgen.1001023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bajpai R, et al. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature. 2010;463:958–62. doi: 10.1038/nature08733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schnetz MP, et al. Genomic distribution of CHD7 on chromatin tracks H3K4 methylation patterns. Genome research. 2009;19:590–601. doi: 10.1101/gr.086983.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nature reviews. Genetics. 2012;13:343–57. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–9. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Alimova I, et al. Targeting the enhancer of zeste homologue 2 in medulloblastoma. International journal of cancer. Journal international du cancer. 2012 doi: 10.1002/ijc.27455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Oberoi J, et al. Structural basis for the assembly of the SMRT/NCoR core transcriptional repression machinery. Nature structural & molecular biology. 2011;18:177–84. doi: 10.1038/nsmb.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chin L, Hahn WC, Getz G, Meyerson M. Making sense of cancer genomic data. Genes & development. 2011;25:534–55. doi: 10.1101/gad.2017311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Satow R, Kurisaki A, Chan TC, Hamazaki TS, Asashima M. Dullard promotes degradation and dephosphorylation of BMP receptors and is required for neural induction. Developmental cell. 2006;11:763–74. doi: 10.1016/j.devcel.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 113.Kim Y, et al. A conserved phosphatase cascade that regulates nuclear membrane biogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:6596–601. doi: 10.1073/pnas.0702099104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Han S, et al. Nuclear envelope phosphatase 1-regulatory subunit 1 (formerly TMEM188) is the metazoan Spo7p ortholog and functions in the lipin activation pathway. The Journal of biological chemistry. 2012;287:3123–37. doi: 10.1074/jbc.M111.324350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cogen PH, et al. Involvement of multiple chromosome 17p loci in medulloblastoma tumorigenesis. American journal of human genetics. 1992;50:584–9. [PMC free article] [PubMed] [Google Scholar]
- 116.Cogen PH, Daneshvar L, Metzger AK, Edwards MS. Deletion mapping of the medulloblastoma locus on chromosome 17p. Genomics. 1990;8:279–85. doi: 10.1016/0888-7543(90)90283-z. [DOI] [PubMed] [Google Scholar]
- 117.Biegel JA, Burk CD, Barr FG, Emanuel BS. Evidence for a 17p tumor related locus distinct from p53 in pediatric primitive neuroectodermal tumors. Cancer research. 1992;52:3391–5. [PubMed] [Google Scholar]
- 118.Adamson DC, et al. OTX2 is critical for the maintenance and progression of Shh-independent medulloblastomas. Cancer research. 2010;70:181–91. doi: 10.1158/0008-5472.CAN-09-2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pfister S, et al. Outcome prediction in pediatric medulloblastoma based on DNA copy-number aberrations of chromosomes 6q and 17q and the MYC and MYCN loci. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:1627–36. doi: 10.1200/JCO.2008.17.9432. [DOI] [PubMed] [Google Scholar]
- 120.Mendrzyk F, et al. Genomic and protein expression profiling identifies CDK6 as novel independent prognostic marker in medulloblastoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005;23:8853–62. doi: 10.1200/JCO.2005.02.8589. [DOI] [PubMed] [Google Scholar]
- 121.Li M, et al. Multiple CDK/CYCLIND genes are amplified in medulloblastoma and supratentorial primitive neuroectodermal brain tumor. Cancer genetics. 2012;205:220–31. doi: 10.1016/j.cancergen.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 122.Le Guezennec X, Bulavin DV. WIP1 phosphatase at the crossroads of cancer and aging. Trends in biochemical sciences. 2010;35:109–14. doi: 10.1016/j.tibs.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 123.Castellino RC, et al. Medulloblastomas overexpress the p53-inactivating oncogene WIP1/PPM1D. Journal of neuro-oncology. 2008;86:245–56. doi: 10.1007/s11060-007-9470-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Doucette TA, et al. WIP1 enhances tumor formation in a sonic hedgehog-dependent model of medulloblastoma. Neurosurgery. 2012;70:1003–10. doi: 10.1227/NEU.0b013e31823e5332. discussion 1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Huppi K, et al. The identification of microRNAs in a genomically unstable region of human chromosome 8q24. Molecular cancer research : MCR. 2008;6:212–21. doi: 10.1158/1541-7786.MCR-07-0105. [DOI] [PubMed] [Google Scholar]
- 126.Beck-Engeser GB, et al. Pvt1-encoded microRNAs in oncogenesis. Retrovirology. 2008;5:4. doi: 10.1186/1742-4690-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Guan Y, et al. Amplification of PVT1 contributes to the pathophysiology of ovarian and breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13:5745–55. doi: 10.1158/1078-0432.CCR-06-2882. [DOI] [PubMed] [Google Scholar]
- 128.Engelender S, et al. Synphilin-1 associates with alpha-synuclein and promotes the formation of cytosolic inclusions. Nature genetics. 1999;22:110–4. doi: 10.1038/8820. [DOI] [PubMed] [Google Scholar]
- 129.Chung KK, et al. Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nature medicine. 2001;7:1144–50. doi: 10.1038/nm1001-1144. [DOI] [PubMed] [Google Scholar]
- 130.Eyal A, Engelender S. Synphilin isoforms and the search for a cellular model of lewy body formation in Parkinson’s disease. Cell cycle. 2006;5:2082–6. doi: 10.4161/cc.5.18.3209. [DOI] [PubMed] [Google Scholar]
- 131.Pfister S, et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. The Journal of clinical investigation. 2008;118:1739–49. doi: 10.1172/JCI33656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jones DT, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer research. 2008;68:8673–7. doi: 10.1158/0008-5472.CAN-08-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Malkin D, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–8. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- 134.Srivastava S, Zou ZQ, Pirollo K, Blattner W, Chang EH. Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature. 1990;348:747–9. doi: 10.1038/348747a0. [DOI] [PubMed] [Google Scholar]
- 135.Stephens PJ, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Boveri T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. Journal of cell science. 2008;121(Suppl 1):1–84. doi: 10.1242/jcs.025742. [DOI] [PubMed] [Google Scholar]
- 137.Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nature reviews. Genetics. 2012;13:189–203. doi: 10.1038/nrg3123. [DOI] [PubMed] [Google Scholar]
- 138.Storchova Z, Pellman D. From polyploidy to aneuploidy, genome instability and cancer. Nature reviews. Molecular cell biology. 2004;5:45–54. doi: 10.1038/nrm1276. [DOI] [PubMed] [Google Scholar]
- 139.Fujii Y, Hongo T, Hayashi Y. Chromosome analysis of brain tumors in childhood. Genes, chromosomes & cancer. 1994;11:205–15. doi: 10.1002/gcc.2870110402. [DOI] [PubMed] [Google Scholar]
- 140.Tomita T, Das L, Radkowski MA. Bone metastases of medulloblastoma in childhood; correlation with flow cytometric DNA analysis. Journal of neurooncology. 1990;8:113–20. doi: 10.1007/BF00177833. [DOI] [PubMed] [Google Scholar]
- 141.Giangaspero F, et al. “Desmoplastic” versus “classic” medulloblastoma: comparison of DNA content, histopathology and differentiation. Virchows Archiv. A, Pathological anatomy and histopathology. 1991;418:207–14. doi: 10.1007/BF01606058. [DOI] [PubMed] [Google Scholar]
- 142.Zerbini C, et al. Prognostic factors in medulloblastoma, including DNA ploidy. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1993;11:616–22. doi: 10.1200/JCO.1993.11.4.616. [DOI] [PubMed] [Google Scholar]
- 143.Keles GE, et al. Establishment and characterization of four human medulloblastoma-derived cell lines. Oncology research. 1995;7:493–503. [PubMed] [Google Scholar]
- 144.Rajcan-Separovic E, et al. Interphase fluorescence in situ hybridization and DNA flow cytometry analysis of medulloblastomas with a normal karyotype. Cancer genetics and cytogenetics. 2002;133:94–7. doi: 10.1016/s0165-4608(01)00558-1. [DOI] [PubMed] [Google Scholar]
- 145.Davoli T, de Lange T. Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer cell. 2012;21:765–76. doi: 10.1016/j.ccr.2012.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fujiwara T, et al. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005;437:1043–7. doi: 10.1038/nature04217. [DOI] [PubMed] [Google Scholar]
- 147.Castedo M, et al. Selective resistance of tetraploid cancer cells against DNA damage-induced apoptosis. Annals of the New York Academy of Sciences. 2006;1090:35–49. doi: 10.1196/annals.1378.004. [DOI] [PubMed] [Google Scholar]
- 148.Rello-Varona S, et al. Preferential killing of tetraploid tumor cells by targeting the mitotic kinesin Eg5. Cell cycle. 2009;8:1030–5. doi: 10.4161/cc.8.7.7950. [DOI] [PubMed] [Google Scholar]
- 149.Vitale I, et al. Inhibition of Chkl kills tetraploid tumor cells through a p53-dependent pathway. PloS one. 2007;2:e1337. doi: 10.1371/journal.pone.0001337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Strahm B, Malkin D. Hereditary cancer predisposition in children: genetic basis and clinical implications. International journal of cancer. Journal international du cancer. 2006;119:2001–6. doi: 10.1002/ijc.21962. [DOI] [PubMed] [Google Scholar]
- 151.Searles Nielsen S, et al. Family cancer history and risk of brain tumors in children: results of the SEARCH international brain tumor study. Cancer causes & control: CCC. 2008;19:641–8. doi: 10.1007/s10552-008-9128-7. [DOI] [PubMed] [Google Scholar]
- 152.Marth GT, et al. The functional spectrum of low-frequency coding variation. Genome biology. 2011;12:R84. doi: 10.1186/gb-2011-12-9-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 155.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nature reviews. Cancer. 2011;11:726–34. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nature reviews. Genetics. 2011;12:87–98. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Curtis C, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–52. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Parmigiani G, et al. Design and analysis issues in genome-wide somatic mutation studies of cancer. Genomics. 2009;93:17–21. doi: 10.1016/j.ygeno.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–24. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Premsrirut PK, et al. A rapid and scalable system for studying gene function in mice using conditional RNA interference. Cell. 2011;145:145–58. doi: 10.1016/j.cell.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zuber J, et al. Toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi. Nature biotechnology. 2011;29:79–83. doi: 10.1038/nbt.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Copeland NG, Jenkins NA. Harnessing transposons for cancer gene discovery. Nature reviews. Cancer. 2010;10:696–706. doi: 10.1038/nrc2916. [DOI] [PubMed] [Google Scholar]
- 163.Wu X, et al. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature. 2012;482:529–33. doi: 10.1038/nature10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ellison DW, et al. Definition of disease-risk stratification groups in childhood medulloblastoma using combined clinical, pathologic, and molecular variables. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29:1400–7. doi: 10.1200/JCO.2010.30.2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.von Hoff K, et al. Long-term outcome and clinical prognostic factors in children with medulloblastoma treated in the prospective randomised multicentre trial HIT’91. European journal of cancer. 2009;45:1209–17. doi: 10.1016/j.ejca.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 166.Taylor RE, et al. Outcome for patients with metastatic (M2-3) medulloblastoma treated with SIOP/UKCCSG PNET-3 chemotherapy. European journal of cancer. 2005;41:727–34. doi: 10.1016/j.ejca.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 167.Packer RJ. Childhood brain tumors: accomplishments and ongoing challenges. Journal of child neurology. 2008;23:1122–7. doi: 10.1177/0883073808320758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gilbertson RJ. Mapping cancer origins. Cell. 2011;145:25–9. doi: 10.1016/j.cell.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes & development. 2000;14:994–1004. [PMC free article] [PubMed] [Google Scholar]
- 170.Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22:103–14. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]