

Abstract
The complete 154-kbp linear double-stranded genomic DNA sequence of herpes simplex virus 2 (HSV-2), consisting of two extended regions of unique sequences bounded by a pair of inverted repeat elements, was published in 1998 and since then has been widely employed in a wide range of studies. Throughout the HSV-2 genome are scattered 150 microsatellites (also referred to as short tandem repeats) of 1- to 6-nucleotide motifs, mainly distributed in noncoding regions. Microsatellites are considered reliable markers for genetic mapping to differentiate herpesvirus strains, as shown for cytomegalovirus and HSV-1. The aim of this work was to characterize 12 polymorphic microsatellites within the HSV-2 genome by use of 3 multiplex PCR assays in combination with length polymorphism analysis for the rapid genetic differentiation of 56 HSV-2 clinical isolates and 2 HSV-2 laboratory strains (gHSV-2 and MS). This new system was applied to a specific new HSV-2 variant recently identified in HIV-1-infected patients originating from West Africa. Our results confirm that microsatellite polymorphism analysis is an accurate tool for studying the epidemiology of HSV-2 infections.
INTRODUCTION
Herpes simplex virus 2 (HSV-2), classified together with HSV-1 in the Alphaherpesvirinae subfamily, is a highly prevalent sexually transmitted infection worldwide. This virus shows prevalence rates ranging from 15 to 20% in Europe and the United States to 90% in some sub-Saharan African populations (1). After primary infection, HSV-2 has the ability to establish lifelong latent infections in the host sensory sacral ganglia and may reactivate frequently, with clinical manifestations varying from asymptomatic viral shedding to painful genital vesicular lesions. The symptoms are usually self-limiting in immunocompetent individuals, whereas severe disseminated disease, sometimes debilitating, may occur in immunocompromised patients. HSV-2 may also cause neonatal herpes after vertical transmission from mother to newborn (2, 3).
The HSV-2 genome consists of a linear double-stranded genomic DNA sequence of 154 kbp (strain HG52; GenBank accession no. Z86099), composed of two extended regions of unique sequences (i.e., unique long [UL] and unique short [US]) bounded by a pair of terminal repeat and internal inverted repeat elements and over 74 open reading frames (4, 5). The occurrence of endogenous reinfection or superinfection in genital HSV-2 infections has been previously documented using genomic analysis, i.e., restriction fragment length polymorphism (RFLP) analysis or multilocus sequencing assays based on the analysis of several DNA regions, yielding specific patterns of subgenomic products to distinguish unrelated HSV-2 strains (6–8). The main advantage of RFLP analysis over PCR-based and sequencing processes is that no prior nucleotide sequence data are required. However, this method remains relatively tedious to perform (9, 10). Herpesvirus genomes are quite stable and exhibit limited sequence variation (11). However, they have been shown to contain several sites with short tandem repeats of 1- to 6-nucleotide motifs known as microsatellites. Homopolymers of at least 6 consecutive nucleotides are the most abundant class of microsatellites in all viral genomes examined so far (12). Microsatellites are further classified as perfect (identical repeats), interrupted (repeats interrupted by point mutations), or compound (adjacent perfect microsatellites with different motifs) microsatellites (13, 14). Genetic variation within these elements has been reported to explain microsatellite instability through recombination processes or DNA polymerase slippage during DNA replication (15, 16). As hypervariable genomic regions, microsatellites have proven to be useful as molecular markers for epidemiological studies of viral strain diversity. To date, the identification and genetic mapping of polymorphic microsatellite markers have been reported to have high discriminatory power and high throughput for human cytomegalovirus (HCMV), HSV-1, and HIV infections (17–19). The aims of this work were to establish the map and to characterize microsatellites within the full-length genome of HSV-2 and to confirm that the analysis of microsatellite polymorphism constitutes an accurate tool for the mapping of molecular profiles of HSV-2 clinical isolates in different epidemiological situations.
MATERIALS AND METHODS
Viral strains and cells.
A total of 56 HSV-2 clinical isolates and 2 laboratory strains (gHSV-2 and MS) were included in the present study. The HSV-2 isolates were selected retrospectively from viral isolates obtained in cell culture and stored in the laboratory between 2005 and 2012, in order to obtain a range of viruses collected from both immunocompetent and immunosuppressed (mainly HIV-infected) patients and recovered from different body sites (i.e., anal, genital, buttock, ear, bronchoalveolar lavage fluid, and back specimens). This selection allowed comparison of isolates from epidemiologically unrelated patients (isolates 1 to 43), sequential isolates corresponding to recurrent episodes in the same individuals, for which resistance to antivirals was investigated (isolates 44 to 46), and isolates recently described and belonging to a new HSV-2 genetic variant (designated HSV-2v; isolates 47 to 50) (20). All HSV-2 isolates were recovered from patients originating from European countries (mainly France) except for isolates 47 to 50, which were obtained from patients from West Africa (20). Both HSV-2 clinical isolates and laboratory strains were propagated in Vero cell cultures, as described previously (21). The determination of HSV type was performed by means of a specific real-time PCR (artus HSV-1/2 kit; Qiagen, Courtaboeuf, France) or an immunofluorescence assay using type-specific monoclonal antibodies (Trinity Biotech Plc, Wicklow, Ireland). The susceptibilities to acyclovir (ACV) and foscarnet (FOS) of HSV-2 laboratory strains and clinical isolates were determined by plaque reduction assays and genotypic assays, as described previously (21). Viruses were considered to be resistant at 50% effective concentration (EC50) values of ≥7 μM and 330 μM for ACV and FOS, respectively (21, 22).
Identification of HSV-2 microsatellites by computer study.
Microsatellite identification within the whole genome of HSV-2 reference strain HG52 (GenBank accession no. Z86099) was performed using the MSatFinder program (5) (http://bioinformatics.rri.sari.ac.uk/bioinformatics/docs/documentation/msatfinder/msatfinder_manual.html). Results included only short microsatellites with at least four trinucleotide, five dinucleotide, or nine mononucleotide repeat units, as described previously (18, 23).
HSV-2 microsatellite characterization by conventional sequencing.
Viral DNA was extracted from 200 μl of viral stock (laboratory strains and clinical isolates) by using the QIAamp DNA blood minikit (Qiagen) or the MagNA Pure compact system (Roche Diagnostics, Meylan, France), according to the manufacturer's instructions. The purified nucleic acids were eluted in 100 μl of elution buffer and stored at −20°C until further analysis. PCR-amplified DNA fragments obtained with 19 different PCR systems encompassing reiterated regions corresponding to 22 microsatellites (including 6 trinucleotide, 3 dinucleotide, and 13 mononucleotide repeat units) were used to classify each microsatellite as monomorphic or polymorphic and to evaluate the number of distinct variants (Table 1). Primer design was performed using Primer3 software (24). Singleplex PCRs were performed with 10 μl of DNA extract using the proofreading enzyme Expand High Fidelity (Roche Diagnostics), in a mixture containing 10× buffer, 1.5 mM MgCl2, 800 μM deoxynucleoside triphosphates (dNTPs), and 300 μM forward and reverse primers. PCR product sizes ranged from 138 to 616 bp (Table 1). Amplification conditions included an initial denaturation step of 2 min at 96°C, 45 cycles of 1 min at 96°C, 1 min at 52.5°C, and 2 min at 68°C, and a final extension step of 2 min at 68°C. All PCRs were performed using an Eppendorf thermal cycler (Eppendorf, Le Pecq, France). Amplified products were sequenced with the Prism BigDye Terminator cycle sequencing ready reaction kit (Applied Biosystems, Courtaboeuf, France) and were analyzed with an ABI Prism 3730 automated sequencer (Applied Biosystems). All nucleotide sequences were compared with that of HSV-2 reference strain HG52 (GenBank accession no. Z86099) using SeqScape v2.5 software (5).
Table 1.
Characteristics of selected HSV-2 microsatellites and primer pairs used for conventional sequencing and length polymorphism analysis
| Microsatellite | Primer sequence (5′ to 3′)a | Expected size (bp)b | Target repeat | Positionc | Locus | No. of variant alleles (no. of variants)d |
|---|---|---|---|---|---|---|
| HSV-2 M20e | F, AAGAGTGTGTCCGTGTTGACAG | 184 | (G)9 | 5968–5976 | TRL | 6 (7) |
| R, CCCCCTCTCTATCAAAGTTCC | ||||||
| HSV-2 M28e | F, GAAATGCCCACAGCAACAC | 138 | (C)9 | 7900–7908 | TRL | 4 (5) |
| R, CACGCCCTTCCATTAAACAC | ||||||
| HSV-2 M36 | F, CCAGAACGCACGCATACA | 394 | (GC)6 | 20519–20530 | UL8 | 1 |
| R, GGGCGGTGAACTTTAGCAC | ||||||
| HSV-2 M37/M38e | F, ACCCGATCTACGACGAAGTG | 444 | (GT)6 | 24669–24680 | Between UL10 and UL11 | 3 (8) |
| R, CATCGCGTCACACCAGAC | (T)11 | 24680–24690 | 4 (8) | |||
| HSV-2 M42 | F, CTTCATGCAGGAGATCATCG | 242 | (CCA)4 | 34042–34053 | UL15 | 1 |
| R, CCAGGGCGAAGATAATGAAA | ||||||
| HSV-2 M43e | F, AGTGTTGTCCGTTGGTTGG | 163 | (AC)5 | 34981–34990 | Between UL15 and UL18 | 2 (5) |
| R, GAGCGTGTGGTTTGTGTTTATT | ||||||
| HSV-2 M50/M51e | F, ATTATTAACGCCCACGATGC | 498 | (GGT)4 | 50806–50817 | Between UL25 and UL26 | 2 (5) |
| R, AAACCCGGCCACGTAGAT | (C)12 | 50987–50998 | 5 (5) | |||
| HSV-2 M55e | F, GGAAGGCCCGGAAGACTAC | 218 | (C)13 | 53293–53305 | Between UL26 and UL27 | 6 (9) |
| R, TATAAATAAAAAGACACCGATGTTCAA | ||||||
| HSV-2 M63/M64e | F, ACGGAGGGGCTAAGTATCG | 420 | (G)9 | 84804–84812 | Between UL37 and UL38 | 7 (13) |
| R, GCTTGGTTTTCATGGTTTTCC | (C)9 | 85019–85027 | 5 (13) | |||
| HSV-2 M61 | F, AGCTCTTCCAGGGGCTTG | 279 | (G)9 | 78380–78388 | UL36 | 1 |
| R, CCACGACTGTCGGTTTCAC | ||||||
| HSV-2 M66 | F, CCGGTTGTCATCCTGGAG | 261 | (C)9 | 86512–86520 | Between UL38 and UL39 | 5 |
| R, CATGAATCCCATTTGCATGT | ||||||
| HSV-2 M72e | F, TGGTGTTTACGAACGAGTTTGA | 364 | (T)9 | 108945–108953 | Between UL50 and UL51 | 4 (5) |
| R, CTACACCGGACCCACGTC | ||||||
| HSV-2 M74 | F, GTCAAGCGCCTCCTCAAG | 340 | (GCG)4 | 111600–111611 | UL52 | 1 |
| R, CGTTGCGGTTCACGTACA | ||||||
| HSV-2 M88/M89 | F, CGTGCATGCGTTGTGATT | 461 | (C)9 | 121274–121282 | IRL | 4 |
| R, CAATAAAGTTTTGTGATGCTTTTGA | (CCT)4 | 121531–121542 | 2 | |||
| HSV-2 M102 | F, GGGGAGAGTCGCTGATGAC | 382 | (GCT)4 | 123475–123486 | RL2 | 1 |
| R, GGGCTCCCGTCTCTGTCT | ||||||
| HSV-2 M124 | F, ACAAGGCGTTGCCACTATG | 616 | (GAT)4 | 134042–134053 | US1 | 1 |
| R, GATCCGAAAGAGCTGGTTGA | ||||||
| HSV-2 M125e | F, CGCGTCCGATACAACAAAC | 483 | (G)19 | 140744–140762 | Between US4 and US5 | 8 (9) |
| R, TCGAATCGGAGTATGGTGGT | ||||||
| HSV-2 M128 | F, AGCGGTCTGTACACCCTGTC | 540 | (C)10 | 144416–144425 | US8 | 1 |
| R, AGCGGTCTGTACACCCTGTC |
F, forward; R, reverse.
Expected length of the PCR fragment according to the reference HSV-2 complete sequence (strain HG52; GenBank accession no. Z86099).
Positions in the genome according to the numbering of the same reference sequence.
Numbers of variant alleles among 2 laboratory strains (gHSV-2 and MS) and a panel of 13 clinical isolates, as determined by conventional sequencing. The numbers in parentheses indicate the numbers of sequence variants observed by length polymorphism analysis.
Microsatellite selected for multiplex PCR assay.
HSV-2 microsatellite characterization by length polymorphism analysis.
Twelve polymorphic microsatellites (corresponding to 9 loci) localized throughout the complete genome of HSV-2, i.e., M20, M28, M37, M38, M43, M50, M51, M55, M63, M64, M72, and M125, were selected for length polymorphism analysis (Fig. 1 and Table 1). This choice of microsatellite markers was based on both their polymorphic character and the size of the generated amplicons (to avoid any potential size range overlapping), in order to yield multiplex sets that were as discriminative as possible (Table 1). Three multiplex PCR systems were developed to amplify these microsatellites. Forward primers of each set were labeled with 6-carboxyfluorescein dye at the 5′ end. Three different primer pair combinations were tested to identify the optimal combination. Once suitable multiplex PCR protocols had been defined, primer pair concentrations were adapted as necessary to obtain relatively equal amplicon productions, with visualization under UV light on agarose gels stained with ethidium bromide. Multiplex PCRs were carried out using an Eppendorf thermal cycler (Eppendorf) with a reaction mixture containing 10 μl of DNA extract, the proofreading enzyme Expand High Fidelity (Roche Diagnostics), 10× buffer, 1.5 mM MgCl2, 800 μM dNTPs, and forward and reverse primers with the following concentrations: 800 nM M20 primers, 800 nM M72 primers, and 1,280 nM M37 and M38 primers for multiplex 1; 800 nM M28 primers, 800 nM M50 and M51 primers, and 800 nM M55 primers for multiplex 2; 800 nM M43 primers, 1,200 nM M63 and M64 primers, and 1,280 nM M125 primers for multiplex 3. All multiplex PCRs were performed with the same cycling conditions, namely, an initial denaturation step of 2 min at 96°C, 45 cycles of 1 min at 96°C, 1 min at 52.5°C, and 2 min at 68°C, and a final extension step of 2 min at 68°C. One microliter of the amplified multiplex PCR products was diluted with sterile water (1:200) and analyzed with an ABI Prism 3730 automated capillary sequencer (Applied Biosystems) with the GeneScan 600LIZ internal size standard. Amplicon sizes were scored using Peak Scanner v.1.0 software (Applied Biosystems). In order to assess the specificity of this multiplex assay, labeled amplicons obtained from single PCRs were secondarily mixed together and analyzed according to the protocol described below for the first 13 HSV-2 clinical isolates. Each isolate was tested by multiplex PCRs and analyzed in triplicate (intra-assay precision) and in independent assays (interassay variability).
Fig 1.
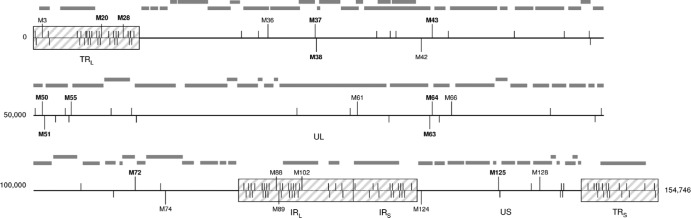
Linear map of genomic distribution of the 150 HSV-2 microsatellites (mononucleotide, dinucleotide, and trinucleotide repeats) in reference strain HG52. The HSV-2 genome (reference strain HG52; GenBank accession no. Z86099) is a linear double-stranded DNA of 154,746 bp that contains at least 74 genes (gray rectangles). It is composed of two covalently linked segments termed the unique long (UL) and unique short (US) sequences. Each segment is bracketed by terminal repeat (TR) and internal inverted repeat (IR) sequences (TRL, IRL, IRS, and TRS) (hatched boxes). Microsatellites are indicated by vertical lines. The names (Mx) of the 23 microsatellites characterized in this study among 2 laboratory strains and 13 clinical isolates are indicated. Microsatellites used for multiplex PCR assays are indicated in bold.
Neighbor-joining phylogenetic tree and principal component analysis.
The genetic relationships among HSV-2 isolates based on the frequencies of amplicon sizes at the different microsatellite loci were analyzed using two different approaches. First, phylogenetic trees were constructed using the neighbor-joining clustering method, and the genetic divergence between HSV-2 isolates was calculated according to genetic distances using Population 1.2.32 software (http://www.bioinformatics.org/project/?group_id=84). Second, as an alternative approach to represent the genetic relationships among HSV-2 isolates, principal component analysis (PCA) was applied using gene frequencies at the loci to represent the data on a two-dimensional map and to identify trends. A scattergram of the score data was examined for visualization of the geometric relationships among the HSV-2 isolates. PCA using microsatellite data was performed using the XLSTAT program (XLSTAT, New York, NY).
RESULTS
Microsatellite mapping within the HSV-2 complete genome.
In silico analysis allowed the identification of 150 different microsatellites (66 mononucleotide, 11 dinucleotide, and 73 trinucleotide repeats), numbered HSV-2 M1 to HSV-2 M150, which were located predominantly within noncoding regions of the HSV-2 genome (strain HG52; GenBank accession no. Z86099). Indeed, 129 microsatellites were found to be distributed in intergenic or noncoding inverted repeat regions, whereas 21 were localized in open reading frames (Fig. 1). The maximal numbers of repeat units were 12 and 8 for dinucleotide [M30-(TC)12 and M78-(GA)12] and trinucleotide [M116-(TCG)8 and M140-(ACG)8] motifs, respectively. As expected, poly(C/G) tracts (89.4%) were more abundant than poly(A/T) mononucleotide repeat sequences, because HSV-2 genomes demonstrate GC contents of around 70%. M125-(G)19 has been identified as the longest mononucleotide tract within the HSV-2 reference sequence. All of the microsatellites were perfect microsatellites except for M25/M26, M37/M38, M78/M79, and M82/M83, which were compound microsatellites (Fig. 1 and Table 1).
Characterization of HSV-2 microsatellites.
Twenty-two microsatellites were selected and surveyed using 19 PCR systems in order to perform length polymorphism analyses of a set of 2 laboratory strains (gHSV-2 and MS) and 13 clinical isolates (isolates 1 to 13). These microsatellites were localized along the HSV-2 genome, mainly within noncoding regions, and were composed of mononucleotide, dinucleotide, and trinucleotide units (Table 2). Sequence analyses allowed the identification of 7 monomorphic microsatellites (M36, M42, M61, M74, M102, M124, and M128) and 15 polymorphic microsatellites (M20, M28, M37, M38, M43, M50, M51, M55, M63, M64, M66, M72, M88, M89, and M125). Regarding the microsatellite variability, 2 to 8 variants (mean, 4.3 variants) among the 22 microsatellites studied were evidenced (Table 1). The laboratory strains gHSV-2 and MS showed distinct nucleotide patterns concerning these target repeats, in comparison with the 13 clinical isolates (Table 2).
Table 2.
Determination of HSV-2 microsatellite haplotypes for 2 laboratory strains and 56 clinical isolates
| Virus | Sample site | Clinical contexta | Antiviral susceptibility | Length of PCR products (bp) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M20 | M28 | M37/M38 | M43 | M50/M51 | M55 | M63/M64 | M72 | M125 | ||||
| Reference strain HG52 | 184 | 138 | 444 | 163 | 498 | 218 | 420 | 364 | 488 | |||
| Laboratory strains | ||||||||||||
| gHSV-2 | ACV-S/FOS-S | 181 | 132 | 440 | 157 | 493 | 210 | 406 | 360 | 469 | ||
| MS | ACV-S/FOS-S | 179 | 132 | 435 | 159 | 494 | 200 | 418 | 361 | 470 | ||
| Clinical isolates | ||||||||||||
| 1 | Genital | NA | ND | 180 | 133 | 442 | 157 | 493 | 214 | 409 | 361 | 480 |
| 2 | Anal | NA | ND | 180 | 132 | 442 | 157 | 493 | 209 | 419 | 360 | 472 |
| 3 | Genital | IC | ND | 182 | 132 | 439 | 157 | 493 | 211 | 413 | 362 | 469 |
| 4 | Genital | NA | ND | 181 | 132 | 440 | 157 | 490 | 211 | 415 | 361 | 468 |
| 5 | Genital | Pregnancy | ND | 180 | 132 | 438 | 157 | 494 | 209 | 421 | 361 | 474 |
| 6 | Genital | Elderly | ND | 181 | 132 | 440 | 159 | 494 | 200 | 412 | 362 | 471 |
| 7 | Genital | NA | ND | 179 | 135 | 440 | 157 | 493 | 212 | 422 | 361 | 469 |
| 8 | Genital | NA | ND | 177 | 133 | 437 | 159 | 493 | 270 | 405 | 360 | 471 |
| 9 | Anal | ICU | ND | 183 | 132 | 438 | 158 | 494 | 200 | 422 | 362 | 469 |
| 10 | Genital | HIV | ND | 180 | 132 | 438 | 160 | 494 | 211 | 416 | 363 | 471 |
| 11 | Genital | HIV | ND | 180 | 133 | 441 | 157 | 493 | 212 | 414 | 361 | 473 |
| 12 | Genital | HIV | ND | 179 | 134 | 440 | 157 | 492 | 214 | 412 | 362 | 472 |
| 13 | Genital | HIV | ND | 179 | 132 | 439 | 157 | 492 | 217 | 418 | 362 | 471 |
| 14 | Genital | NA | ND | 184 | 133 | 439 | 159 | 493 | 225 | 422 | 361 | 475 |
| 15 | Genital | Pregnancy | ND | 179 | 133 | 438 | 157 | 493 | 212 | 421 | 361 | 479 |
| 16 | Anal | SOT | ND | 179 | 134 | 438 | 157 | 496 | 214 | 417 | 360 | 473 |
| 17 | Anal | CLL | ND | 177 | 132 | 437 | 158 | 492 | 199 | 410 | 361 | 469 |
| 18 | Genital | IC | ND | 181 | 134 | 441 | 157 | 491 | 214 | 410 | 360 | 479 |
| 19 | Genital | IC | ND | 181 | 134 | 436 | 159 | 493 | 197 | 411 | 360 | 471 |
| 20 | Genital | IC | ND | 182 | 132 | 441 | 150 | 493 | 202 | 422 | 359 | 468 |
| 21 | Genital | IC | ND | 182 | 132 | 438 | 159 | 490 | 200 | 418 | 361 | 470 |
| 22 | Genital | IC | ND | 180 | 133 | 443 | 157 | 492 | 212 | 411 | 360 | 474 |
| 23 | BAL fluid | IC | ND | 178 | 136 | 438 | 157 | 495 | 214 | 417 | 361 | 474 |
| 24 | Genital | IC | ND | 180 | 133 | 437 | 157 | 494 | 211 | 420 | 362 | 473 |
| 25 | Buttock | IC | ND | 182 | 132 | 438 | 159 | 493 | 198 | 408 | 360 | 475 |
| 26 | Buttock | IC | ND | 182 | 132 | 438 | 159 | 494 | 211 | 414 | 363 | 475 |
| 27 | Genital | IC | ND | 180 | 136 | 440 | 157 | 491 | 211 | 415 | 361 | 477 |
| 28 | Genital | IC | ND | 181 | 135 | 440 | 159 | 497 | 197 | 416 | 359 | 470 |
| 29 | Anal | IC | ND | 180 | 132 | 439 | 157 | 492 | 198 | 416 | 361 | 470 |
| 30 | Genital | IC | ND | 181 | 136 | 442 | 157 | 493 | 209 | 415 | 359 | 475 |
| 31 | Genital | IC | ACV-S | 181 | 132 | 441 | 157 | 493 | 210 | 416 | 364 | 470 |
| 32 | Genital | HIV | ND | 180 | 133 | 439 | 159 | 490 | 200 | 413 | 361 | 474 |
| 33 | Buttock | HIV | ND | 181 | 132 | 438 | 159 | 489 | 202 | 417 | 360 | 476 |
| 34 | Genital | HIV | ND | 179 | 132 | 440 | 159 | 492 | 201 | 415 | 361 | 469 |
| 35 | Genital | HIV | ND | 181 | 132 | 439 | 159 | 493 | 209 | 415 | 362 | 471 |
| 36 | Anal | HIV | ND | 183 | 136 | 440 | 157 | 480 | 196 | 411 | 356 | 466 |
| 37 | Genital | HIV | ND | 179 | 132 | 437 | 159 | 493 | 199 | 411 | 362 | 471 |
| 38 | Genital | HIV | ND | 180 | 132 | 439 | 159 | 494 | 200 | 416 | 362 | 469 |
| 39 | Buttock | HIV | ND | 179 | 132 | 438 | 158 | 493 | 213 | 412 | 360 | 468 |
| 40 | Buttock | HIV | ND | 182 | 133 | 439 | 157 | 492 | 211 | 420 | 363 | 473 |
| 41 | Genital | HIV | ND | 180 | 134 | 439 | 157 | 490 | 212 | 413 | 360 | 475 |
| 42 | Genital | HIV | ND | 179 | 132 | 438 | 158 | 492 | 214 | 414 | 361 | 475 |
| 43 | Ear | HIV | ND | 180 | 132 | 439 | 157 | 493 | 209 | 415 | 360 | 471 |
| 44-1b | Back | HIV | ND | 179 | 132 | 438 | 159 | 493 | 188 | 416 | 361 | 474 |
| 44-2 | Genital | HIV | ACV-R/FOS-S | 179 | 132 | 437 | 159 | 493 | 188 | 423 | 360 | 469 |
| 44-3 | Genital | HIV | ACV-R/FOS-R | 179 | 132 | 437 | 159 | 493 | 188 | 413 | 360 | 470 |
| 44-4 | Genital | HIV | ACV-R/FOS-R | 179 | 132 | 438 | 159 | 493 | 188 | 413 | 361 | 469 |
| 45-1c | Genital | HIV | ACV-R/FOS-S | 180 | 132 | 438 | 159 | 494 | 200 | 412 | 361 | 473 |
| 45-2 | Genital | HIV | ACV-R/FOS-S | 180 | 132 | 438 | 159 | 494 | 200 | 412 | 361 | 472 |
| 46-1d | Buttock | HIV | ACV-R/FOS-R | 180 | 131 | 436 | 157 | 494 | 211 | 420 | 360 | 468 |
| 46-2 | Buttock | HIV | ND | 180 | 131 | 436 | 157 | 494 | 212 | 419 | 360 | 471 |
| 46-3 | Buttock | HIV | ACV-R/FOS-S | 180 | 131 | 436 | 157 | 494 | 212 | 418 | 360 | 467 |
| 47 | Genital | HIV | ACV-S/FOS-S | 183 | 133 | 439 | 157 | 492 | 199 | 403 | 360 | 470 |
| 48 | Genital | HIV | ACV-S/FOS-S | 181 | 139 | 439 | 157 | 509 | 199 | 409 | 356 | 469 |
| 49 | Genital | HIV | ACV-S/FOS-S | 184 | 133 | 441 | 159 | 492 | 199 | 411 | 361 | 469 |
| 50 | Genital | HIV | ACV-S/FOS-S | 179 | 139 | 438 | 157 | 493 | 212 | 418 | 361 | 476 |
| Total range | 177–184 | 131–139 | 435–443 | 150–160 | 480–509 | 188–270 | 403–423 | 356–364 | 466–480 | |||
NA, not available; BAL, bronchoalveolar lavage; ACV, acyclovir; FOS, foscarnet; CLL, chronic lymphocytic leukemia; IC, immunocompetent; ICU, intensive care unit hospitalization; ND, not determined; R, resistant; S, sensitive; SOT, solid-organ transplantation.
Sequential isolates 2, 3, and 4 for patient 44 were obtained 41, 42, and 48 months, respectively, after the first isolate.
The second isolate for patient 45 was obtained 12 months after the first isolate.
Sequential isolates 2 and 3 for patient 46 were obtained 48 and 51 months, respectively, after the first isolate.
Multiplex assay for differentiation of HSV-2 clinical isolates.
For the rapid mapping of genetic profiles of HSV-2 clinical isolates by length polymorphism analysis, 3 distinct multiplex PCR systems, amplifying 12 polymorphic microsatellites, were developed by mixing primer sets as follows: (i) M20, M72, and M37/M38, (ii) M28, M50/M51, and M55, and (iii) M43, M63/M64, and M125. Each HSV-2 clinical isolate was thereby characterized by its own microsatellite haplotype corresponding to the combination of the PCR product lengths obtained with the 3 multiplex assays (Fig. 2). For the 2 laboratory strains and the 13 isolates investigated above, the numbers of size variants ranged from 5 to 13 repeats (mean, 7.7 repeats) (Table 2). As expected, the microsatellite haplotypes for the clinical isolates obtained from distinct individuals were all different (Table 1). The interassay reproducibility was evaluated by performing the multiplex PCRs in triplicate with a gHSV-2 laboratory strain sample in 10 independent reactions. No variation in haplotypes was observed. The investigation of microsatellite haplotype stability after each of 16 passages of the gHSV-2 laboratory strain and 2 clinical isolates (isolates 14 and 15) (Table 2) in Vero cell cultures detected minor variations over time (2 months) within microsatellite M63/M64 only, as illustrated in Table 3. These data emphasize the stability of HSV-2 microsatellites during viral replication. This strategy, based on the analysis of 12 combined microsatellites, yields, in theory, a discriminatory power allowing the identification of at least 106 to 107 different haplotypes, considering the numbers of variants described for each of these microsatellites (Table 1). As a whole, the multiplex assays in combination with length polymorphism analyses permitted rapid accurate identification of HSV-2 strains.
Fig 2.

Electropherograms of multiplex PCR products used to differentiate HSV-2 strains. The haplotype was defined as the combination of the lengths of 9 amplicons (corresponding to 12 microsatellites), labeled at the 5′ end with 6-carboxyfluorescein, obtained from 3 multiplex assays and analyzed by polyacrylamide capillary electrophoresis. The sizes of the amplicons are indicated in base pairs (bp).
Table 3.
Influence of HSV-2 serial propagation in cell culture on microsatellite stability
| Virus | Time of cell propagation (days) | Length of PCR products (bp) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| M20 | M28 | M37/M38 | M43 | M50/M51 | M55 | M63/M64 | M72 | M125 | ||
| Laboratory strain gHSV-2 | 0 | 181 | 132 | 440 | 157 | 493 | 210 | 406 | 360 | 469 |
| 30 | 181 | 132 | 440 | 157 | 493 | 210 | 404 | 360 | 469 | |
| 60 | 181 | 132 | 440 | 157 | 493 | 210 | 406 | 360 | 469 | |
| Clinical isolates | ||||||||||
| 14 | 0 | 184 | 133 | 439 | 159 | 493 | 225 | 422 | 361 | 475 |
| 30 | 184 | 133 | 439 | 159 | 493 | 225 | 423 | 361 | 475 | |
| 60 | 184 | 133 | 439 | 159 | 493 | 225 | 422 | 361 | 475 | |
| 15 | 0 | 179 | 133 | 438 | 157 | 493 | 212 | 421 | 361 | 479 |
| 30 | 179 | 133 | 438 | 157 | 493 | 212 | 422 | 361 | 479 | |
| 60 | 179 | 133 | 438 | 157 | 493 | 212 | 421 | 361 | 479 | |
Differentiation of HSV-2 clinical isolates.
The multiplex PCR method was applied to characterize HSV-2 clinical isolates corresponding to different epidemiological situations, i.e., (i) isolates from epidemiologically unrelated patients (isolates 14 to 43), (ii) isolates corresponding to recurrent episodes in the same individuals, for which HSV-2 resistance to antivirals was investigated (isolates 44 to 46), and (iii) isolates recently described and considered new HSV-2 genetic variants (designated HSV-2v; isolates 47 to 50) (Table 2). As observed previously for the first 13 clinical isolates investigated, the microsatellite haplotypes for 30 isolates recovered from unrelated patients were all different. Microsatellite haplotypes for recurrent HSV-2 isolates that exhibited drug resistance (isolates 44 and 46) remained quite stable over time despite variations in antiviral susceptibility. Thus, only microsatellites M37/M38, M55, M63/M64, M72, and M125 exhibited length polymorphisms ranging from 1 to 10 bp. Of note, the widest polymorphism was observed for M63/M64 (Table 2). Microsatellite haplotypes for HSV-2v isolates appeared to be more variable than those for classic HSV-2 isolates (Table 2).
In order to illustrate the results for the HSV-2 haplotypes obtained for all laboratory strains (n = 2) and clinical isolates (n = 56) presented above, microsatellite allele frequencies were used to generate an unrooted phylogenetic tree using the neighbor-joining algorithm and PCA was performed (Fig. 3 and 4). The results of both analyses showed that all HSV-2 strains were genetically distinguishable and converged to the lack of evidence for a close genetic relationship among them. On the PCA chart, the laboratory strains gHSV-2 and MS clustered together with the majority of HSV-2 clinical isolates. However, reference strain HG52 and isolates 20 and 36 were distinguishable from the rest of the grouped isolates. Among the newly described HSV-2v isolates, only isolate 48 did not cluster together with the majority of HSV-2 isolates.
Fig 3.

Phylogenetic tree based on 12 HSV-2 microsatellites, depicting the genetic relationships among the 56 clinical isolates and 2 laboratory strains investigated. The neighbor-joining relationship unrooted tree was constructed based on Nei's distance matrix derived from microsatellite allele frequencies (37). The numbers of HSV-2 isolates, presented in Table 1, are indicated beside the corresponding branches. The solid bar beside the phylogenetic tree indicates length based on similarity. The predicted haplotype of HSV-2 reference strain HG52 (GenBank accession number Z86099) was included, as well as the haplotypes of laboratory strains gHSV-2 and MS.
Fig 4.
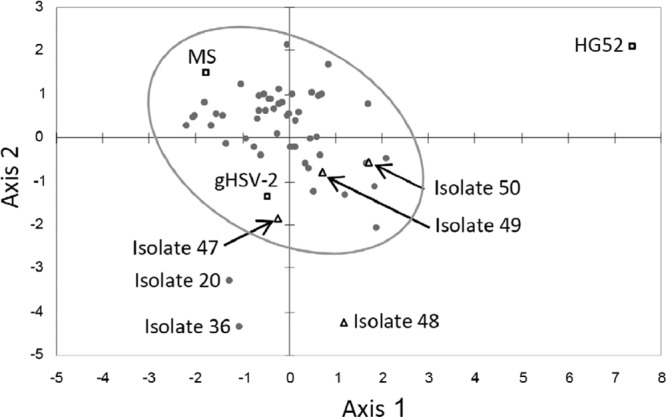
Global principal component analysis of the HSV-2 microsatellite haplotypes of the 56 clinical isolates and 2 laboratory strains investigated. Isolates are depicted as follows: □, HSV-2 reference strain (HG52; GenBank accession number Z86099) and HSV-2 laboratory strains gHSV-2 and MS; △, new genetic variants of HSV-2 described previously (20) (isolates 47 to 50); ●, remaining HSV-2 isolates.
DISCUSSION
This study, aiming to describe and to characterize HSV-2 short microsatellites, provides for the first time a map of numerous microsatellites across the whole viral genome, characterized by a high level of length polymorphism. HSV-1 and HSV-2, two closely related viruses belonging to the Simplexvirus genus in the Alphaherpesvirinae subfamily, constitute important human pathogens with different infection profiles. HSV-1 often is associated with orolabial lesions, whereas HSV-2 primarily causes genital infections. Despite their high degree of genomic identity, the two viruses do not have similar microsatellite features in terms of abundance, density, and motifs. Deback et al. (18) previously identified 79 microsatellites throughout the HSV-1 genome, whereas this work reports 150 microsatellites within the HSV-2 genome (using the same filtering options for research), located mainly in noncoding regions and with overrepresentation of homopolymeric repeats of guanosine or cytosine (Fig. 1 and Table 1). The latter point may be attributable to the high GC content (around 70%) of the HSV-2 genome (25, 26).
Microsatellites among viruses have higher mutation rates than do the eukaryotic DNA sequences and therefore are thought to be the source of genetic diversity (27). Moreover, these microsatellites seem to play important roles in the evolution of viruses, in terms of epidemiology, pathophysiology, or environmental conditions. Length variations within microsatellites have been implicated in the virulence of avian influenza virus and encephalomyocarditis virus (28, 29). Regarding herpesviruses, genetic variations of microsatellites within the pseudorabies virus genome have been shown to be involved in attenuation of the vaccine strain (30). Microsatellite markers are also useful for epidemiological studies, as previously reported for the precise characterization of HCMV or HSV-1 strains (18, 19, 23, 31). Furthermore, telomeric repeats located in the human herpesvirus 6 genome have been shown to facilitate viral DNA integration into host cell chromosomes (32, 33). Mutations in HSV-2 intragenic microsatellites have been reported to have clinical impact. Thus, HSV resistance to ACV may result from insertions or deletions in homopolymers located in the UL23 gene (encoding thymidine kinase) (34), and variations in the human antibody response to HSV may be associated with insertions or deletions in homopolymers located in the US4 gene (encoding glycoprotein G) (35). As a whole, the characterization of microsatellites within viral genomes may improve our knowledge regarding the epidemiology and pathogenesis of viruses.
In this study, a new multiplex assay was developed in order to differentiate HSV-2 isolates according to polymorphic microsatellite markers. In the manner of multiplex assays used for HSV-1 strains (18, 31), microsatellite analysis is shown to be a reliable and highly discriminative method. Microsatellite length variations are probably generated by DNA polymerase slippage during DNA replication or recombination processes (15). To minimize nonspecific artifacts of PCR amplification, a proofreading Taq polymerase was used in the multiplex assay reported here. The short-term stability of microsatellite profiles was assessed by monitoring 2 distinct clinical isolates (isolates 14 and 15) and one HSV-2 laboratory strain (gHSV-2) propagated in vitro in Vero cell cultures for 8 weeks. Our multiplex system demonstrated that HSV-2 strains can undergo microevolutionary events for particular microsatellites without effects on antiviral susceptibility. The phylogenetic reconstructions using microsatellite haplotypes demonstrated a clear distinction between HSV-2 strains. Indeed, all clinical isolates exhibited distinct patterns, with the exception of isolates recovered from the same patient (Fig. 3 and Table 2). This study demonstrates the potential of microsatellites to provide accurate polymorphic molecular markers that can be used to address medical questions regarding HSV-2, such as potential nosocomial infections, mother-to-child transmission, multiple recurrent infections, or exogenous reinfection.
Among the 12 microsatellites used in the multiplex assay, M63/M64 seemed to show a higher level of variation. This intergenic microsatellite is located within a noncoding region, between the UL37 gene (encoding a tegument phosphoprotein) and the UL38 gene (encoding the capsid protein VP19C). This is in accordance with a previous result indicating that this region of the HSV-2 genome exhibits a higher degree of variation than do others (36). However, the reasons for this higher level of polymorphism remain unknown.
Recently, we reported the description of a new genetic variant, designated HSV-2v, that was isolated from HIV-infected African patients and exhibited high levels of divergence within the DNA polymerase (UL30) and glycoprotein G (US4) genes, in comparison with classic HSV-2 isolates (20). Phylogenetic analysis based on UL30 and US4 sequences demonstrated that HSV-2v isolates clustered in a genetic group distinct from that of classic HSV-2 isolates. Preliminary data indicated that HSV-2v might represent zoonosis or might result from a recombination mechanism (20). In this study, the analysis of microsatellite polymorphisms did not reveal any specific clustering profile for HSV-2v isolates. Thus, the global principal component analysis demonstrated that only one of these viruses (isolate 48) was clearly distinguishable from the classic HSV-2 isolates (Fig. 4). Further studies using next-generation sequencing technology are now warranted to assess the origin of this new variant.
In conclusion, the analysis of microsatellite polymorphisms using a newly developed multiplex PCR assay allowed the precise characterization of HSV-2 isolates. The promising preliminary data presented herein should have implications for further epidemiological investigations (e.g., to distinguish unrelated strains, to demonstrate superinfection rather than reactivation, or to demonstrate nosocomial infections) and HSV-2 evolution studies, as reported previously for HSV-1 and HCMV (18, 19, 23, 31).
ACKNOWLEDGMENTS
We thank Christel Brunet for helpful advice and excellent technical assistance.
This work was supported in part by the Association pour la Recherche sur les Infections Virales (ARIV).
Footnotes
Published ahead of print 21 August 2013
REFERENCES
- 1. Paz-Bailey G, Ramaswamy M, Hawkes SJ, Geretti AM. 2007. Herpes simplex virus type 2: epidemiology and management options in developing countries. Sex. Transm. Infect. 83:16–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gupta R, Warren T, Wald A. 2007. Genital herpes. Lancet 370:2127–2137 [DOI] [PubMed] [Google Scholar]
- 3. Whitley R, Kimberlin DW, Prober CG. 2007. Pathogenesis and disease, chapter 32. In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom: http://www.ncbi.nlm.nih.gov/books/NBK47449/ [PubMed] [Google Scholar]
- 4. Hayward GS, Jacob RJ, Wadsworth SC, Roizman B. 1975. Anatomy of herpes simplex virus DNA: evidence for four populations of molecules that differ in the relative orientations of their long and short components. Proc. Natl. Acad. Sci. U. S. A. 72:4243–4247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McGeoch DJ, Moss HW, McNab D, Frame MC. 1987. DNA sequence and genetic content of the HindIII l region in the short unique component of the herpes simplex virus type 2 genome: identification of the gene encoding glycoprotein G, and evolutionary comparisons. J. Gen. Virol. 68:19–38 [DOI] [PubMed] [Google Scholar]
- 6. Buchman TG, Roizman B, Nahmias AJ. 1979. Demonstration of exogenous genital reinfection with herpes simplex virus type 2 by restriction endonuclease fingerprinting of viral DNA. J. Infect. Dis. 140:295–304 [DOI] [PubMed] [Google Scholar]
- 7. Maitland NJ, Smith IW, Peutherer JF, Robertson DH, Jones KW. 1982. Restriction endonuclease analysis of DNA from genital isolates of herpes simplex virus type 2. Infect. Immun. 38:834–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schmidt OW, Fife KH, Corey L. 1984. Reinfection is an uncommon occurrence in patients with symptomatic recurrent genital herpes. J. Infect. Dis. 149:645–646 [DOI] [PubMed] [Google Scholar]
- 9. Abba MC, Golijow CD. 2004. Herpes simplex virus genotyping: multiple optional PCR-based RFLP systems and a non-isotopic single-strand conformation polymorphism method. J. Virol. Methods 118:73–76 [DOI] [PubMed] [Google Scholar]
- 10. Madhavan HN, Priya K, Bagyalakshmi R. 2003. Phenotypic and genotypic methods for the detection of herpes simplex virus serotypes. J. Virol. Methods 108:97–102 [DOI] [PubMed] [Google Scholar]
- 11. Baines J, Pellett P. 2007. Genetic comparison of human alphaherpesvirus genomes, chapter 5. In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom: http://www.ncbi.nlm.nih.gov/books/NBK47393/ [Google Scholar]
- 12. Szpara ML, Tafuri YR, Parsons L, Shamim SR, Verstrepen KJ, Legendre M, Enquist LW. 2011. A wide extent of inter-strain diversity in virulent and vaccine strains of alphaherpesviruses. PLoS Pathog. 7:e1002282. 10.1371/journal.ppat.1002282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schlotterer C. 2000. Evolutionary dynamics of microsatellite DNA. Chromosoma 109:365–371 [DOI] [PubMed] [Google Scholar]
- 14. Merkel A, Gemmell N. 2008. Detecting short tandem repeats from genome data: opening the software black box. Brief. Bioinform. 9:355–366 [DOI] [PubMed] [Google Scholar]
- 15. Ellegren H. 2004. Microsatellites: simple sequences with complex evolution. Nat. Rev. Genet. 5:435–445 [DOI] [PubMed] [Google Scholar]
- 16. Tachida H, Iizuka M. 1992. Persistence of repeated sequences that evolve by replication slippage. Genetics 131:471–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen M, Tan Z, Jiang J, Li M, Chen H, Shen G, Yu R. 2009. Similar distribution of simple sequence repeats in diverse completed human immunodeficiency virus type 1 genomes. FEBS Lett. 583:2959–2963 [DOI] [PubMed] [Google Scholar]
- 18. Deback C, Boutolleau D, Depienne C, Luyt CE, Bonnafous P, Gautheret-Dejean A, Garrigue I, Agut H. 2009. Utilization of microsatellite polymorphism for differentiating herpes simplex virus type 1 strains. J. Clin. Microbiol. 47:533–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Walker A, Petheram SJ, Ballard L, Murph JR, Demmler GJ, Bale JF., Jr 2001. Characterization of human cytomegalovirus strains by analysis of short tandem repeat polymorphisms. J. Clin. Microbiol. 39:2219–2226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Burrel S, Abrao EP, Desire N, Seang S, Caumes E, Agut H, Boutolleau D. 2013. Detection of a new variant of herpes simplex virus type 2 among HIV-1-infected individuals. J. Clin. Virol. 57:267–269 [DOI] [PubMed] [Google Scholar]
- 21. Burrel S, Deback C, Agut H, Boutolleau D. 2010. Genotypic characterization of UL23 thymidine kinase and UL30 DNA polymerase of clinical isolates of herpes simplex virus: natural polymorphism and mutations associated with resistance to antivirals. Antimicrob. Agents Chemother. 54:4833–4842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Thi TN, Deback C, Malet I, Bonnafous P, Ait-Arkoub Z, Agut H. 2006. Rapid determination of antiviral drug susceptibility of herpes simplex virus types 1 and 2 by real-time PCR. Antiviral Res. 69:152–157 [DOI] [PubMed] [Google Scholar]
- 23. Davis CL, Field D, Metzgar D, Saiz R, Morin PA, Smith IL, Spector SA, Wills C. 1999. Numerous length polymorphisms at short tandem repeats in human cytomegalovirus. J. Virol. 73:6265–6270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rozen S, Skaletsky H. 2000. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 132:365–386 [DOI] [PubMed] [Google Scholar]
- 25. Duffy KE, Quail MR, Nguyen TT, Wittrock RJ, Bartus JO, Halsey WM, Leary JJ, Bacon TH, Sarisky RT. 2002. Assessing the contribution of the herpes simplex virus DNA polymerase to spontaneous mutations. BMC Infect. Dis. 2:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morfin F, Thouvenot D. 2003. Herpes simplex virus resistance to antiviral drugs. J. Clin. Virol. 26:29–37 [DOI] [PubMed] [Google Scholar]
- 27. Xu X, Peng M, Fang Z. 2000. The direction of microsatellite mutations is dependent upon allele length. Nat. Genet. 24:396–399 [DOI] [PubMed] [Google Scholar]
- 28. Hahn H, Palmenberg AC. 1995. Encephalomyocarditis viruses with short poly(C) tracts are more virulent than their mengovirus counterparts. J. Virol. 69:2697–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Perdue ML, Garcia M, Senne D, Fraire M. 1997. Virulence-associated sequence duplication at the hemagglutinin cleavage site of avian influenza viruses. Virus Res. 49:173–186 [DOI] [PubMed] [Google Scholar]
- 30. Szpara ML, Tafuri YR, Parsons L, Shamim SR, Verstrepen KJ, Legendre M, Enquist LW. 2011. A wide extent of inter-strain diversity in virulent and vaccine strains of alphaherpesviruses. PLoS Pathog. 7:e1002282. 10.1371/journal.ppat.1002282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deback C, Luyt CE, Lespinats S, Depienne C, Boutolleau D, Chastre J, Agut H. 2010. Microsatellite analysis of HSV-1 isolates: from oropharynx reactivation toward lung infection in patients undergoing mechanical ventilation. J. Clin. Virol. 47:313–320 [DOI] [PubMed] [Google Scholar]
- 32. Arbuckle JH, Medveczky MM, Luka J, Hadley SH, Luegmayr A, Ablashi D, Lund TC, Tolar J, De Meirleir K, Montoya JG, Komaroff AL, Ambros PF, Medveczky PG. 2010. The latent human herpesvirus-6A genome specifically integrates in telomeres of human chromosomes in vivo and in vitro. Proc. Natl. Acad. Sci. U. S. A. 107:5563–5568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kaufer BB, Jarosinski KW, Osterrieder N. 2011. Herpesvirus telomeric repeats facilitate genomic integration into host telomeres and mobilization of viral DNA during reactivation. J. Exp. Med. 208:605–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Piret J, Boivin G. 2011. Resistance of herpes simplex viruses to nucleoside analogues: mechanisms, prevalence, and management. Antimicrob. Agents Chemother. 55:459–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liljeqvist JA, Svennerholm B, Bergstrom T. 1999. Herpes simplex virus type 2 glycoprotein G-negative clinical isolates are generated by single frameshift mutations. J. Virol. 73:9796–9802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martin ET, Koelle DM, Byrd B, Huang ML, Vieira J, Corey L, Wald A. 2006. Sequence-based methods for identifying epidemiologically linked herpes simplex virus type 2 strains. J. Clin. Microbiol. 44:2541–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nei M, Tajima F, Tateno Y. 1983. Accuracy of estimated phylogenetic trees from molecular data. II. Gene frequency data. J. Mol. Evol. 19:153–170 [DOI] [PubMed] [Google Scholar]