

The many facets of fatigue
Many definitions of fatigue have been proposed. One that is often cited refers to fatigue as “an overwhelming, debilitating, and sustained sense of exhaustion that decreases one’s ability to carry out daily activities, including the ability to work effectively and to function at one’s usual level in family or social roles” (1). There are at least two dimensions in fatigue, “I cannot do it, I am exhausted” versus “I do not feel like doing it, it is not worth it”. The first dimension is relatively easy to characterize as it is usually associated with obvious physical signs. The second dimension is more difficult to characterize and is usually referred to as central fatigue or “the failure to initiate and/or sustain attentional tasks and physical activities requiring self motivation” (2). Central fatigue rarely occurs alone, it is often associated with sleep disorders, pain, and affective and cognitive alterations.
Fatigue is highly prevalent in the general population, with rates around 20% (3). Approximately one third of patients’ complaints in medical general practice relate to fatigue (4). The prevalence of fatigue increases dramatically, up to and above 50%, in a number of medical conditions that involve dysregulation of the immune system, such as cancer, chronic infection, autoimmune diseases and neurological diseases (5). Fatigue in the physically ill patient is one of the most common and earliest non-specific symptoms of disease and can persist long after the medical condition has resolved.
In practice fatigue is self-reported. Because subjective fatigue correlate very poorly with objective measures of physical activity and performance (6), several questionnaires have been developed to measure the overall severity of fatigue and in some cases its dimensions. However, what is often forgotten while processing these scores is that fatigue is a product of the patients’ representation of their condition and is part of the perceptions patients form about their illness. These perceptions vary according to time and depend on the way patients can mobilize their resources and cope with this particular threat to their integrity that their illness represents (7, 8). Patients do not remain passive but are always forming expectations about possible futures and actions. This aspect is captured by the concept of self-efficacy, a cognitive construct implicating one’s self-perception about one’s performance ability. Fatigued patients with a history of breast cancer or multiple sclerosis have a poor self-efficacy that accounts for their inability to engage in physical exercise despite its potential benefits (9).
In view of the large body of data that is available on the relationship between fatigue and inflammation and the negative impact of inflammation on motivated behavior, we propose that fatigue in subjects with inflammation is a feeling that relates to the lack of motivation to deploy resources and engage in high effort performance to cope with their situation. This is in contrast to depression that has a helplessness component together with self-depreciation, sadness, and anhedonia.
Inflammation, sickness, incentive motivation, and fatigue
The conceptualization of inflammation-induced fatigue requires an evolutionary perspective. In a seminal paper, Hart suggested the behavior of sick animals is not a maladaptive response or the effect of debilitation but an organized, evolved strategy to facilitate the role of fever in combating viral and bacterial infections (10). Subsequent studies have focused on the mechanistic aspect of inflammation-induced sickness and demonstrated that it is triggered by activation of a limited set of innate immune receptors known as toll-like receptors and common to plants and animals by evolutionary conserved structures of pathogens (11). The interaction of toll-like receptors with their ligands results in the de novo production of proinflammatory cytokines. In addition to their local role in coordinating the mounting and regulation of an immune response, proinflammatory cytokines produced by activated innate immune cells signal the brain via a number of immune-to-brain communication pathways. In response, the brain forms a cellular and molecular image of the peripheral inflammatory response that organizes behavior and metabolism. In essence, activation of the immune-to-brain communication pathways ultimately allows caring for the ill body to impose new behavioral priorities.
In motivational terms the motivation of sickness competes with other internally or externally driven motivational states (e.g., hunger, exploration, sex) and takes precedence unless competing motivational stimuli become more important for survival. Motivational competition between sickness and other motivational states can be demonstrated by varying the intensity of the triggering stimuli. For instance lactating mice injected with lipopolysaccharide stayed curled in a corner of the cage and failed to display typical maternal care behavior such as upright crouched nursing posture in response to solicitations from their pups (12). However, if the maternal motivation of lipopolysaccharide-treated dams was challenged by removing the pups from the nest and dispersing them in the cage, they overcame their lethargy and engaged in pup retrieving although they were slower in doing so than saline-injected dams. When tested at thermoneutral environmental temperatures lipopolysaccharide-treated dams did not engage in nest building when the nest was removed and replaced by cotton wool. However, they built a near perfect nest when exposed to a 6°C environment.
The reluctance of sick individuals to engage in motivated behaviors that are unrelated to sickness has been studied mainly in the context of food motivation. In general animals that are made sick by administration of lipopolysaccharide or proinflammatory cytokines are less likely to consume food if effort is required to obtain it than if the food is freely available. The greater the necessary effort the more sensitive to disruption their behavior is. For example intraperitoneal administration of interleukin-1(IL-1)β decreased home cage consumption of sweetened milk to a greater extent in ad libitum fed mice than in food-restricted mice (13). Similarly, IL-1β-treated mice that had to repeatedly nose poke to get a small amount of sweetened milk stopped responding earlier when more nose pokes were required.. Finally, rats that had to deploy effort to obtain highly preferred carbohydrate food pellets in a concurrent performance task with laboratory chow freely available decreased responding when treated with IL-1β while simultaneously increasing chow consumption (14). These results indicate that inflammation reduces incentive motivation.
This approach to fatigue is characteristic of the prevailing strategy in biological psychiatry which aims to specify more quantifiable behaviors and neurobiological measures as an alternative to the current clinically-based classifications of mental disorders (15). Although fatigue is not a mental disorder it is a symptom complex that still requires deconstruction its full range of variation. Of the research domain criteria proposed by NIH, the most relevant to fatigue would be the behavioral function “sustained responsiveness to reward” (15). As discussed below, this behavioral function has the advantage of being a validated construct with a specifiable neural circuit.
Inflammation and fatigue: Clinical findings
At the clinical level there has been little attempt thus far at deconstructing fatigue when studying its relationship with inflammation, with the sole exception a preliminary study in which inflammation was more related to physical than to mental fatigue in patients with advanced cancer (16). Most of what we know about the role of proinflammatory cytokines in the development and severity of fatigue comes from cross-sectional clinical studies in physically ill patients suffering from fatigue although causality studies are now emerging.
Associations between fatigue and inflammatory markers (primarily IL-6, tumor necrosis factor-alpha (TNFα) and C-reactive protein, an acute phase protein) have been documented in various medical conditions, including cancer, viral infections, chronic inflammation, autoimmunity, neurological diseases, and mood disorders (17–19). Fatigue develops in a large proportion of cancer patients receiving chemotherapy and/or radiation therapy. Fatigued cancer patients have elevated circulating levels of biomarkers of inflammation although this association is more consistently observed in longitudinal than in cross-sectional studies (20).
The measurement of circulating concentrations of cytokines represents the main limitation of the present studies on fatigue and inflammation. As cytokines are autocrine and paracrine communication factors, their circulating levels have little functional value and represent mostly spillover from the site of cytokine production and action. Alternative strategies are available. They are based on in vitro measurements of cytokines produced by peripheral blood mononuclear cells or specific immune cell populations in response to well identified immune stimuli (21). They often include an assessment of glucocorticoid receptor function as cortisol is the main endogenous brake on the production of proinflammatory cytokines (22). However, these assays require equipment and expertise that are not easily accessible for most clinical researchers.
Compelling evidence for a causal link between inflammation and fatigue comes from studies conducted in individuals receiving cytokine inducers such as lipopolysaccharide in an experimental setting or recombinant cytokines such as interferon-alpha (IFNα) for the treatment of hepatitis C virus infection, malignant melanoma, or kidney cancer. For example, moderate to severe fatigue develops in up to 80% of cancer patients treated chronically with IFNα, as early as the first week of treatment (23). Further evidence for a causal relationship between immune activation and fatigue comes from studies in which treatment with an antagonist of TNFα significantly reduced fatigue in patients with rheumatoid arthritis or psoriasis (24, 25).
In IFN-α-treated cancer patients, physical fatigue appears earlier than central fatigue, which is concomitant to the occurrence of mood and cognitive symptoms (23). The various dimensions of fatigue do not respond in the same way to treatment. For instance, preventive administration of paroxetine, a selective serotonin reuptake inhibitor, does not block the development of IFNα-induced fatigue (23). In contrast, this treatment is effective in preventing depressed mood and the related symptom of lassitude in the same population as well as in depressed patients. Similarly, data obtained in fatigued patients with Parkinson disease indicate that central fatigue and physical fatigue are independent symptoms that require separate treatment interventions (26).
Neural basis of inflammation-induced fatigue
There have been only a limited number of studies on the neural mechanisms of inflammation-induced fatigue, in contrast to a large number on inflammation-induced sickness and depression (11, 27). Peripheral cytokines can affect central neurotransmition indirectly by modulating the bioavailability of amino acid precursors of neurotransmitters (Fig. 1). In addition, peripherally released cytokines activate immune-to-brain communication pathways, enabling the brain to be informed about immune events even in the absence of blood-brain-barrier disturbances (Box 1). Specifically, peripheral cytokines induce the production and release of inflammatory mediators including prostaglandins and cytokines by endothelial cells, macrophages and microglia in the central nervous system. These inflammatory mediators can influence neurons directly or indirectly by modifying astrocyte, oligodendrocyte, and endothelial cell functions (Fig. 1).
Fig. 1. Peripheral and central mechanisms of inflammation-associated central fatigue.
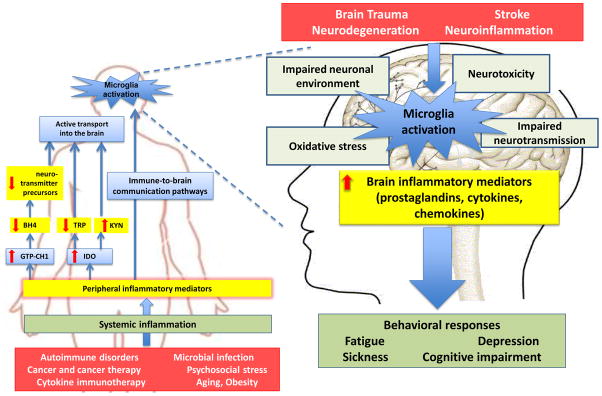
Systemic inflammation, which can be caused by a number of factors, involves both innate immune cells and T lymphocytes. The proinflammatory cytokines that are produced de novo by these cells affect the bioavailability of amino acid precursors of neurotransmitters. Specifically, peripheral proinflammatory cytokines activate guanosine-triphosphate cyclohydrolase-1 (GTP-CH1), which mediates synthesis of neopterin by macrophages. This results in a relative deficit in tetrahydrobiopterin (BH4), an essential cofactor of aromatic amino acid hydroxylase enzymes used in the synthesis of dopamine, norepinephrine and serotonin. BH4 is also a co-factor for the synthesis of nitric oxide by inducible nitric oxide synthase. Proinflammatory cytokines also activate indoleamine 2,3 dioxygenase (IDO) in macrophages and dendritic cells, which degrades tryptophan (TRP) along the kynurenine (KYN) pathway. Kynurenine competes with tryptophan for entry into the brain. Kynurenine is further metabolized by activated microglia into 3-hydroxy kynurenine and quinolinic acid, which are both potent radical donors. Quinolinic acid acts also as an agonist of N-methyl-D-aspartic acid (NMDA) receptors and promotes neurotoxicity. The conversion of kynurenine into kynurenic acid, which acts as an antagonist of NMDA receptors, takes place in astrocytes. However, in conditions of inflammation this potentially neuroprotective pathway is less effective than the pathway leading to quinolinic acid.
Peripheral inflammatory mediators activate immune-to-brain communication pathways including afferent nerves. This leads to the local synthesis of inflammatory mediators that affect neuronal function and structure directly or via impairment of the neuronal environment, reduction of the synthesis of neurotrophic factors, and oxidative stress. These effects are rarely sufficient to cause neurotoxicity but they can easily potentiate the neurotoxic activity of a number of other factors. Activation of the pituitary-adrenal axis by proinflammatory cytokines under the combined effect of corticotrophin-releasing hormone and vasopressin (not shown in the graph) should normally contribute to down-regulation of the inflammatory response both at the periphery and in the central nervous system via the production of cortisol and the anti-inflammatory properties of vasopressin. However, this effect can be compromised by the development of cortisol resistance during inflammation. Adverse behavioral responses are the ultimate consequence of activation of these pathways. Red arrows signify the direction of change in a specific inflammatory mediator, enzyme or molecule following systemic inflammation, processes at similar levels, (e.g. both peripheral and central causes of inflammation) highlighted with a common color.
Box 1. Immune-to-Brain Communication Pathways.
Several communication pathways are involved in the transmission of the peripheral inflammatory message to the brain, including: a humoral pathway by which circulating pathogen-associated molecular patterns such as lipopolysaccharide engage Toll-like receptors on macrophage-like cells in the circumventricular organs, which results in the local production of cytokines that propagate from the blood side to the brain side of the blood-brain barrier (54–56); saturable transport systems allowing the passage of cytokines across the blood-brain barrier (57); and a neural pathway that bypasses the blood-brain barrier (55). In this last case, peripheral inflammatory mediators activate sensory nerves that innervate the site of inflammation and relay the immune message to their sites of primary and secondary projection in the brain.
The role of brain endothelial cells and perivascular macrophages in the transmission of the peripheral immune message to the central nervous system has received much attention, as they are the primary sites where activation of arachidonic cascades takes place (58). However, most of the cytokines that are expressed in the brain in response to systemic immune stimuli are produced by microglia. Adriano Fontana was the first to show that interleukin (IL)-1 is produced in the brain in response to endotoxin and to propose that it could mediate the pyrogenic action of endotoxin (59). The synthesis of IL-1β and other cytokines in the brain in response to non-septic doses of intra-peritoneal lipopolysaccharide was subsequently demonstrated at the mRNA level by reverse transcriptase polymerase chain reaction (60, 61). Receptors for IL-1 and other proinflammatory cytokines were also identified in the brain and pituitary using autoradiography, RT-PCR, and immunohistochemistry. Furthermore, light and electron microscopic analysis of the brains of endotoxin-treated rats revealed the presence of IL-1β-producing cells in the brain in the form of macrophages in the meninges and choroid plexus and microglia (62). Involvement of microglia in the response to systemic lipopolysaccharide has since been confirmed by various approaches. Parenchymal microglia, for instance, express the inhibitory factor kappaB alpha, a proxy for activation of the nuclear factor kappa B signaling pathway, in response to systemic lipopolysaccharide (63). More recently, ex vivo isolation of microglial cells from the brain of adult animals has been used to assess the ability of these cells to produce inflammatory mediators in response to systemic lipopolysaccharide (e.g., (64)).
The possibility that cytokines expressed in the brain in response to systemic inflammation mediate the many facets of the host response to immune stimulation has been investigated at length. There is ample evidence from pharmacological studies with cytokine antagonists administered into the brain and from genetic studies using cytokine or cytokine receptor knock-out mice that cytokines produced in situ underlie the brain response to systemic inflammation (11, 65, 66).
The evidence for an impact of inflammation on dopamine, norepinephrine and serotonin neurotransmission is summarized in Box 2. There is already a vast literature on the participation of the fronto-striatal dopaminergic neurocircuitry in reward-based decision making (28, 29). Dysfunction of this network is responsible for the reduced anticipation and motivation that are at the core of anhedonia in major depressive disorders. As mentioned above, fatigue is associated with alterations in sustained response to reward. This implies that the neural circuit involved in fatigue overlaps with the neuronal network that underlies reward-based decision making. The basic elements of the fronto-striatal network including the basal ganglia and frontal cortex have been found to be the targets of inflammatory mediators (30–33) as well as the site of significant alterations in activity and function in chronic inflammatory conditions (e.g., multiple sclerosis and Parkinson’s disease) (34–36).
Box 2. The neurochemical basis of fatigue.
Most of the studies of the effects of inflammation on brain neurotransmitters focus on dopamine, norepinephrine and serotonin. Dopaminergic neurotransmission is very sensitive to inflammation. At the periphery, the production of neopterin and nitric oxide during inflammation consumes tetrahydrobiopterin to the detriment of the hydroxylase enzymes that use this compound as a cofactor (67). This results in the decreased bioavailability of dihydroxyphenylalanine and tyrosine for the synthesis of dopamine. In the brain, microglia activation negatively affects dopaminergic neurotransmission and sensitizes dopaminergic neurons to neurotoxins (30, 68). Inflammatory mediators can also inhibit D2 dopaminergic receptor activation indirectly by activating striatal adenosine A2A receptors (14, 69). In addition, cytokines can enhance dopamine transporter activity, resulting in decreasing synaptic availability of dopamine. Most of these studies have been done in condition of acute inflammation so that the generalization to chronic inflammation is not possible. Chronic administration of IFN-α to rhesus monkeys has been found to decrease D2 binding and striatal dopamine release in association with reduced sucrose consumption (70).
Fatigue and decreased vigilance are directly linked to impaired brain norepinephrine transmission (71). In addition to the peripheral effect of inflammation on the bioavailability of norepinephrine precursors noted above, there is evidence from discrete lesion studies in rats injected with the cytokine inducer lipopolysaccharide that the inflammation-induced reduction in exploratory behavior is mediated by catecholaminergic projections from the ventrolateral medulla and nucleus tractus solitarius to the ventral tegmental area, hypothalamus, dorsal striatum and hippocampus (72).
Serotoninergic neurotransmission can be impaired during inflammation because of cytokine-induced increase in serotonin transporter activity and increased metabolism of tryptophan to kynurenine in response to activation of indoleamine 2,3 dioxygenase (11, 73, 74). This last reaction is ultimately responsible for the formation of neurotoxic kynurenine metabolites such as 3-hydroxy kynurenine and quinolinic acid. There is evidence that kynurenine metabolites are responsible for the development of inflammation-induced depression (75). Association studies also support a role for these compounds in fatigue in cancer patients (76) and elderly subjects (77) but the causality has not yet been tested.
Finally, the development of lipopolysaccharide-induced lethargy has been attributed to a GABA-mediated inhibition of orexin containing neurons in the perifornical lateral hypothalamic area (78, 79). The involvement of orexin in fatigue appears to have some degree of specificity, as centrally administered orexin counteracted lipopolysaccharide-induced lethargy but not anorexia in rats (79).
Consistent with this model, the increase in depressed mood that is caused by the administration of a low dose of endotoxin to healthy volunteers was associated with decreased ventral striatum reactivity to monetary reward cues (37). Using functional magnetic resonance imaging (fMRI), the ventral striatum response to hedonic reward was also found to be reduced in hepatitis C patients treated with IFNα and this effect was correlated with scores of fatigue and depression (38). Although these results are strongly suggestive of an association between inflammation and decreased sensitivity to reward, it is still necessary to qualify the precise nature of this deficit. It should be possible to measure inflammation-induced impairment in motivation and reward-based decision making in fatigued patients using the Effort Expenditure for Rewards Task, a translational measure of reward motivation (39) developed as a human analog of the concurrent performance task used in rodents (Fig. 2) (14).
Fig. 2.

A model for deconstructing fatigue in neurobehavioral units associated with dysfunction of the fronto-striatal network in response to microglia activation and activation of the anterior insula by interoceptive visceral afferents. Note that the connection from the insula to the ventral striatum involves the anterior cingulate cortex (not shown).
The ventral striatum plays a key role in mediating the rewarding aspects of stimuli through its dopaminergic innervation (40, 41). It allows the learning of goal-directed responses as the selection of appropriate actions requires the assessment of the incentive value of response outcomes. Once fully learned, goal-directed responses normally become habits. Behavior is then driven by contextual cues through stimulus-response associations rather than by response outcomes. The predominance of stimulus-response over response-outcome associations is made possible by a switch from the ventral to the dorsal striatum in rodents (caudate to putamen in humans) (41). Prefrontal cortical regions are required for the updating of response-outcome associations necessary for formation of new habits (42). Formation and maintenance of habits have primarily been studied in the context of drug addiction because of the compulsive nature of drug seeking. However, these findings can be fruitfully applied to fatigue. We propose here that inflammation-induced impairment in fronto-striatal circuits impairs the formation of habits and therefore render even simple everyday activities effortful for fatigued patients (Fig. 2). This would account for the cognitive fatigability of fatigued patients. This hypothesis could be tested by comparing the ability of fatigued and non-fatigued subjects to perform a conditional associative learning task in which there is normally a gradual transition from goal-directed actions to habitual responses (43). The emergence of habitual responses should be more vulnerable to fatigue than the early phase dominated by goal-directed actions.
As noted above, fatigue refers not only to the diminished capacity to engage in self-motivated behavior but is also a feeling. Awareness of fatigue is triggered by interoceptive stimuli arising from the activation of visceral afferents that monitor the condition of tissues of the body (Fig. 2). As proposed by Craig (44, 45), this interoceptive system in primates is composed of autonomic afferent fibers that project to lamina I neurons and the nucleus tractus solitarius, relay in the parabrachial nucleus and ventromedial and mediodorsal nucleus of the primate thalamus, and ultimately terminate in the limbic sensory (insula) and motor (cingulate) cortices. In other mammals including rodents interoceptive inputs do not project to the thalamus. Input-output loops operating at different levels of organization of the interoceptive sensation system are at the origin of somato-autonomic reflexes. According to this view, awareness of fatigue would take place in the insula while its motivational dimension would be dependent on the anterior cingulate cortex via its output to the basal ganglia (Fig. 2).
This mode of representation of internal feelings matches the results of functional imaging studies. For example, Harrison and colleagues studied the interaction between systemic inflammation produced by typhoid vaccination (as measured by increased circulating IL-6) and the enhanced cognitive demands of a Stroop task that required processing of incongruent versus congruent stimuli (46). Typhoid vaccination activated afferent interoceptive fibers within the vagus nerve and spinal lamina I pathway. This information reached the cingulate and prefrontal cortex via the right medial thalamus, and also the dorsal mid/posterior insula. Inflammation-associated fatigue, as measured by the Profile of Mood States questionnaire, was correlated positively with activity changes in the mid/posterior insula bilaterally. Of note, activation of the insula was not due to efferent autonomic changes as it remained significant after accounting for changes in blood pressure.
An alternative explanation for this pattern of results is that in these experiments subjects had to make a choice while their default bias would have been to opt for doing nothing (47). The default attitude of status quo would normally decrease activity in the anterior insula and avoid the generation of anticipatory somatic markers of risky, aversive events as a result of the activation of the insula (48, 49). In this context, activation of the insula would be the consequence of switching away from the default and running into the risk of trying to do something that would be inappropriate, with potential risk proportional to fatigue severity. This would explain why activation of the insula was also correlated with mental confusion in subjects who had received typhoid vaccination (46). As the insula is also involved in processing information about risk and uncertainty (50), the decreased ability to rely on habitual behavior to respond to environmental demands could add to contextual complexity and ambiguity and further enhance insula activation (Fig. 2).
Conclusion and perspectives
There is a wide consensus that inflammation plays a key role in the development and persistence of fatigue in patients suffering from physical illness. We have seen that this hypothesis is supported by a number of clinical studies demonstrating associations between fatigue and biomarkers of inflammation and by preclinical studies in which animals exposed to inflammatory stimuli behave in a way reminiscent of fatigue, i.e., they display reduced motor activity and incentive motivation. Among the interventions that are proposed to alleviate fatigue there are several treatments aimedat counteracting inflammation. However, their clinical efficacy remains dubious (Box#3). The subjective, patient-reported nature of fatigue has made it difficult to identify mechanisms and targets for treatment. This is not to say that patient-reported outcomes are useless. Provided their psychometric properties have been carefully validated, symptom assessment scales represent valuable tools for describing symptom trajectories and capturing the impact of treatments on patient functioning and well-being (51, 52). This information can be used to inform clinical decisions and lower treatment burden (thus increasing compliance rates for prescribed treatments), develop more tolerable drugs, and test and approve new treatment methods. However, here we have tried to show that a better understanding of fatigue requires more than consideration of patient-reported outcomes. Specifically, this will necessitate deconstructing fatigue in a number of objectively defined constructs or endophenotypes that correspond to “changes in well-defined behavioral or cognitive processes associated with discrete deficits in defined neural systems” (53). Such a task is at hand as we already have an etiological factor for inducing fatigue, in the form of inflammation. It is now important to establish and validate basic neurobehavioral units of fatigue in order to be able to draw fruitful parallels between animal and human studies of fatigue.
Box 3. In search of a cure for fatigue.
Despite all the claims that are made, there is no effective cure for fatigue. Numerous strategies have been put forward to ameliorate symptoms of fatigue, but with mitigated success. Clinicaltrials.gov lists nearly 2,000 clinical trials for fatigue, with most for cancer-related fatigue, arthritis, and multiple sclerosis. In view of the key role of inflammation in the onset of fatigue, several pharmacological and nutritional strategies can be proposed to limit inflammation and its effect on the brain.
Targeting proinflammatory cytokines with anti-cytokine strategies, particularly TNFα, improves quality of life and relieves symptoms of fatigue. Most of the studies using anti-cytokine strategies have been carried out in patients with rheumatoid arthritis, as taming the inflammatory process is the primary outcome. Alternative approaches based on modulation of inflammation by dietary supplements targeting NF-kappaB activation or oxidative stress have achieved impressive positive results in animal models (80, 81). However, controlled clinical trials are very difficult to run and the low bioavailability of these supplements represents a major obstacle. The second-generation tetracycline minocycline has the advantage of targeting inflammation both at the periphery and in the brain, where it down-regulates microglia activation (82), but its possible anti-fatigue efficacy is not yet documented.
The possibility of inhibiting the consequences of activation of indoleamine 2,3 dioxygenase and guanosine triphosphate (GTP)-cyclo hydrolase 1 on the generation of kynurenine and the relative deficit of tetrahydrobiopterin is worth considering. However, clinical trials of 1-methyl tryptophan, a competitive inhibitor of indoleamine 2,3 dioxygenase, are still at a very early stage where the emphasis is on tolerability and side effects. Tetrahydrobiopterin supplementation is currently being considered only in the context of cardiac dysfunction.
In terms of neurotransmission, serotoninergic neurotransmission could be targeted via specific serotonin reuptake inhibitors (SSRIs), especially in view of the upregulation of the serotonin transporter by inflammation. However, SSRIs do not alleviate the symptoms of fatigue in depressed patients, and the same applies to cytokine-induced fatigue in somatic patients. Targeting dopamine with the norepinephrine-dopamine reuptake inhibitor bupropion is more effective, although the usefulness of this drug in non-depressed patients remains to be assessed. Psychostimulants such as methylphenidate can provide some short-term relief for fatigue. The efficacy of modafinil or its R-enantiomer armodafinil requires further investigation since most of the available results have been obtained in non-controlled studies. Amantadine is a non-competitive antagonist of NMDA receptors with a weak dopaminergic activity that is widely used for the treatment of fatigue in multiple sclerosis and Parkinson’s disease although its efficacy is still debated. It was initially developed as an anti-viral drug and has some inhibitory effects on microglial activation.
Holistic approaches (exercise, acupuncture, yoga, tai-chi, meditation) are often advocated for the treatment of fatigue. Besides its direct action on muscle physiology and cardiovascular function, physical exercise induces brain neurotrophic factors, which could account for its positive effects on fatigue and depression (83–85). Cognitive behavioral therapy has been claimed to improve the symptoms of fatigue in patients with chronic fatigue syndrome and fibromyalgia. Fatigued patients view themselves as fatigued and unable to engage in any physical activity, which contributes to the perpetuation of their feelings of fatigue (86). This can be aggravated by negative thoughts of anxiety and helplessness.
In most of the studies that have been conducted on the treatment of fatigue, there has been no attempt to dissociate those components of fatigue that respond to therapy from those that are unaffected. Further studies are needed to investigate the effects of therapies targeting specific pathophysiological pathways to ultimately impact the specific dimensions of fatigue.
Highlights.
Despite its prevalence the pathophysiology of fatigue remains elusive
We focus on the etiology of fatigue in chronic inflammatory diseases, cancer or neuropathologies
Fatigue can be decomposed into mental and physical aspects
Convergent data from many disciplines point to the importance of inflammation in the pathophysiology of fatigue
Peripheral inflammation can ultimately disrupt monoaminergic neurotranmission
Resulting alteration in fronto-striatal/insular networks underlies aspects of fatigue
Acknowledgments
Research reported here was supported by the National Institute of Neurological Diseases and Stroke of the National Institutes of Health under award numbers RO1NS073939 and RO1NS074999. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. RD is supported by MDACC research funds. AK is supported by a STARS award of the University of Texas System.
Footnotes
Conflict of interest
R. Dantzer works as a consultant for Ironwood Pharma, Cambridge, MA
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Riley WT, Rothrock N, Bruce B, Christodolou C, Cook K, Hahn EA, et al. Patient-reported outcomes measurement information system (PROMIS) domain names and definitions revisions: further evaluation of content validity in IRT-derived item banks. Qual Life Res. 2010;19(9):1311–21. doi: 10.1007/s11136-010-9694-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaudhuri A, Behan PO. Fatigue and basal ganglia. J Neurol Sci. 2000;179(S 1–2):34–42. doi: 10.1016/s0022-510x(00)00411-1. [DOI] [PubMed] [Google Scholar]
- 3.Kroenke K, Price RK. Symptoms in the community. Prevalence, classification, and psychiatric comorbidity. Arch Intern Med. 1993;153(21):2474–80. [PubMed] [Google Scholar]
- 4.van’t Leven M, Zielhuis GA, van der Meer JW, Verbeek AL, Bleijenberg G. Fatigue and chronic fatigue syndrome-like complaints in the general population. Eur J Public Health. 2010;20(3):251–7. doi: 10.1093/eurpub/ckp113. [DOI] [PubMed] [Google Scholar]
- 5.Kroenke K, Stump T, Clark DO, Callahan CM, McDonald CJ. Symptoms in hospitalized patients: outcome and satisfaction with care. Am J Med. 1999;107(5):425–31. doi: 10.1016/s0002-9343(99)00268-5. [DOI] [PubMed] [Google Scholar]
- 6.Leavitt VM, DeLuca J. Central fatigue: issues related to cognition, mood and behavior, and psychiatric diagnoses. PM & R: the journal of injury, function, and rehabilitation. 2010;2(5):332–7. doi: 10.1016/j.pmrj.2010.03.027. [DOI] [PubMed] [Google Scholar]
- 7.Bandura A. Health promotion by social cognitive means. Health education & behavior: the official publication of the Society for Public Health Education. 2004;31(2):143–64. doi: 10.1177/1090198104263660. [DOI] [PubMed] [Google Scholar]
- 8.McAndrew LM, Musumeci-Szabo TJ, Mora PA, Vileikyte L, Burns E, Halm EA, et al. Using the common sense model to design interventions for the prevention and management of chronic illness threats: from description to process. British journal of health psychology. 2008;13(Pt 2):195–204. doi: 10.1348/135910708X295604. [DOI] [PubMed] [Google Scholar]
- 9.McAuley E, White SM, Rogers LQ, Motl RW, Courneya KS. Physical activity and fatigue in breast cancer and multiple sclerosis: psychosocial mechanisms. Psychosomatic medicine. 2010;72(1):88–96. doi: 10.1097/PSY.0b013e3181c68157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hart BL. Biological basis of the behavior of sick animals. Neuroscience and biobehavioral reviews. 1988;12(2):123–37. doi: 10.1016/s0149-7634(88)80004-6. [DOI] [PubMed] [Google Scholar]
- 11.Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9(1):46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aubert A, Goodall G, Dantzer R, Gheusi G. Differential effects of lipopolysaccharide on pup retrieving and nest building in lactating mice. Brain, behavior, and immunity. 1997;11(2):107–18. doi: 10.1006/brbi.1997.0485. [DOI] [PubMed] [Google Scholar]
- 13.Larson SJ, Romanoff RL, Dunn AJ, Glowa JR. Effects of interleukin-1beta on food-maintained behavior in the mouse. Brain, behavior, and immunity. 2002;16(4):398–410. doi: 10.1006/brbi.2001.0634. [DOI] [PubMed] [Google Scholar]
- 14.Nunes E, PR, Estrada A, Epling B, Hart E, Lee C, et al. Effort-related motivational effects of the proinflammatory cytokine interleukin 1-beta: studies with the concurrent fixed ratio 5/chow feeding choice task. Psychopharmacology. 2013 doi: 10.1007/s00213-013-3285-4. accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cuthbert BN, Insel TR. Toward the future of psychiatric diagnosis: the seven pillars of RDoC. BMC medicine. 2013;11:126. doi: 10.1186/1741-7015-11-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Raaf PJ, Sleijfer S, Lamers CH, Jager A, Gratama JW, van der Rijt CC. Inflammation and fatigue dimensions in advanced cancer patients and cancer survivors: an explorative study. Cancer. 2012;118(23):6005–11. doi: 10.1002/cncr.27613. [DOI] [PubMed] [Google Scholar]
- 17.Miller AH, Ancoli-Israel S, Bower JE, Capuron L, Irwin MR. Neuroendocrine-immune mechanisms of behavioral comorbidities in patients with cancer. J Clin Oncol. 2008;26(6):971–82. doi: 10.1200/JCO.2007.10.7805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heesen C, Nawrath L, Reich C, Bauer N, Schulz KH, Gold SM. Fatigue in multiple sclerosis: an example of cytokine mediated sickness behaviour? J Neurol Neurosurg Psychiatry. 2006;77(1):34–9. doi: 10.1136/jnnp.2005.065805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bower JE, Lamkin DM. Inflammation and cancer-related fatigue: mechanisms, contributing factors, and treatment implications. Brain Behav Immun. 2013;30 (Suppl):S48–57. doi: 10.1016/j.bbi.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saligan LN, Kim HS. A systematic review of the association between immunogenomic markers and cancer-related fatigue. Brain Behav Immun. 2012;26(6):830–48. doi: 10.1016/j.bbi.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.ter Wolbeek M, van Doornen LJ, Kavelaars A, van de Putte EM, Schedlowski M, Heijnen CJ. Longitudinal analysis of pro- and anti-inflammatory cytokine production in severely fatigued adolescents. Brain, behavior, and immunity. 2007;21(8):1063–74. doi: 10.1016/j.bbi.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 22.van Zuiden M, Geuze E, Maas M, Vermetten E, Heijnen CJ, Kavelaars A. Deployment-related severe fatigue with depressive symptoms is associated with increased glucocorticoid binding to peripheral blood mononuclear cells. Brain, behavior, and immunity. 2009;23(8):1132–9. doi: 10.1016/j.bbi.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 23.Capuron L, Gumnick JF, Musselman DL, Lawson DH, Reemsnyder A, Nemeroff CB, et al. Neurobehavioral effects of interferon-alpha in cancer patients: phenomenology and paroxetine responsiveness of symptom dimensions. Neuropsychopharmacology. 2002;26(5):643–52. doi: 10.1016/S0893-133X(01)00407-9. [DOI] [PubMed] [Google Scholar]
- 24.Yount S, Sorensen MV, Cella D, Sengupta N, Grober J, Chartash EK. Adalimumab plus methotrexate or standard therapy is more effective than methotrexate or standard therapies alone in the treatment of fatigue in patients with active, inadequately treated rheumatoid arthritis. Clin Exp Rheumatol. 2007;25(6):838–46. [PubMed] [Google Scholar]
- 25.Tyring S, Gottlieb A, Papp K, Gordon K, Leonardi C, Wang A, et al. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet. 2006;367(9504):29–35. doi: 10.1016/S0140-6736(05)67763-X. [DOI] [PubMed] [Google Scholar]
- 26.Lou JS. Physical and mental fatigue in Parkinson’s disease: epidemiology, pathophysiology and treatment. Drugs Aging. 2009;26(3):195–208. doi: 10.2165/00002512-200926030-00002. [DOI] [PubMed] [Google Scholar]
- 27.Capuron L, Miller AH. Immune system to brain signaling: neuropsychopharmacological implications. Pharmacol Ther. 2011;130(2):226–38. doi: 10.1016/j.pharmthera.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kurniawan IT, Guitart-Masip M, Dolan RJ. Dopamine and effort-based decision making. Frontiers in neuroscience. 2011;5:81. doi: 10.3389/fnins.2011.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salamone JD, Correa M, Nunes EJ, Randall PA, Pardo M. The behavioral pharmacology of effort-related choice behavior: dopamine, adenosine and beyond. Journal of the experimental analysis of behavior. 2012;97(1):125–46. doi: 10.1901/jeab.2012.97-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hirsch EC, Vyas S, Hunot S. Neuroinflammation in Parkinson’s disease. Parkinsonism & related disorders. 2012;18 (Suppl 1):S210–2. doi: 10.1016/S1353-8020(11)70065-7. [DOI] [PubMed] [Google Scholar]
- 31.Hernandez-Romero MC, Delgado-Cortes MJ, Sarmiento M, de Pablos RM, Espinosa-Oliva AM, Arguelles S, et al. Peripheral inflammation increases the deleterious effect of CNS inflammation on the nigrostriatal dopaminergic system. Neurotoxicology. 2012;33(3):347–60. doi: 10.1016/j.neuro.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 32.Herrera AJ, Castano A, Venero JL, Cano J, Machado A. The single intranigral injection of LPS as a new model for studying the selective effects of inflammatory reactions on dopaminergic system. Neurobiology of disease. 2000;7(4):429–47. doi: 10.1006/nbdi.2000.0289. [DOI] [PubMed] [Google Scholar]
- 33.Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2000;20(16):6309–16. doi: 10.1523/JNEUROSCI.20-16-06309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chaudhuri A, Behan PO. Fatigue in neurological disorders. Lancet. 2004;363(9413):978–88. doi: 10.1016/S0140-6736(04)15794-2. [DOI] [PubMed] [Google Scholar]
- 35.DeLuca J, Genova HM, Capili EJ, Wylie GR. Functional neuroimaging of fatigue. Physical medicine and rehabilitation clinics of North America. 2009;20(2):325–37. doi: 10.1016/j.pmr.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 36.Roelcke U, Kappos L, Lechner-Scott J, Brunnschweiler H, Huber S, Ammann W, et al. Reduced glucose metabolism in the frontal cortex and basal ganglia of multiple sclerosis patients with fatigue: a 18F-fluorodeoxyglucose positron emission tomography study. Neurology. 1997;48(6):1566–71. doi: 10.1212/wnl.48.6.1566. [DOI] [PubMed] [Google Scholar]
- 37.Eisenberger NI, Berkman ET, Inagaki TK, Rameson LT, Mashal NM, Irwin MR. Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol Psychiatry. 2010;68(8):748–54. doi: 10.1016/j.biopsych.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Capuron L, Pagnoni G, Drake DF, Woolwine BJ, Spivey JR, Crowe RJ, et al. Dopaminergic mechanisms of reduced basal ganglia responses to hedonic reward during interferon alfa administration. Arch Gen Psychiatry. 2012;69(10):1044–53. doi: 10.1001/archgenpsychiatry.2011.2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Treadway MT, Bossaller NA, Shelton RC, Zald DH. Effort-based decision-making in major depressive disorder: a translational model of motivational anhedonia. Journal of abnormal psychology. 2012;121(3):553–8. doi: 10.1037/a0028813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yin HH, Knowlton BJ. The role of the basal ganglia in habit formation. Nature reviews Neuroscience. 2006;7(6):464–76. doi: 10.1038/nrn1919. [DOI] [PubMed] [Google Scholar]
- 41.Everitt BJ, Robbins TW. From the ventral to the dorsal striatum: Devolving views of their roles in drug addiction. Neuroscience and biobehavioral reviews. 2013 doi: 10.1016/j.neubiorev.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 42.Ashby FG, Turner BO, Horvitz JC. Cortical and basal ganglia contributions to habit learning and automaticity. Trends in cognitive sciences. 2010;14(5):208–15. doi: 10.1016/j.tics.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hadj-Bouziane F, Benatru I, Brovelli A, Klinger H, Thobois S, Broussolle E, et al. Advanced Parkinson’s disease effect on goal-directed and habitual processes involved in visuomotor associative learning. Frontiers in human neuroscience. 2012;6:351. doi: 10.3389/fnhum.2012.00351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Craig AD. Significance of the insula for the evolution of human awareness of feelings from the body. Annals of the New York Academy of Sciences. 2011;1225:72–82. doi: 10.1111/j.1749-6632.2011.05990.x. [DOI] [PubMed] [Google Scholar]
- 45.Craig AD. How do you feel--now? The anterior insula and human awareness. Nature reviews Neuroscience. 2009;10(1):59–70. doi: 10.1038/nrn2555. [DOI] [PubMed] [Google Scholar]
- 46.Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Dolan RJ, et al. Neural origins of human sickness in interoceptive responses to inflammation. Biological psychiatry. 2009;66(5):415–22. doi: 10.1016/j.biopsych.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anderson CJ. The psychology of doing nothing: forms of decision avoidance result from reason and emotion. Psychological bulletin. 2003;129(1):139–67. doi: 10.1037/0033-2909.129.1.139. [DOI] [PubMed] [Google Scholar]
- 48.Preuschoff K, Quartz SR, Bossaerts P. Human insula activation reflects risk prediction errors as well as risk. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28(11):2745–52. doi: 10.1523/JNEUROSCI.4286-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu R, Mobbs D, Seymour B, Calder AJ. Insula and striatum mediate the default bias. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30(44):14702–7. doi: 10.1523/JNEUROSCI.3772-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Singer T, Critchley HD, Preuschoff K. A common role of insula in feelings, empathy and uncertainty. Trends in cognitive sciences. 2009;13(8):334–40. doi: 10.1016/j.tics.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 51.Eton DT, Elraiyah TA, Yost KJ, Ridgeway JL, Johnson A, Egginton JS, et al. A systematic review of patient-reported measures of burden of treatment in three chronic diseases. Patient related outcome measures. 2013;4:7–20. doi: 10.2147/PROM.S44694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cleeland CS, Fisch MJ, Dunn AJ, editors. Measurement, Mechanisms, and Management. Cambridge: Cambridge University Press; 2011. Cancer Symptom Science. [Google Scholar]
- 53.Robbins TW, Gillan CM, Smith DG, de Wit S, Ersche KD. Neurocognitive endophenotypes of impulsivity and compulsivity: towards dimensional psychiatry. Trends in cognitive sciences. 2012;16(1):81–91. doi: 10.1016/j.tics.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 54.Rivest S. Molecular insights on the cerebral innate immune system. Brain Behav Immun. 2003;17(1):13–9. doi: 10.1016/s0889-1591(02)00055-7. [DOI] [PubMed] [Google Scholar]
- 55.Konsman JP, Parnet P, Dantzer R. Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci. 2002;25(3):154–9. doi: 10.1016/s0166-2236(00)02088-9. [DOI] [PubMed] [Google Scholar]
- 56.Quan N. Immune-to-brain signaling: how important are the blood-brain barrier-independent pathways? Mol Neurobiol. 2008;37(2–3):142–52. doi: 10.1007/s12035-008-8026-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Banks WA, Erickson MA. The blood-brain barrier and immune function and dysfunction. Neurobiol Dis. 2010;37(1):26–32. doi: 10.1016/j.nbd.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 58.Saper CB, Romanovsky AA, Scammell TE. Neural circuitry engaged by prostaglandins during the sickness syndrome. Nat Neurosci. 2012;15(8):1088–95. doi: 10.1038/nn.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fontana A, Weber E, Dayer JM. Synthesis of interleukin 1/endogenous pyrogen in the brain of endotoxin-treated mice: a step in fever induction? Journal of immunology. 1984;133(4):1696–8. [PubMed] [Google Scholar]
- 60.Gatti S, Bartfai T. Induction of tumor necrosis factor-alpha mRNA in the brain after peripheral endotoxin treatment: comparison with interleukin-1 family and interleukin-6. Brain Res. 1993;624(1–2):291–4. doi: 10.1016/0006-8993(93)90090-a. [DOI] [PubMed] [Google Scholar]
- 61.Laye S, Parnet P, Goujon E, Dantzer R. Peripheral administration of lipopolysaccharide induces the expression of cytokine transcripts in the brain and pituitary of mice. Brain Res Mol Brain Res. 1994;27(1):157–62. doi: 10.1016/0169-328x(94)90197-x. [DOI] [PubMed] [Google Scholar]
- 62.Van Dam AM, Bauer J, Tilders FJ, Berkenbosch F. Endotoxin-induced appearance of immunoreactive interleukin-1 beta in ramified microglia in rat brain: a light and electron microscopic study. Neuroscience. 1995;65(3):815–26. doi: 10.1016/0306-4522(94)00549-k. [DOI] [PubMed] [Google Scholar]
- 63.Laflamme N, Lacroix S, Rivest S. An essential role of interleukin-1beta in mediating NF-kappaB activity and COX-2 transcription in cells of the blood-brain barrier in response to a systemic and localized inflammation but not during endotoxemia. J Neurosci. 1999;19(24):10923–30. doi: 10.1523/JNEUROSCI.19-24-10923.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Corona AW, Huang Y, O’Connor JC, Dantzer R, Kelley KW, Popovich PG, et al. Fractalkine receptor (CX3CR1) deficiency sensitizes mice to the behavioral changes induced by lipopolysaccharide. J Neuroinflammation. 2010;7:93. doi: 10.1186/1742-2094-7-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Imeri L, Opp MR. How (and why) the immune system makes us sleep. Nature reviews Neuroscience. 2009;10(3):199–210. doi: 10.1038/nrn2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rivest S. Interactions between the immune and neuroendocrine systems. Prog Brain Res. 2010;181:43–53. doi: 10.1016/S0079-6123(08)81004-7. [DOI] [PubMed] [Google Scholar]
- 67.Neurauter G, Schrocksnadel K, Scholl-Burgi S, Sperner-Unterweger B, Schubert C, Ledochowski M, et al. Chronic immune stimulation correlates with reduced phenylalanine turnover. Curr Drug Metab. 2008;9(7):622–7. doi: 10.2174/138920008785821738. [DOI] [PubMed] [Google Scholar]
- 68.Deleidi M, Hallett PJ, Koprich JB, Chung CY, Isacson O. The Toll-like receptor-3 agonist polyinosinic:polycytidylic acid triggers nigrostriatal dopaminergic degeneration. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30(48):16091–101. doi: 10.1523/JNEUROSCI.2400-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hanff TC, Furst SJ, Minor TR. Biochemical and anatomical substrates of depression and sickness behavior. The Israel journal of psychiatry and related sciences. 2010;47(1):64–71. [PubMed] [Google Scholar]
- 70.Felger JC, Mun J, Kimmel HL, Nye JA, Drake DF, Hernandez CR, et al. Chronic Interferon-alpha Decreases Dopamine 2 Receptor Binding and Striatal Dopamine Release in Association with Anhedonia-Like Behavior in Nonhuman Primates. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2013;38(11):2179–87. doi: 10.1038/npp.2013.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Blier P, Briley M. The noradrenergic symptom cluster: clinical expression and neuropharmacology. Neuropsychiatric disease and treatment. 2011;7(Suppl 1):15–20. doi: 10.2147/NDT.S19613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gaykema RP, Goehler LE. Ascending caudal medullary catecholamine pathways drive sickness-induced deficits in exploratory behavior: brain substrates for fatigue? Brain, behavior, and immunity. 2011;25(3):443–60. doi: 10.1016/j.bbi.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.O’Connor JC, Lawson MA, Andre C, Moreau M, Lestage J, Castanon N, et al. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Molecular psychiatry. 2009;14(5):511–22. doi: 10.1038/sj.mp.4002148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.O’Connor JC, Lawson MA, Andre C, Briley EM, Szegedi SS, Lestage J, et al. Induction of IDO by bacille Calmette-Guerin is responsible for development of murine depressive-like behavior. Journal of immunology. 2009;182(5):3202–12. doi: 10.4049/jimmunol.0802722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Walker AK, Budac DP, Bisulco S, Lee AW, Smith RA, Beenders B, et al. NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2013;38(9):1609–16. doi: 10.1038/npp.2013.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kurz K, Fiegl M, Holzner B, Giesinger J, Pircher M, Weiss G, et al. Fatigue in patients with lung cancer is related with accelerated tryptophan breakdown. PloS one. 2012;7(5):e36956. doi: 10.1371/journal.pone.0036956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Capuron L, Schroecksnadel S, Feart C, Aubert A, Higueret D, Barberger-Gateau P, et al. Chronic low-grade inflammation in elderly persons is associated with altered tryptophan and tyrosine metabolism: role in neuropsychiatric symptoms. Biol Psychiatry. 2011;70(2):175–82. doi: 10.1016/j.biopsych.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 78.Gaykema RP, Goehler LE. Lipopolysaccharide challenge-induced suppression of Fos in hypothalamic orexin neurons: their potential role in sickness behavior. Brain, behavior, and immunity. 2009;23(7):926–30. doi: 10.1016/j.bbi.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Grossberg AJ, Zhu X, Leinninger GM, Levasseur PR, Braun TP, Myers MG, Jr, et al. Inflammation-induced lethargy is mediated by suppression of orexin neuron activity. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31(31):11376–86. doi: 10.1523/JNEUROSCI.2311-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rahman I, Biswas SK, Kirkham PA. Regulation of inflammation and redox signaling by dietary polyphenols. Biochem Pharmacol. 2006;72(11):1439–52. doi: 10.1016/j.bcp.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 81.Calder PC. Fatty acids and inflammation: the cutting edge between food and pharma. Eur J Pharmacol. 2011;668 (Suppl 1):S50–8. doi: 10.1016/j.ejphar.2011.05.085. [DOI] [PubMed] [Google Scholar]
- 82.Zemke D, Majid A. The potential of minocycline for neuroprotection in human neurologic disease. Clin Neuropharmacol. 2004;27(6):293–8. doi: 10.1097/01.wnf.0000150867.98887.3e. [DOI] [PubMed] [Google Scholar]
- 83.Dal Bello-Haas V, Florence JM. Therapeutic exercise for people with amyotrophic lateral sclerosis or motor neuron disease. Cochrane Database Syst Rev. 2013;5:CD005229. doi: 10.1002/14651858.CD005229.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cramp F, Byron-Daniel J. Exercise for the management of cancer-related fatigue in adults. Cochrane Database Syst Rev. 2012;11:CD006145. doi: 10.1002/14651858.CD006145.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Payne C, Wiffen PJ, Martin S. Interventions for fatigue and weight loss in adults with advanced progressive illness. Cochrane Database Syst Rev. 2012;1:CD008427. doi: 10.1002/14651858.CD008427.pub2. [DOI] [PubMed] [Google Scholar]
- 86.Knoop H, Prins JB, Moss-Morris R, Bleijenberg G. The central role of cognitive processes in the perpetuation of chronic fatigue syndrome. J Psychosom Res. 2010;68(5):489–94. doi: 10.1016/j.jpsychores.2010.01.022. [DOI] [PubMed] [Google Scholar]