

Abstract
Antiviral therapy for cytomegalovirus (CMV) plays an important role in the clinical management of solid organ and hematopoietic stem cell transplant recipients. However, CMV antiviral therapy can be complicated by drug resistance associated with mutations in the phosphotransferase UL97 and the DNA polymerase UL54. We have developed an amplicon-based high-throughput sequencing strategy for detecting CMV drug resistance mutations in clinical plasma specimens using a microfluidics PCR platform for multiplexed library preparation and a benchtop next-generation sequencing instrument. Plasmid clones of the UL97 and UL54 genes were used to demonstrate the low overall empirical error rate of the assay (0.189%) and to develop a statistical algorithm for identifying authentic low-abundance variants. The ability of the assay to detect resistance mutations was tested with mixes of wild-type and mutant plasmids, as well as clinical CMV isolates and plasma samples that were known to contain mutations that confer resistance. Finally, 48 clinical plasma specimens with a range of viral loads (394 to 2,191,011 copies/ml plasma) were sequenced using multiplexing of up to 24 specimens per run. This led to the identification of seven resistance mutations, three of which were present in <20% of the sequenced population. Thus, this assay offers more sensitive detection of minor variants and a higher multiplexing capacity than current methods for the genotypic detection of CMV drug resistance mutations.
INTRODUCTION
Human cytomegalovirus (CMV) is an important cause of severe systemic and tissue-invasive disease in immunocompromised patients (reviewed in reference 1), such as solid organ transplant (SOT) and hematopoietic stem cell transplant (HSCT) recipients. In these patients, prophylactic or preemptive treatment with antiviral agents for CMV significantly reduces the rates of CMV disease, the risk of other viral and bacterial infections, and all-cause mortality (2–5), but these treatments place patients at risk for the development of CMV drug resistance (6). In addition to anti-CMV therapy lasting for ≥3 months, other risk factors for drug resistance are intensive immunosuppression, multiple episodes of CMV reactivation, high viral counts, and suboptimal antiviral concentrations due to poor compliance or low drug absorption (7).
The number of antiviral drugs approved for CMV treatment is limited and includes ganciclovir (GCV) and its prodrug valganciclovir (VCV), foscarnet (FOS), and cidofovir (CDV). Intravenous GCV and oral VCV are used as first-line agents for both prophylaxis and treatment, largely because of their lower toxicities than those of FOS and CDV (8–13). All of the available CMV drugs ultimately target the viral DNA polymerase UL54; however, GCV and VCV also require phosphorylation by the phosphotransferase UL97 for their antiviral activity (6). Thus, mutations in the UL97 and UL54 genes can confer resistance to GCV and VCV, while CDV and FOS resistance is only associated with mutations in UL54. Large numbers of sequence variants have been identified in UL97 and UL54, but only a relatively small number of these lead to drug resistance in phenotypic assays (6). Phenotypically confirmed resistance mutations in UL97 cluster in hot spots within codons 405 to 607 of the gene, with >80% of the GCV-resistant clinical isolates carrying one of the seven frequently encountered drug resistance mutations (DRMs): M460V, M460I, H520Q, C592G, A594V, L595S, and C603W (6). In contrast, resistance mutations in the CMV polymerase can be found throughout the UL54 gene, particularly the exonuclease and polymerase domains, and many of these confer cross-resistance to more than one drug (6, 7).
The rates of CMV drug resistance vary between 5% and 12.5% for SOT recipients and are <5% for HSCT recipients (6, 14–17). In HIV-infected patients, GCV resistance has been reported in >20% of patients before the availability of highly active antiretroviral therapy (HAART) and in 9% after and is associated with CD4+ T-cell counts of <50 cells/μl (18–21). Continued administration of a drug to which resistance has developed can lead to an accumulation of multiple drug resistance mutations (6), increased morbidity, and, in SOT recipients, shortened graft survival (15, 16). On the other hand, therapy modification following the identification of GCV resistance is associated with more rapid viral clearance (3), further emphasizing the importance of timely resistance detection. Standardized protocols for selecting alternative therapy do not currently exist; however, several SOT and HSCT expert panels have proposed management algorithms (11, 22) that advocate genotypic resistance testing when the CMV viral load rises or when CMV-related disease develops in the setting of CMV antiviral treatment that has lasted >2 weeks.
The current gold standard for genotypic detection of CMV drug resistance is Sanger sequencing of PCR-amplified UL97 and UL54 gene segments. However, this approach often fails for viral loads of <1,000 CMV copies/ml of patient plasma, and it cannot detect mutants that are present in <10 to 20% of the viral population. In contrast, next-generation sequencing (NGS) methods have an improved ability to detect mixed populations and have been employed for the detection of HIV drug resistance (23, 24). NGS approaches have also been used to specifically assess the impact of low-abundance variants on treatment outcomes in HIV-infected patients (23). Moreover, two recent studies have successfully used NGS platforms to study CMV intrahost population diversity in clinical specimens (25, 26). A specific advantage of 454 pyrosequencing over most of the other currently available NGS technologies is the length of the sequencing reads (up to 1,000 bp with the GS FLX Titanium XL+ instrument and 400 bp with the 454 GS Junior sequencer).
Here, we report the development and evaluation of an NGS assay for the genotypic detection of CMV drug resistance in clinical samples using the Roche 454 GS Junior sequencer, a benchtop high-throughput pyrosequencing platform. The assay accuracy, error rates, and the ability to detect low-abundance variants were assessed using plasmid clones of the UL54 and UL97 genes. The assay was further evaluated using cultured CMV isolates and archived plasma specimens with known DRMs, followed by the testing of 48 clinical plasma specimens with a wide range of viral loads. Using this approach, we have achieved reproducible detection of well-characterized drug resistance mutations (DRMs) that are present in ≤10% of the viral population.
MATERIALS AND METHODS
Ethics.
This study was reviewed and approved by the institutional review board of Stanford University.
Patients and clinical specimens.
This study utilized clinical specimens submitted to the Stanford Hospital & Clinics (SHC) Clinical Virology Laboratory between April 2012 and March 2013 for CMV quantitative measurement. Clinical data were collected from the SHC and Lucile Packard Children's Hospital (LPCH) electronic medical records. Specimens were selected based on a recent history of CMV drug resistance testing, rising viral counts in the setting of ongoing viral therapy, and exposure to multiple CMV drugs. In some cases, multiple samples per patient were tested. The relevant patient data are summarized in Table 1. Where available, the CMV serologic data were obtained (see Table S1 in the supplemental material). Additional specimen characteristics for the samples used in the study, including viral loads and the outcomes of standard genotypic resistance testing, if performed, can be found in Table S1 in the supplemental material. DNA extracts from four CMV culture isolates from patients with well-characterized phenotypic and genotypic resistance to multiple drugs were selected from prior publications (27–30), and three plasma specimens with prior conventional Sanger sequencing genotype data and associated clinical ganciclovir resistance were provided from the Oregon Health & Science University archives.
Table 1.
Patient demographics and diagnoses
| Patient characteristic | Value |
|---|---|
| Age (mean [SD]) (yr) | 53.3 (15.6) |
| No. of patients with: | |
| HSCT | 23 |
| SOT | 9 |
| HIV/AIDS | 1 |
| Other | 1 |
| No. of unique patients | 34 |
| Total no. of specimens | 48 |
DNA extraction and viral load estimation.
For the clinical plasma specimens, DNA was extracted from 1.0 ml plasma using the Qiagen virus/bacteria midi kit on the QIAsymphony SP device and eluted in 90 μl of Tris-EDTA (TE) buffer (Qiagen, Germantown, MD). Viral loads were measured using artus CMV PCR reagents (Qiagen, Germantown, MD) on the Rotor-Gene Q, as previously described (31). For the reference control, cultured CMV strain AD169 (ATCC VR-538) (American Type Culture Collection, Manassas, VA) was diluted in defibrinated human plasma (SeraCare, Milford, MA) at 1:1,000 (high copy number) and 1:100,000 (low copy number), processed by the same protocol as the clinical specimens, and the viral load in copies/ml plasma was measured with the artus CMV PCR. Viral loads estimated for the low-copy AD169 control are typically in the range of 2,700 to 3,400 copies/ml plasma.
Primer design.
Primers targeting UL97 and UL54 were designed using NCBI Primer-BLAST and the CMV Merlin strain reference sequence (NC_006273.1) (Tables 2 and 3). Polymorphic regions were avoided by aligning all primers to 22 CMV sequences from GenBank (see http://www.ncbi.nlm.nih.gov/genomes/VIRUSES/VIAL/10359/10359.html). A nested approach was used, such that the UL54 and UL97 genes were initially amplified by long-range PCR and the resulting products were used as a template in a four-primer library preparation step as outlined in Fig. 1. To avoid allelic dropout, two distinct long-range primer sets for each gene were designed (Table 2). Primers for amplicon generation and bar coding with the Fluidigm Access Array (Fluidigm, South San Francisco, CA) were designed according to the manufacturer's specifications. Briefly, for target-specific enrichment (Table 3), two primers were designed such that primer 1 included a 5′-extension containing the 454 consensus sequence 1 (CS1) (ACA CTG ACG ACA TGG TTC TAC A) followed by the CMV-specific sequence, while primer 2 included a 5′-extension containing CS2 (TAC GGT AGC AGA GAC TTG GTC T) followed by the CMV-specific sequence. The second primer set for the 4-primer Access Array step contained bar codes and 454 adapters: forward bar code primer (CGT ATC GCC TCC CTC GCG CCA TCA GNN NNN NNN NNA CAC TGA CGA CAT GGT TCT ACA) and reverse bar code primer (CTA TGC GCC TTG CCA GCC CGC TCA GNN NNN NNN NNT ACG GTA GCA GAG ACT TGG TCT), where the Ns represent the Fluidigm 10-bp nucleotide bar code sequences. All primer pairs were validated for 4-primer PCR conditions using CMV strain AD169 DNA as a template. Seventeen primer sets were selected after primer validation: four sets spanning codons 399 to 664 of UL97 and 13 sets spanning codons 212 to 311 and 356 to 1042 of UL54 (Table 3).
Table 2.
Primers for long-range PCR
| Gene | Primer name | Sequence | Reference positiona | Product length (bp) |
|---|---|---|---|---|
| UL54 | UL54(long)F-1 | CGCGTCGCCGTTGCACGTAG | 78015–78034 | 4,106 |
| UL54(long)R-1 | AGTGTCGGTAGCCGTTTTTGCGA | 82098–82120 | ||
| UL54(long)F-2 | AGTCCACGCCGCCTCATCTC | 78071–78090 | 3,882 | |
| UL54(long)R-2 | GTGAGAGGCTGACAGCTTACGA | 81931–81952 | ||
| UL97 | UL97(long)F-1 | GCCACCTTGGTGGACTCGGT | 141612–141631 | 2,257 |
| UL97(long)R-1 | CTTTCGGCGCACCACCAGCA | 143849–143868 | ||
| UL97(long)F-2 | GCAATCCCCGTCACGCCTCTG | 141686–141706 | 2,259 | |
| UL97(long)R-2 | GGACGCGGAACGTGACGGTT | 143925–143944 |
CMV strain Merlin NCBI accession no. NC_006273.1.
Table 3.
Target-specific primers for Access Array PCR
| Gene and primer name | Primer 1a | Primer 2b | Nucleotides | Amino acids |
|---|---|---|---|---|
| UL54 | ||||
| Pol_D23 | ACGCACACGGTGCAGGTACAGATCGTCGCG | GTGGAGCCCGTCAAGCTGGAGTT | 604–880 | 950–1042 |
| Pol_E44 | CACCAGATCCACGCCCTTCATG | GGATTCGGAGGAGAGCAG | 803–1078 | 884–975 |
| Pol_F45 | GCGTCAGGCCACGAAAGCGGAC | GTGCCGTGCGCGAATGCATGC | 985–1355 | 792–915 |
| Pol_F54 | CGAAAGCGGACAAACACGCTGTC | GAATGCATGCGCGAGTGTCAAG | 997–1343 | 796–911 |
| Pol_G45 | TGGGCAGACACGGCATCATG | CACGGTGTTTGAGCCCGAGG | 1253–1613 | 706–825 |
| Pol_G54 | CGACCGATGCGCGTAATGCTG | AGGTGGGTTACTACAACGAC | 1226–1596 | 712–834 |
| Pol_H65 | CACCAGCAGGGTGGAGTAGCAGAG | TGTGGCGGCTATGTTTCAGATGTC | 1540–1807 | 641–730 |
| Pol_I46 | CGGCGCCGTGATTGTCGTTGG | CACCATTAATTTTCACTACGAG | 1686–2055 | 559–681 |
| Pol_I54 | CTGGTACGAAACCGCCGCAGTAC | CGGACAGCAGATCCGTATCTAC | 1662–1980 | 584–689 |
| Pol_J56 | GCAACACTATTCGTTTCGGGCAC | CCAAGGTGTATATTGCGGGTTC | 1912–2287 | 481–606 |
| Pol_K23 | CTCGGCATTAGCCACGAAACAAC | GCAGCGCTTCTGCAAGTTGCCTAC | 2163–2419 | 437–522 |
| Pol_L11 | ACCCACGGCGGGGCTGTGTAAA | GCTGCACTTCGGAGGGTGTGA | 2399–2663 | 356–443 |
| Pol_N22 | GAATGACAATGTCATCGGACTTCTCGGCGCAG | GACCCGTGCCCGATTTGCAGTGTGTGTC | 2795–3094 | 212–311 |
| UL97 | ||||
| Pt_G45 | CGTGCACCGCGGTCTGCTCAC | CGGCATGGGTCGGAAAGCAG | 1194–1561 | 399–520 |
| Pt_G65 | CGGTACATCGACGTTTCCACAC | CGGCATGGGTCGGAAAGCAG | 1248–1561 | 416–520 |
| Pt_H22 | CAGCCTCAGCGAGCCCTATCCGGATTAC | GTCGGAGCAGTGCGTGAGCTTGCCGTTC | 1474–1787 | 492–596 |
| Pt_I23 | GACCGGCGCGGTCTGGACGAGGTGCGCATG | CCAACAGACGCTCCACGTTCTTTCTG | 1735–1994 | 579–665 |
Primer 1 is preceded by consensus sequence 1 (CS1) (ACACTGACGACATGGTTCTACA).
Primer 2 is preceded by CS2 (TACGGTAGCAGAGACTTGGTCT).
Fig 1.

CMV drug resistance mutation testing approach. Long-range PCR, followed by library preparation and bar coding using the Fluidigm Access Array, is shown relative to the UL97 (A) and UL54 (B) genes and known drug resistance mutations across the genes (not drawn to scale). This approach produces 4 overlapping amplicons for UL97 and 13 for UL54, which were pooled and subjected to 454 pyrosequencing using the Roche GS Junior sequencer.
Long-range PCR.
The UL54 and UL97 genes were amplified using the LongAmp Hot Start Taq DNA polymerase (New England BioLabs, Ipswich, MA), according to the manufacturer's instructions. Two multiplexed PCRs were run simultaneously with distinct primer pairs for UL97 and UL54 (Table 2). Each 50-μl reaction mixture contained LongAmp Hot Start Taq 2× master mix, 400 nM each primer, 3% dimethyl sulfoxide (DMSO), and 20 μl of extracted nucleic acid as a template. The reactions were run in a DNA Engine PTC-200 (Bio-Rad, Hercules, CA), using the following cycling parameters: 94°C for 30 s, 30 cycles of 94°C for 30 s, 60°C for 30 s, and 65°C for 4 min 15 s, and a final extension for 10 min at 65°C. The PCR products were pooled, and a 15-μl aliquot was electrophoresed on a 1% agarose gel and stained with ethidium bromide to ascertain the presence of visible bands for both genes.
Library preparation.
Sequencing libraries were prepared on the 48.48 Access Array (Fluidigm, South San Francisco, CA) using long-range PCR products as a template. Up to 24 specimens were included in each array. A corresponding number of bar-coded primers were selected from the 96 Access Array bar code library for the 454 FLX Titanium sequencer (Roche 454 Life Sciences, Branford, CT). The reaction mixtures were assembled on the integrated fluidic circuit (IFC) using IFC Controller AX (Fluidigm), and amplification was performed using the FC1 cycler (Fluidigm) according to the manufacturer's instructions. The primers (1 μM) were distributed in the primer half-plate of the IFC according to the setup outlined in Table S2 in the supplemental material. Each sample was loaded in duplicate in the sample half-plate of the IFC. The PCR products from each sample were automatically pooled and harvested using a separate IFC Controller AX and examined by capillary electrophoresis on the QIAxcel Advanced system (Qiagen, Germantown, MD). The amplicons were purified twice using Agencourt AMPure XP magnetic beads (Beckman Coulter, Brea, CA). The amplicon lengths were assessed using the Agilent 2100 Expert bioanalyzer (Agilent Technologies, Santa Clara, CA). DNA concentrations were estimated using Quant-iT PicoGreen double-stranded DNA (dsDNA) reagents (Life Technologies, Carlsbad, CA) according to the manufacturer's instructions. The amplicon concentrations were calculated according to the Amplicon Library Preparation Method manual, GS Junior Titanium series (Roche 454 Life Sciences), using the following equation: amplicon molecules/μl = [(sample DNA concentration) × 6.022 × 1023/[656.6 × 109 × (amplicon length)], where the sample DNA concentration was measured in ng/μl and the amplicon length was measured in bp. The amplicon concentrations were adjusted to 1 × 109 molecules/μl before storage or downstream use in emulsion PCR.
Emulsion PCR and sequencing.
Water-in-oil emulsion PCR and the enrichment of clonally amplified beads were performed using the GS Junior Titanium emulsion PCR (emPCR) (Lib-A) kit (Roche 454 Life Sciences, Branford, CT) according to the manufacturer's instructions. The purified Access Array-generated library was mixed at 0.6 DNA molecules per bead to achieve the recommended 5 to 20% enrichment of the amplified beads. Sequencing was performed using the GS Junior instrument and the GS Junior Titanium sequencing kit (Roche 454 Life Sciences) in the forward and reverse directions, using ∼5 × 106 enriched beads. All sequencing runs were examined against quality-control benchmarks preset by the manufacturer (see the 454 Sequencing System software manual version 2.5).
Data analysis.
Sequencing data were obtained as standard flowgram format (SFF) files from the GS run processor after quality-control filtering and read trimming using the manufacturer's default parameters (see 454 Sequencing System software manual version 2.5). The SFF data files were further analyzed using the GS Amplicon Variant Analyzer (AVA) version 2.7 (Roche 454 Life Sciences, Branford, CT). The reads were trimmed using the Fluidigm blueprint, demultiplexed based on the multiplex identifiers (MIDs) for each sample, and mapped to the reference FASTA sequences of UL54 and UL97 from the Merlin strain (NC_006273). Variant calls were made using the default AVA settings. To facilitate data analysis, CMV variants were classified into one of three categories: accepted, rejected, or putative. Accepted variants represent phenotypically proven DRMs as reported in the literature (see Tables S3 and S4 in the supplemental material). The rejected category contains silent mutations, differences between CMV strains based on alignments of the Merlin sequence (NC_006273) with the sequences of 21 other strains deposited in GenBank, and variants that have been shown in the literature to be drug sensitive in phenotypic assays (6, 32). Variants that did not fall into either of these categories were designated as putative. The read depths and coverage results for all variants were exported from the AVA for further analysis. Calculations of average observed error rates for plasmid clones were performed by obtaining the mean percentage of variants reported by the AVA software for all sequenced positions that fell within a given category, i.e., nucleotide substitutions or insertion/deletions (indels) in the homopolymer or nonhomopolymer regions. A homopolymer region was defined as a repeat of ≥3 of the same nucleotide with nonidentical flanking nucleotides (33), which defined as such generated 692 homopolymer and 2,466 nonhomopolymer positions. For nucleotides where no variant was reported by the AVA, the error value was considered to be zero.
Plasmids.
Full-length UL97 and UL54 genes were cloned into pCR 2.1 Topo vector (Invitrogen, Grand Island, NY) according to the manufacturer's instructions. The templates used for cloning consisted of DNA from the wild-type (WT) reference strain AD169 and a cultured mutant isolate (MUT) from a patient with multiple known DRMs in UL97 (M460V) and UL54 (G841A and P522A) (28). The target-specific primers used for cloning were as follows: UL54(long)F-1 and UL54(long)R-1 for UL54 (Table 2), and UL97(long)F-1 (Table 2) and GGT ACA AAC CCA CCG GCG GG for UL97. Each 50-μl reaction mixture contained LongAmp Hot Start Taq 2× master mix, 400 nM each primer, 3% DMSO, and 20 μl of extracted nucleic acids. The long-range PCR amplification was performed as described above. Plasmids were purified using the GeneJET plasmid miniprep kit (Fermentas, Glen Burnie, MD), performed according to the manufacturer's instructions. The DNA concentrations were measured using the Quant-iT PicoGreen dsDNA reagents. The sequences of the cloned UL97 and UL54 genes were confirmed using Sanger sequencing. The plasmid concentrations were calculated using the following formula: plasmid copies/μl = [(sample DNA concentration) × 6.022 × 1023/[656.6 × 109 × (plasmid length)], where the sample DNA concentration was measured in ng/μl and the plasmid length was measured in bp. Equal copy numbers of the corresponding UL54 and UL97 plasmids were mixed to prepare the WT and MUT plasmid templates. To estimate an equivalent of viral copies/ml plasma for plasmids, the following calculation was performed: plasmid copies/ml plasma = (plasmid copies/μl) × 90 μl eluate, as 90 μl is the elution volume for nucleic acids extracted from 1 ml of human plasma using the QIAsymphony protocol described above. This calculation produces an equivalent of the viral load for the plasmid samples and was used as the basis for preparing two different dilutions of plasmid mixes that were equivalent to 1,000 or 10,000 viral copies/ml plasma. These dilutions were used as a template for long-range PCR and the subsequent assay steps in order to estimate the error rates at the two copy number equivalents. The detection of minor variants was assessed by sequencing MUT and WT plasmid mixes at 5%, 10%, and 20% of MUT. These mixes were prepared at the same copy number equivalents as above, 1,000 and 10,000 copies/ml plasma. Each dilution was processed and sequenced on two separate occasions.
Sanger sequencing.
Clinical specimens and isolates in which DRMs were identified were also tested by Sanger sequencing. Briefly, one amplicon covering a detected DRM was amplified using 1 μl of long-range PCR template, 400 nM the corresponding CS1- or CS2-tagged primers, PCR master mix (Fermentas, Glen Burnie, MD), and 3% DMSO. The following cycling conditions were used: 95°C for 2 min, 30 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 30 s, and a final extension for 10 min at 72°C. The PCR products were sequenced by bidirectional dideoxynucleotide termination sequencing using CS1 (forward) and CS2 (reverse) primers (Elim Biopharmaceuticals, Inc., Hayward, CA). The sequences were aligned to the reference using Sequencher software (version 5.1; Gene Codes Corporation, Ann Arbor, MI), and chromatograms were examined manually for secondary peaks at positions where variants were identified by NGS.
Statistical analysis.
The error rates associated with the assay were determined by comparing the variant output from the AVA software to Sanger sequences of the WT or MUT plasmid clones as described above. AVA reads in which the sequence differed from the expected Sanger sequence were termed noise_reads. A generalized linear model (34) was used to model the dependency of the number of noise_reads on the covariates using the R language for data analysis and graphing (35) and the function glm.nb from the package MASS (36). The following covariates were included in the model: total_reads (the number of reads covering a given nucleotide position), treated as a continuous variable, copy number (1,000 or 10,000 copies/ml plasma), treated as a two-level factor, and Is-homopolymer (location in homopolymer or nonhomopolymer region), treated as a two-level variable. Orientation (whether the error was detected in the forward or reverse sequencing direction), specimen type (WT or MUT), and gene (UL54 or UL97) were also examined but were not included in the model because the number of noise_reads did not appear to be strongly dependent on these variables.
RESULTS
Assay design and optimization.
We designed a nested amplification strategy followed by 454 pyrosequencing, as outlined in Fig. 1. Briefly, two multiplexed PCRs were performed to amplify the UL54 and UL97 genes using nucleic acids extracted from clinical plasma specimens or the cultured CMV AD169 reference strain. The purpose of this step was to enrich viral DNA from the clinical specimens with low viral loads, as preliminary experiments showed variable results when the 4-primer PCR was performed directly on nucleic acids from clinical specimens with viral loads of ≤1,000 copies/ml plasma. As an additional quality-control measure, aliquots from pooled long-range PCR products from each specimen were examined for visible UL54 and UL97 bands by agarose gel electrophoresis before proceeding to library preparation. The performance of the long-range PCR was first tested using serial 1:3 dilutions of the cultured AD169 reference strain from 1,200 to 44 copies/ml plasma as a template, all of which produced bands at the expected sizes for UL97 and UL54, even at the lowest dilutions (see Fig. S1 in the supplemental material). Next, 62 clinical plasma specimens were tested with CMV viremia ranging from 253 to 2,191,011 copies/ml plasma. Forty-eight samples (77.4%) produced UL97 and UL54 amplification products that could be visualized, whereas 14 specimens, all with viral loads of <2,400 copies/ml plasma, produced either no band for either gene (n = 12) or only a UL97 band (n = 2). Among the 19 specimens with viral loads of ≤1,000 copies/ml plasma, 13 (68.4%) were amplified successfully, indicating that most specimens in this range of viremia can be used in the assay. The lowest viral load for which the two genes were successfully amplified was 394 copies/ml plasma.
The regions of UL54 and UL97 that were targeted by primers in the library preparation step were selected based on the locations of phenotypically validated drug resistance mutations (6, 32, 37, 38). The optimization of these primer sets was performed using the AD169 strain to ensure that all amplicons produced bands of similar intensities when examined by agarose gel electrophoresis. Ultimately, 13 primer sets targeting UL54 and 4 primer sets targeting UL97 were selected (Fig. 1).
Sequencing reads and coverage.
A maximum of 24 specimens were amplified and sequenced at a time, producing 2,278 to 3,735 reads per sample and an average coverage of ∼140 reads per nucleotide for areas covered by a single amplicon and up to ∼580 reads per nucleotide in regions with overlapping amplicons (Fig. 2). When only 8 specimens were processed and sequenced at one time, coverage increased to an average of ∼650 reads per nucleotide for areas covered by a single amplicon and ∼2,600 reads per nucleotide in regions with overlapping amplicons (see Fig. S2 in the supplemental material). The degree of read coverage per nucleotide did not correlate with the viral loads of the clinical specimens (data not shown), suggesting that when the long-range PCR is successful, it provides sufficient template in the 4-primer Access Array step, even for specimens with low starting viral counts. Additionally, the read coverages per amplicon in the forward and reverse directions were equivalent for all 17 amplicons (see Fig. S3 in the supplemental material).
Fig 2.

Average read coverage per nucleotide when 24 specimens are sequenced. The data are presented as the mean number of sequencing reads per nucleotide from 24 patient specimens processed and sequenced in a single run. Also shown are the locations of the known drug resistance mutations (DRMs) that were considered in this study and the overlapping amplicons generated during library preparation for the UL54 (A) and UL97 (B) genes.
Error rates.
In order to quantify errors due to amplification and sequencing, the NGS results for two sets of plasmid clones were compared to those from Sanger sequencing, with the assumption that discrepancies between the methods represent errors. Such errors may originate at the level of long-range PCR, library preparation, sequencing, or sequence alignment. For the purpose of these experiments, the full-length UL54 and UL97 genes from the reference AD169 strain and a clinical isolate known to carry multiple resistance mutations (M460V in UL97 and G841A and P522A in UL54) were cloned. Plasmid samples containing WT UL54 and UL97 or MUT UL54 and UL97 were used for long-range PCR and the NGS assay, prepared at two different dilutions simulating viral loads of 1,000 or 10,000 copies/ml plasma. The overall error rate observed for all plasmid samples was 0.189%, representing the average percent error across all nucleotide positions that were sequenced for both genes. Since 454 pyrosequencing is associated with more frequent errors in homopolymer regions (24, 33), we examined the average error rates for indels in the homopolymer and nonhomopolymer regions and found substantially higher average insertional error rates in the homopolymer regions (Fig. 3A). Similar results were observed for the two plasmid dilutions, although the samples with 10,000 copies/ml showed modestly less error than those with 1,000 copies/ml (data not shown). In contrast, the average error rates for deletions and substitutions did not markedly differ between the two types of sequences (Fig. 3A).
Fig 3.

Error rates for plasmid clones in homopolymer and nonhomopolymer regions. (A) Observed error rates per error type and sequence context. UL54 and UL97 plasmid clones mixed at 1,000 copies/ml plasma were processed and sequenced in duplicate. All variant reads called by the GS amplicon variant analyzer compared to Sanger sequencing of the plasmids were considered errors and were calculated as the percentage of the total reads covering a given nucleotide. The data are presented as the average percent error for substitutions, insertions, and deletions in the homopolymer and nonhomopolymer regions. The averages from two independent experiments are shown. (B) Threshold for minor variant detection relative to total read coverage based on empirical error rates for plasmid clones. The covariates included in the negative binomial model were total_reads (number of reads covering a given nucleotide position), copy number (1,000 or 10,000 copies/ml plasma), and location in homopolymer versus nonhomopolymer regions. The average from two independent experiments is shown.
Next, we used the observed error rates at all positions to generate a statistical model that could be used to distinguish true variants from errors. The distribution of error reads (termed noise_reads in the model) indicated a strong departure from the Poisson model (data not shown); therefore, a gamma-Poisson mixture equivalent to a negative binomial distribution was assumed for the noise_reads variable. Significant covariates included copy number, occurrence within a homopolymer region, and total number of reads covering a specific nucleotide position. To obtain the detection thresholds at which a given percentage of variants would be considered significant (33), we used the model to plot the predicted error as a percentage of the total reads covering a given position (Fig. 3B). Interestingly, the predictions for a given number of total reads were not markedly different between the homopolymer and nonhomopolymer regions (Fig. 3B) or between 1,000 and 10,000 copies/ml plasma (data not shown). An examination of known CMV DRMs (see Tables S3 and S4 in the supplemental material) reveals that the majority of known DRMs were substitutions, which exhibited similar error rates in the homopolymer and nonhomopolymer regions (Fig. 3A), and the small number of deletions associated with resistance (see Tables S3 and S4 in the supplemental material) occurred in nonhomopolymer regions. Thus, a single model combining the homopolymer and nonhomopolymer regions may be adequate.
Detection of minor variants in mixed plasmid populations.
To assess the ability of the assay to detect minor variants, the WT and MUT plasmids were mixed in different ratios, such that the MUT plasmids contributed 5, 10, or 20% to the plasmid population. In addition to the 3 known DRMs in the MUT plasmids, 30 other positions in UL54 and UL97 were identified by Sanger sequencing where the MUT sequence was different from the WT and would constitute a minor variant. These positions represent CMV strain differences, previously characterized single nucleotide polymorphisms, and amino acid substitutions that have been shown to be drug sensitive in phenotypic assays (6, 32, 39). The plasmid mixes were prepared at two concentrations (representing the equivalents of 1,000 and 10,000 viral copies/ml plasma), and the NGS protocol was performed in duplicate for each mix, beginning at the long-range PCR step. Minor variants were missed most frequently in plasmid mixes with the lowest contribution of MUT plasmid tested at the low copy equivalent. For example, at 1,000 copies/ml, the 5% and 10% MUT variants had variable detection results, such that within a given experiment, a mean of 14 minor variants were not detected out of the 33 possible variants (range, 3 to 29). This indicates that at the low starting copy numbers, minor variants are not reliably represented, which is consistent with previous reports (40, 41). The inaccuracy and imprecision of minor variant quantitation at low starting viral loads were evident when the mean percentage of variants for all 33 positions was considered for each dilution and each replicate (Fig. 4). The replicates at 10,000 copies/ml (Fig. 4A) more closely approximated the expected variant ratio than the replicates at 1,000 copies/ml (Fig. 4B).
Fig 4.

Minor variant detection in mixed plasmid samples. Plasmid clones of UL54 and UL97 derived from wild-type (WT) AD169 and a patient specimen containing multiple resistance mutations (MUT) were mixed at different proportions, such that MUT plasmids represented 5, 10, or 20% of the mixture. There were 33 UL54 and UL97 nucleotide positions where MUT plasmids represented a minority population. The mixing studies were performed at two dilutions, representing 10,000 (A) and 1,000 (B) copies/ml plasma. Each mixture was tested on two separate occasions, indicated as 1 (red) and 2 (blue) on the graphs. The data are presented as box whisker plots of the interquartile ranges (1.5 interquartile range [IQR], Tukey method) of the 33 UL54 and UL97 nucleotide positions for each replicate. The black lines represent the expected MUT percentages.
Drug resistance mutations in clinical specimens.
To evaluate the ability of the assay to detect drug resistance mutations, we tested four archived CMV isolates obtained from patients with confirmed phenotypic and genotypic resistance to multiple drugs and three archived plasma samples associated with known genotypic and clinical GCV resistance. A review of the sequencing data from these experiments demonstrated that the majority of sequence variations represented silent mutations. After such variants were filtered out, only 5 to 15 variants for UL54 and <5 variants for UL97 remained for further analysis, representing known DRMs as well as putative variants. In all cases, the NGS assay correctly identified the resistance mutations that had been previously detected in each of the cultured isolates and archived plasma specimens by Sanger sequencing (Table 4), indicating that the NGS strategy can detect high-abundance variants as efficiently as traditional methods.
Table 4.
CMV drug resistance mutations detected in archived clinical isolates and specimens
| Gene name and sample no. | DRMa | % variants | Predicted drug resistance pattern | Original genotypeb | Resistance phenotypec | Source or reference |
|---|---|---|---|---|---|---|
| UL54 | ||||||
| OHSU-E65 | G841A | 88.3 | GCVr CDVr FOSr | G841A | GCVr, FOSi, CDVnd | 28 |
| P522A | 90.0 | GCVr CDVr | P522A | |||
| L501I | 5.7 | GCVr CDVr | Not detected | |||
| OHSU-E724 | L802M | 69.1 | FOSr (low-level GCVr CDVr) | L802 M | GCVr, FOSr, CDVr | 29 |
| F412C | 94.0 | GCVr CDVr | F412C | |||
| D588N | 10.5 | FOSr (low-level GCVr CDVr) | Not detected | |||
| OHSU-E749 | P522S | 87.6 | GCVr CDVr | P522S | GCVr, FOSi, CDVnd | 30 |
| P522A | 8.6 | GCVr CDVr | Not detected | |||
| OHSU-E1178 | A987G | 99.6 | GCVr CDVr | A987G | GCVr, FOSnd, CDVnd | 27 |
| A834P | 97.4 | GCVr CDVr FOSr | A834P | |||
| UL97 | ||||||
| OHSU-E65 | M460V | 100.0 | GCVr | M460V | GCVr | 28 |
| OHSU-E724 | C603W | 93.9 | GCVr | C603W | GCVr | 29 |
| OHSU-E749 | C607F | 97.3 | GCVr | C607F | GCVr | 30 |
| OHSU-E1145 | C603W | 99.7 | GCVr | C603W | No culture isolated | S. Choue |
| OHSU-E1162 | L595S | 56.7 | GCVr | L595S mix | No culture isolated | S. Choue |
| A594V | 15.1 | GCVr | A594V possible mix | |||
| OHSU-E1369 | C603W | 94.2 | GCVr | C603W | No culture isolated | S. Choue |
| OHSU-E1178 | L595S | 94.4 | GCVr | L595S | GCVr | 27 |
DRM, drug resistance mutation.
Predicted amino acid change determined by conventional Sanger sequencing.
Result of plaque reduction testing of original culture isolate. r, resistant; i, intermediate; nd, not done.
Genotypic testing done on plasma specimen without accompanying culture.
Unpublished.
Interestingly, for four of the specimens, the NGS assay also identified low-abundance resistance mutations that had not been reported by conventional sequencing (Table 4). Isolate OHSU-E724 demonstrated a minor subpopulation of these low-abundance resistance mutations with D588N in UL54, which is associated with FOS resistance and lower levels of GCV and CDV cross-resistance (42, 43). This mutation was reproducibly detected at similar levels in two independent sequencing experiments where different DNA dilutions were used for long-range PCR (10.5% at a 1:2,000 dilution and 10.6% at a 1:20 dilution), suggesting that this finding was not due to amplification or sequencing artifact. Furthermore, the low levels at which the mutation was detected are consistent with prior reports of reduced viral fitness associated with this mutation (43). For isolate OHSU-E65, a minor population was found to harbor the amino acid substitution L501I, which is known to confer resistance to GCV and CDV (44). This mutation was identified in 5.2% and 6.3% of the viral population at the 1:20 and 1:2,000 dilutions, respectively. An isolate from the same patient obtained 5 months earlier demonstrated a different mutation at the same position, L501F (28). Similarly, isolate OHSU-E749 contained two different mutants at position P522 of UL54: a major P522S mutation (present in 87.6%) and a minor P522A mutation (present in 8.6%), both of which were reproducibly detected at two different sequencing depths and are associated with GCV and CDV resistance (44, 45). Finally, plasma specimen OHSU-E1162 contained a minor subpopulation (15.1%) of the UL97 resistance mutation A594V. Taken together, these results indicate that the NGS assay can reproducibly identify low-prevalence resistance mutations that may be missed by traditional genotyping approaches, although the findings in these particular cases would not have changed the resistance phenotypes that were inferred from the previously diagnosed mutations.
Next, we tested 48 plasma samples that had been submitted to the Stanford Clinical Virology laboratory for CMV viral load testing and could be amplified by long-range PCR. The majority of the specimens were from HSCT and SOT recipients receiving preemptive anti-CMV therapy or long-term prophylaxis (Table 1; see also Table S1 in the supplemental material). The viral loads of the plasma specimens, as well as patient characteristics, such as diagnosis, CMV serology status, CMV drug exposures, and history of testing by Sanger sequencing, can be found in Table S1 in the supplemental material. For 14 out of the 48 specimens that were tested, CMV drug resistance testing by Sanger sequencing had been performed at a reference laboratory within 21 days of when the specimen was tested in our assay. In two cases, the reference laboratory reported drug resistance mutations, H520Q in UL97 for patient 30 and M460V in UL97 for patient 32, both of which were identified as high-abundance DRMs by NGS (Table 5). Interestingly, patient 32 also had another UL97 mutation, L595S, which was present in 25.9% of the population. Lab-developed Sanger sequencing demonstrated a secondary peak at nucleotide 1784, indicating the presence of the expected T-to-C substitution (Fig. 5B). Five of the 14 cases were reported by the reference laboratory as being negative for resistance mutations, and the NGS results were concordant. However, for 7 of the 14 cases, the reference laboratory did not report a result due to a failure to amplify the UL97 and UL54 genes, which could have been related to low viral loads, failed extraction, amplification inhibition, or allelic dropout. In contrast, the NGS assay produced a definitive result in all of these cases and identified no resistance mutations in five cases and a low-abundance resistance mutation in two specimens (Table 5). The low-abundance mutation, C592G in UL97, was identified at 15.6% and 6.4% (Table 5 and Fig. 5D) in specimens from the same patient that were obtained ∼7 weeks apart.
Table 5.
CMV drug resistance mutations detected in clinical plasma specimens
| Gene name and sample no. | DRM | % variants | Resistance pattern | Reference lab genotype |
|---|---|---|---|---|
| UL54 | ||||
| AA7_10 | T503I | 41.7 | GCVr CDVr | Not tested |
| UL97 | ||||
| AA8_14 | M460V | 68.6 | GCVr | M460V |
| L595S | 25.9 | GCVr | ||
| AA7_08 | C592G | 6.4 | GCVr | Unable to amplify |
| AA8_17 | C592G | 15.6 | GCVr | Unable to amplify |
| AA7_10 | C603W | 14.4 | GCVr | Not tested |
| AA5_18 | H520Q | 77.3 | GCVr | H520Q |
Fig 5.
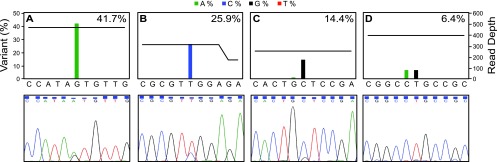
Comparison of next-generation and Sanger sequencing for detecting drug resistance mutations present as minor variants. Sequencing flowgrams (upper panels) and Sanger sequencing electropherograms (lower panels) are shown for four resistance mutations detected in the clinical plasma specimens: T503I in UL54 (A), L595S in UL97 (B), C603W in UL97 (C), and L595S in UL97 (D). The percent variant calls from the amplicon variant analyzer are indicated on the right of each flowgram. The horizontal black line on each flowgram represents the total number of reads covering each mutation and the 5 nucleotides 3′ and 5′ of the mutation.
Known drug resistance mutations were also identified in a clinical specimen that had not been submitted for testing at the reference laboratory. This specimen, with a viral count of 2,980 copies/ml, was from a kidney transplant recipient who was exposed to valganciclovir and was found to have the UL54 mutation T503I in 41.6% and the UL97 mutation C603W in 14.4% of the population (Fig. 5A and C). T503I has been shown to confer resistance to GCV and cross-resistance to CDV (46). Altogether, we identified 6 resistance mutations (5 in UL97 and 1 in UL54) that were present in <50% and >5% of the viral population (Table 5). Lab-developed Sanger sequencing identified secondary peaks for some of these DRMs, although for variants occurring in <20% of the viral population, the signal was not sufficiently above background to make a definitive call (Fig. 5).
We also examined the NGS data for the presence of putative variants in the clinical specimens that were assayed. We focused only on putative variants that were present in >20% of the population and led to missense mutations. This relatively high threshold was deemed appropriate, since the role of such variants in drug resistance is unknown, and they are not currently incorporated in clinical management algorithms, especially when present at low levels. Eleven out of the 48 specimens had 1 to 2 variants that fit these criteria (see Table S1 in the supplemental material) and will require phenotypic confirmation.
DISCUSSION
In this study, we report the development of an NGS assay for the detection of CMV drug resistance mutations. We have targeted regions of the UL54 and UL97 genes containing known drug resistance mutations using a nested PCR approach and overlapping amplicons, which ensures that in most cases, even specimens with viral loads of <1,000 copies/ml can be tested. The sequencing of plasmid clones demonstrated a low overall empirical error rate (0.189%), with higher error rates in the homopolymer regions for insertions and deletions but not substitutions. Error modeling based on observed error rates indicates strong dependence on the total number of reads covering a given position, as was shown previously for HIV (33). Finally, the NGS assay was used to evaluate 48 clinical plasma specimens, five of which were found to have one or more drug resistance mutations.
The detection of drug resistance in CMV has important implications for patient management, as resistance can impact morbidity and graft survival in transplant recipients (15, 16). Given that resistance to current CMV drugs occurs in two well-defined relatively small genomic regions, this virus is well suited for NGS methods that are analogous to the 454 pyrosequencing approaches used in HIV drug resistance testing (reviewed in reference 24). A number of studies have assessed the performance of NGS methods for capturing resistant HIV variants relative to that of Sanger sequencing (23, 33, 47–50) and demonstrated that at least half of the resistance mutations identified in HIV by NGS are missed by Sanger sequencing (23, 49, 51). This has allowed for a more accurate estimation of the rate of transmitted drug resistance to protease and reverse transcriptase inhibitors (23, 51), and it has contributed to increased sensitivity in monitoring the dynamics of viral population changes, specifically the emergence, disappearance, and archiving of drug resistance mutants on and off therapy (52). Three of the seven drug resistance mutations identified in our sample set were present in UL97 in <20% of the viral population. Even though in all of these cases, the patients' viremia was controlled with valganciclovir or ganciclovir, the impact of low-abundance drug-resistant CMV variants on patient outcomes has not been systematically investigated. Assays, such as the one described here, will allow for the prospective monitoring of the abundance of such variants over time and an assessment of their association with virologic failure.
One advantage of NGS platforms is their ability to multiplex the library preparation and sequencing of multiple specimens by bar coding each individual sample with a specific short sequence that can be recognized by the sequence processing software (53, 54). While CMV genotypic drug resistance testing is unlikely to occur on a high-volume scale, this ability to multiplex and bar code may allow for increased laboratory efficiency when used in combination with other NGS assays. Additionally, sample processing and sequencing are performed using benchtop instruments that have become increasingly accessible and can be more affordable than traditional genotyping approaches (55). The workflow of sequencing 24 specimens with this assay requires 5 days, approximately 15 h of hands-on time by a laboratory technologist, and is associated with reagent costs of ∼$70 per specimen. The maximum number of specimens that were simultaneously processed and sequenced in this study was 24, yielding 2,300 to 3,700 reads per sample and an average coverage of ∼300 reads per nucleotide, which is sufficient for a reliable identification of high-abundance variants, as was demonstrated by the sequencing results of archived CMV specimens with known mutations present in >20% of the population.
Despite the low error rates predicted by the statistical model, there was significant variability between the observed and expected percentage of minor variants for the plasmid mixes simulating 1,000 copies/ml. At low viral counts, the number of viral copies that are used for library preparation is small, and a mixed viral population may not be accurately represented in the aliquot used for long-range PCR. While underrepresented variants may be detected by sequencing to a greater depth of reads, variants that are absent from the aliquot due to random sampling will not be detected regardless of the total number of reads. Additionally, stochastic events in early PCR cycles and differential primer annealing efficiencies can lead to the variable amplification of some variants and nonrepresentative final PCR product mixtures (24, 56). Low-abundance variants are particularly susceptible to these effects, as illustrated by a recent study in which amplification and sequencing were performed in quadruplicate for a mixed viral population, yielding a coefficient of variation of 22.8% for the quantitation of minor variants (mean ± standard deviation [SD], 2.426% ± 0.55%) (40). In another study, the use of randomly tagged primers during library preparation was used to ensure that each template received a unique identifier (Primer ID), allowing random errors to be tracked for each individual template (41). The results demonstrated amplification skewing and a wide range of template representation, which was particularly pronounced for low-abundance variants (41).
One approach to address PCR bias in HIV sequencing is to perform multiple independent reverse transcription-PCRs (RT-PCRs) from the same clinical specimen and pool their products to serve as a template for library preparation (57). In fact, the assay described here takes a similar approach by performing two independent long-range PCRs and pooling their products for the 4-primer library preparation step. Performing the entire assay in duplicate or triplicate for low-viral-copy specimens and focusing only on variants that are reproducibly detected may also be helpful. Additionally, the statistical model developed here can be used to make variant calls, depending on the depth of coverage that is achieved for a given base.
A limitation of the assay was its inability to amplify 22.6% of the clinical plasma specimens by long-range PCR. All specimens that failed amplification had viral loads of ≤2,400 copies/ml. One possible explanation is that insufficient amounts of intact UL54 and UL97 DNA are present in such specimens due to the fragmentation that may occur in vivo or during sample processing or storage. Potential ways to address this issue include using larger volumes of template to increase the probability of including intact DNA or reducing the size of the long-range amplicons to increase PCR efficiency. Further clinical studies will be required to directly compare the performance of this assay with that of other CMV genotypic drug resistance tests.
To our knowledge, this is the first report of a next-generation sequencing assay for CMV genotypic drug resistance identification and its application to clinical specimens. Data from NGS studies of CMV drug resistance may ultimately contribute to the development of DRM databases and genotypic interpretation systems similar to those that exist for HIV (58–60). Such tools may be particularly important as new antiviral therapies targeting CMV become available.
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of the Stanford Clinical Virology and Histocompatibility, Immunogenetics, and Disease Profiling laboratories for their continued diligent work and dedication to patient care. In particular, we thank Paolo Libiran for his help with CMV quantitation and Heidi Farina for technical support.
Matthew W. Anderson received honoraria from Roche Diagnostics. The other authors declare no conflicts of interest in the publication of this research.
The analysis of OHSU specimens was supported by Department of Veterans Affairs research funds (grant no. I01-BX00925 to S.C.). The statistical analysis was partially supported by NIH grant R01 GM086884 (to S.P.H.).
Footnotes
Published ahead of print 28 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.01605-13.
REFERENCES
- 1. Boeckh M, Geballe AP. 2011. Cytomegalovirus: pathogen, paradigm, and puzzle. J. Clin. Invest. 121:1673–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baroco AL, Oldfield EC. 2008. Gastrointestinal cytomegalovirus disease in the immunocompromised patient. Curr. Gastroenterol. Rep. 10:409–416 [DOI] [PubMed] [Google Scholar]
- 3. Castagnola E, Cappelli B, Erba D, Rabagliati A, Lanino E, Dini G. 2004. Cytomegalovirus infection after bone marrow transplantation in children. Hum. Immunol. 65:416–422 [DOI] [PubMed] [Google Scholar]
- 4. Owers DS, Webster AC, Strippoli GF, Kable K, Hodson EM. 2013. Pre-emptive treatment for cytomegalovirus viraemia to prevent cytomegalovirus disease in solid organ transplant recipients. Cochrane Database Syst. Rev. 2:CD005133. 10.1002/14651858.CD006133.pub3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hodson EM, Ladhani M, Webster AC, Strippoli GF, Craig JC. 2013. Antiviral medications for preventing cytomegalovirus disease in solid organ transplant recipients. Cochrane Database Syst. Rev. 2:CD003774. 10.1002/14651858.CD003774.pub3 [DOI] [PubMed] [Google Scholar]
- 6. Lurain NS, Chou S. 2010. Antiviral drug resistance of human cytomegalovirus. Clin. Microbiol. Rev. 23:689–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Drew WL. 2010. Cytomegalovirus resistance testing: pitfalls and problems for the clinician. Clin. Infect. Dis. 50:733–736 [DOI] [PubMed] [Google Scholar]
- 8. Kotton CN. 2010. Management of cytomegalovirus infection in solid organ transplantation. Nat. Rev. Nephrol. 6:711–721 [DOI] [PubMed] [Google Scholar]
- 9. Yu MA, Park JM. 2013. Valganciclovir: therapeutic role in pediatric solid organ transplant recipients. Expert Opin. Pharmacother 14:807–815 [DOI] [PubMed] [Google Scholar]
- 10. Biron KK, Stanat SC, Sorrell JB, Fyfe JA, Keller PM, Lambe CU, Nelson DJ. 1985. Metabolic activation of the nucleoside analog 9-[(2-hydroxy-1-(hydroxymethyl)ethoxy]methyl)guanine in human diploid fibroblasts infected with human cytomegalovirus. Proc. Natl. Acad. Sci. U. S. A. 82:2473–2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kotton CN, Kumar D, Caliendo AM, Asberg A, Chou S, Snydman DR, Allen U, Humar A, Transplantation Society International CMV Consensus Group 2010. International consensus guidelines on the management of cytomegalovirus in solid organ transplantation. Transplantation 89:779–795 [DOI] [PubMed] [Google Scholar]
- 12. Crumpacker CS. 1996. Ganciclovir. N. Engl. J. Med. 335:721–729 [DOI] [PubMed] [Google Scholar]
- 13. Field AK, Davies ME, DeWitt C, Perry HC, Liou R, Germershausen J, Karkas JD, Ashton WT, Johnston DB, Tolman RL. 1983. 9-([2-hydroxy-1-(hydroxymethyl)ethoxy]methyl)guanine: a selective inhibitor of herpes group virus replication. Proc. Natl. Acad. Sci. U. S. A. 80:4139–4143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Myhre HA, Haug Dorenberg D, Kristiansen KI, Rollag H, Leivestad T, Asberg A, Hartmann A. 2011. Incidence and outcomes of ganciclovir-resistant cytomegalovirus infections in 1244 kidney transplant recipients. Transplantation 92:217–223 [DOI] [PubMed] [Google Scholar]
- 15. Limaye AP, Corey L, Koelle DM, Davis CL, Boeckh M. 2000. Emergence of ganciclovir-resistant cytomegalovirus disease among recipients of solid-organ transplants. Lancet 356:645–649 [DOI] [PubMed] [Google Scholar]
- 16. Li F, Kenyon KW, Kirby KA, Fishbein DP, Boeckh M, Limaye AP. 2007. Incidence and clinical features of ganciclovir-resistant cytomegalovirus disease in heart transplant recipients. Clin. Infect. Dis. 45:439–447 [DOI] [PubMed] [Google Scholar]
- 17. Hantz S, Garnier-Geoffroy F, Mazeron MC, Garrigue I, Merville P, Mengelle C, Rostaing L, Saint Marcoux F, Essig M, Rerolle JP, Cotin S, Germi R, Pillet S, Lebranchu Y, Turlure P, Alain S, French CMV Resistance Survey Study Group 2010. Drug-resistant cytomegalovirus in transplant recipients: a French cohort study. J. Antimicrob. Chemother. 65:2628–2640 [DOI] [PubMed] [Google Scholar]
- 18. Boivin G, Gilbert C, Gaudreau A, Greenfield I, Sudlow R, Roberts NA. 2001. Rate of emergence of cytomegalovirus (CMV) mutations in leukocytes of patients with acquired immunodeficiency syndrome who are receiving valganciclovir as induction and maintenance therapy for CMV retinitis. J. Infect. Dis. 184:1598–1602 [DOI] [PubMed] [Google Scholar]
- 19. Gilbert C, Handfield J, Toma E, Lalonde R, Bergeron MG, Boivin G. 1998. Emergence and prevalence of cytomegalovirus UL97 mutations associated with ganciclovir resistance in AIDS patients. AIDS 12:125–129 [DOI] [PubMed] [Google Scholar]
- 20. Jabs DA, Dunn JP, Enger C, Forman M, Bressler N, Charache P. 1996. Cytomegalovirus retinitis and viral resistance. Prevalence of resistance at diagnosis, 1994. Cytomegalovirus Retinitis and Viral Resistance Study Group. Arch. Ophthalmol. 114:809–814 [DOI] [PubMed] [Google Scholar]
- 21. Martin BK, Ricks MO, Forman MS, Jabs DA, Cytomegalovirus Retinitis and Viral Resistance Study Group 2007. Change over time in incidence of ganciclovir resistance in patients with cytomegalovirus retinitis. Clin. Infect. Dis. 44:1001–1008 [DOI] [PubMed] [Google Scholar]
- 22. Ljungman P, Reusser P, de la Camara R, Einsele H, Engelhard D, Ribaud P, Ward K, European Group for Blood and Marrow Transplantation 2004. Management of CMV infections: recommendations from the infectious diseases working party of the EBMT. Bone Marrow Transplant. 33:1075–1081 [DOI] [PubMed] [Google Scholar]
- 23. Simen BB, Simons JF, Hullsiek KH, Novak RM, Macarthur RD, Baxter JD, Huang C, Lubeski C, Turenchalk GS, Braverman MS, Desany B, Rothberg JM, Egholm M, Kozal MJ, Terry Beirn Community Programs for Clinical Research on AIDS 2009. Low-abundance drug-resistant viral variants in chronically HIV-infected, antiretroviral treatment-naive patients significantly impact treatment outcomes. J. Infect. Dis. 199:693–701 [DOI] [PubMed] [Google Scholar]
- 24. Vrancken B, Lequime S, Theys K, Lemey P. 2010. Covering all bases in HIV research: unveiling a hidden world of viral evolution. AIDS Rev. 12:89–102 [PubMed] [Google Scholar]
- 25. Görzer I, Guelly C, Trajanoski S, Puchhammer-Stöckl E. 2010. Deep sequencing reveals highly complex dynamics of human cytomegalovirus genotypes in transplant patients over time. J. Virol. 84:7195–7203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Renzette N, Bhattacharjee B, Jensen JD, Gibson L, Kowalik TF. 2011. Extensive genome-wide variability of human cytomegalovirus in congenitally infected infants. PLoS Pathog. 7:e1001344. 10.1371/journal.ppat.1001344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Strasfeld L, Lee I, Tatarowicz W, Villano S, Chou S. 2010. Virologic characterization of multidrug-resistant cytomegalovirus infection in 2 transplant recipients treated with maribavir. J. Infect. Dis. 202:104–108 [DOI] [PubMed] [Google Scholar]
- 28. Erice A, Gil-Roda C, Pérez JL, Balfour HH, Jr, Sannerud KJ, Hanson MN, Boivin G, Chou S. 1997. Antiviral susceptibilities and analysis of UL97 and DNA polymerase sequences of clinical cytomegalovirus isolates from immunocompromised patients. J. Infect. Dis. 175:1087–1092 [DOI] [PubMed] [Google Scholar]
- 29. Chou S, Marousek G, Guentzel S, Follansbee SE, Poscher ME, Lalezari JP, Miner RC, Drew WL. 1997. Evolution of mutations conferring multidrug resistance during prophylaxis and therapy for cytomegalovirus disease. J. Infect. Dis. 176:786–789 [DOI] [PubMed] [Google Scholar]
- 30. Chou S. 2010. Recombinant phenotyping of cytomegalovirus UL97 kinase sequence variants for ganciclovir resistance. Antimicrob. Agents Chemother. 54:2371–2378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Waggoner J, Ho DY, Libiran P, Pinsky BA. 2012. Clinical significance of low cytomegalovirus DNA levels in human plasma. J. Clin. Microbiol. 50:2378–2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hakki M, Chou S. 2011. The biology of cytomegalovirus drug resistance. Curr. Opin. Infect. Dis. 24:605–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang C, Mitsuya Y, Gharizadeh B, Ronaghi M, Shafer RW. 2007. Characterization of mutation spectra with ultra-deep pyrosequencing: application to HIV-1 drug resistance. Genome Res. 17:1195–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McCullagh P, Nelder JA. 1989. Generalized linear model, vol 2 Chapman & Hall/CRC, Boca Raton, FL [Google Scholar]
- 35. Ihaka R, Gentleman R. 1996. R: a language for data analysis and graphics. J. Comput. Graph. Stat. 5:299–314 [Google Scholar]
- 36. Venables WN, Ripley BD. 2002. Modern Applied Statistics with S, 4th ed. Springer, New York, NY [Google Scholar]
- 37. Gilbert C, Azzi A, Goyette N, Lin SX, Boivin G. 2011. Recombinant phenotyping of cytomegalovirus UL54 mutations that emerged during cell passages in the presence of either ganciclovir or foscarnet. Antimicrob. Agents Chemother. 55:4019–4027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chevillotte M, Ersing I, Mertens T, von Einem J. 2010. Differentiation between polymorphisms and resistance-associated mutations in human cytomegalovirus DNA polymerase. Antimicrob. Agents Chemother. 54:5004–5011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Boutolleau D, Burrel S, Agut H. 2011. Genotypic characterization of human cytomegalovirus UL97 phosphotransferase natural polymorphism in the era of ganciclovir and maribavir. Antiviral Res. 91:32–35 [DOI] [PubMed] [Google Scholar]
- 40. Tsibris AM, Korber B, Arnaout R, Russ C, Lo CC, Leitner T, Gaschen B, Theiler J, Paredes R, Su Z, Hughes MD, Gulick RM, Greaves W, Coakley E, Flexner C, Nusbaum C, Kuritzkes DR. 2009. Quantitative deep sequencing reveals dynamic HIV-1 escape and large population shifts during CCR5 antagonist therapy in vivo. PLoS One 4:e5683. 10.1371/journal.pone.0005683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jabara CB, Jones CD, Roach J, Anderson JA, Swanstrom R. 2011. Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc. Natl. Acad. Sci. U. S. A. 108:20166–20171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mousavi-Jazi M, Schloss L, Drew WL, Linde A, Miner RC, Harmenberg J, Wahren B, Brytting M. 2001. Variations in the cytomegalovirus DNA polymerase and phosphotransferase genes in relation to foscarnet and ganciclovir sensitivity. J. Clin. Virol. 23:1–15 [DOI] [PubMed] [Google Scholar]
- 43. Springer KL, Chou S, Li S, Giller RH, Quinones R, Shira JE, Weinberg A. 2005. How evolution of mutations conferring drug resistance affects viral dynamics and clinical outcomes of cytomegalovirus-infected hematopoietic cell transplant recipients. J. Clin. Microbiol. 43:208–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cihlar T, Fuller MD, Cherrington JM. 1998. Characterization of drug resistance-associated mutations in the human cytomegalovirus DNA polymerase gene by using recombinant mutant viruses generated from overlapping DNA fragments. J. Virol. 72:5927–5936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chou S, Marousek G, Li S, Weinberg A. 2008. Contrasting drug resistance phenotypes resulting from cytomegalovirus DNA polymerase mutations at the same exonuclease locus. J. Clin. Virol. 43:107–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chou S, Lurain NS, Thompson KD, Miner RC, Drew WL. 2003. Viral DNA polymerase mutations associated with drug resistance in human cytomegalovirus. J. Infect. Dis. 188:32–39 [DOI] [PubMed] [Google Scholar]
- 47. Mitsuya Y, Varghese V, Wang C, Liu TF, Holmes SP, Jayakumar P, Gharizadeh B, Ronaghi M, Klein D, Fessel WJ, Shafer RW. 2008. Minority human immunodeficiency virus type 1 variants in antiretroviral-naive persons with reverse transcriptase codon 215 revertant mutations. J. Virol. 82:10747–10755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fisher R, van Zyl GU, Travers SA, Kosakovsky Pond SL, Engelbrech S, Murrell B, Scheffler K, Smith D. 2012. Deep sequencing reveals minor protease resistance mutations in patients failing a protease inhibitor regimen. J. Virol. 86:6231–6237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Le T, Chiarella J, Simen BB, Hanczaruk B, Egholm M, Landry ML, Dieckhaus K, Rosen MI, Kozal MJ. 2009. Low-abundance HIV drug-resistant viral variants in treatment-experienced persons correlate with historical antiretroviral use. PLoS One 4:e6079. 10.1371/journal.pone.0006079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Varghese V, Shahriar R, Rhee SY, Liu T, Simen BB, Egholm M, Hanczaruk B, Blake LA, Gharizadeh B, Babrzadeh F, Bachmann MH, Fessel WJ, Shafer RW. 2009. Minority variants associated with transmitted and acquired HIV-1 nonnucleoside reverse transcriptase inhibitor resistance: implications for the use of second-generation nonnucleoside reverse transcriptase inhibitors. J. Acquir. Immune Defic. Syndr. 52:309–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lataillade M, Chiarella J, Yang R, Schnittman S, Wirtz V, Uy J, Seekins D, Krystal M, Mancini M, McGrath D, Simen B, Egholm M, Kozal M. 2010. Prevalence and clinical significance of HIV drug resistance mutations by ultra-deep sequencing in antiretroviral-naive subjects in the CASTLE study. PLoS One 5:e10952. 10.1371/journal.pone.0010952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hedskog C, Mild M, Jernberg J, Sherwood E, Bratt G, Leitner T, Lundeberg J, Andersson B, Albert J. 2010. Dynamics of HIV-1 quasispecies during antiviral treatment dissected using ultra-deep pyrosequencing. PLoS One 5:e11345. 10.1371/journal.pone.0011345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hoffmann C, Minkah N, Leipzig J, Wang G, Arens MQ, Tebas P, Bushman FD. 2007. DNA bar coding and pyrosequencing to identify rare HIV drug resistance mutations. Nucleic Acids Res. 35:e91. 10.1093/nar/gkm435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R. 2008. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat. Methods 5:235–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dudley DM, Chin EN, Bimber BN, Sanabani SS, Tarosso LF, Costa PR, Sauer MM, Kallas EG, O'Connor DH. 2012. Low-cost ultra-wide genotyping using Roche/454 pyrosequencing for surveillance of HIV drug resistance. PLoS One 7:e36494. 10.1371/journal.pone.0036494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mild M, Hedskog C, Jernberg J, Albert J. 2011. Performance of ultra-deep pyrosequencing in analysis of HIV-1 pol gene variation. PLoS One 6:e22741. 10.1371/journal.pone.0022741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vandenbroucke I, Van Marck H, Mostmans W, Van Eygen V, Rondelez E, Thys K, Van Baelen K, Fransen K, Vaira D, Kabeya K, De Wit S, Florence E, Moutschen M, Vandekerckhove L, Verhofstede C, Stuyver LJ. 2010. HIV-1 V3 envelope deep sequencing for clinical plasma specimens failing in phenotypic tropism assays. AIDS Res. Ther. 7:4. 10.1186/1742-6405-7-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tang MW, Liu TF, Shafer RW. 2012. The HIVdb system for HIV-1 genotypic resistance interpretation. Intervirology 55:98–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Woods CK, Brumme CJ, Liu TF, Chui CK, Chu AL, Wynhoven B, Hall TA, Trevino C, Shafer RW, Harrigan PR. 2012. Automating HIV drug resistance genotyping with RECall, a freely accessible sequence analysis tool. J. Clin. Microbiol. 50:1936–1942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zazzi M, Kaiser R, Sönnerborg A, Struck D, Altmann A, Prosperi M, Rosen-Zvi M, Petroczi A, Peres Y, Schülter E, Boucher CA, Brun-Vezinet F, Harrigan PR, Morris L, Obermeier M, Perno CF, Phanuphak P, Pillay D, Shafer RW, Vandamme AM, van Laethem K, Wensing AM, Lengauer T, Incardona F. 2011. Prediction of response to antiretroviral therapy by human experts and by the EuResist data-driven expert system (the EVE study). HIV Med. 12:211–218 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.