

Abstract
The food-borne pathogen Listeria monocytogenes is genetically heterogeneous. Although some clonal groups have been implicated in multiple outbreaks, there is currently no consensus on how “epidemic clones” should be defined. The objectives of this work were to compare the patterns of sequence diversity on two sets of genes that have been widely used to define L. monocytogenes clonal groups: multilocus sequence typing (MLST) and multi-virulence-locus sequence typing (MvLST). Further, we evaluated the diversity within clonal groups by pulsed-field gel electrophoresis (PFGE). Based on 125 isolates of diverse temporal, geographical, and source origins, MLST and MvLST genes (i) had similar patterns of sequence polymorphisms, recombination, and selection, (ii) provided concordant phylogenetic clustering, and (iii) had similar discriminatory power, which was not improved when we combined both data sets. Inclusion of representative strains of previous outbreaks demonstrated the correspondence of epidemic clones with previously recognized MLST clonal complexes. PFGE analysis demonstrated heterogeneity within major clones, most of which were isolated decades before their involvement in outbreaks. We conclude that the “epidemic clone” denominations represent a redundant but largely incomplete nomenclature system for MLST-defined clones, which must be regarded as successful genetic groups that are widely distributed across time and space.
INTRODUCTION
Listeria monocytogenes is a food-borne pathogen that can cause listeriosis, a severe invasive infection with a particularly high (20 to 30%) case fatality rate in persons at risk. Listeriosis is currently regarded as increasing in incidence in Europe, especially in the elderly (1, 2), and can occur in large outbreaks, as illustrated in recent years (3, 4). To assist epidemiological surveillance and outbreak investigations, different strain typing methods have been used, including serotyping and pulsed-field gel electrophoresis (PFGE) (5, 6). Population diversity studies on the global scale have revealed that L. monocytogenes is a genetically heterogeneous species (7–10), and a variety of strain genotyping methods have been used to characterize and classify isolates into four major lineages and clonal groups thereof (9–15). The precise delineation of lineages and clonal groups is a prerequisite to characterize the links between within-species genetic variations and important characteristics, such as pathogenic potential, virulence, or epidemiology.
Given the facultative nature of genetic exchange in bacteria, reproduction is predominantly clonal. In evolutionary biology terms, clones can be defined as groups of isolates that have descended from a common ancestor and accumulate differences among themselves by a predominantly mutational process. As L. monocytogenes is one of the bacterial species with the lowest rate of homologous recombination (9, 16), clones are expected to evolve slowly and to be recognizable over large temporal and geographic scales. As a matter of fact, the discovery of genetically similar isolates involved in either geographically and temporally distant outbreaks or in large, single outbreaks led to the definition of L. monocytogenes epidemic clones (ECs) (6, 8, 16–18). Although the initial ECs were defined primarily based on PFGE, multilocus enzyme electrophoresis, and ribotyping (6, 18), subsequent ECs have been mostly defined on the basis of multi-virulence-locus sequence typing (MvLST) (4, 17, 19). MvLST, which is based on the analysis of six to eight genes, has also been used to redefine previously described epidemic clones (17, 20).
In many bacterial species, multilocus sequence typing (MLST) is used as a reference method for clonal group definition (21, 22). An MLST scheme based on seven housekeeping genes was developed for L. monocytogenes (9, 23). Using this approach, we recognized highly prevalent clones (9, 24), which were defined by using a simple and flexible operational definition: clones are clonal complexes (CCs), i.e., groups that share 6 out of 7 allelic sequences with at least one other member of the group. Currently, it is unknown how MLST clonal complexes and epidemic clones correspond to each other.
The objectives of this study were (i) to determine the phylogenetic position of reference strains of previously defined epidemic clones within the MLST framework, (ii) to compare the patterns of diversity, recombination, and selection of MLST and MvLST genes, and (iii) to estimate the amounts of PFGE diversity within clonal groups or epidemic clones. We found that MLST and MvLST define largely concordant clonal groups and that the current approach of defining epidemic clones based on MvLST is redundant with the MLST nomenclature.
MATERIALS AND METHODS
Strain selection.
A total of 125 Listeria monocytogenes isolates were included (see Table S1 in the supplemental material). First, in order to establish the position of outbreak strains or other reference strains in the international MLST scheme maintained at Institut Pasteur (www.pasteur.fr/mlst), a group of 50 strains was assembled. This reference set included (i) 27 reference strains from well-documented outbreaks, including 19 strains of the outbreak set included in the ILSI collection (25) and 8 genome sequence reference strains corresponding to outbreak strains, (ii) 15 lineage I and II strains from the diversity set of the ILSI collection, (iii) 7 additional reference strains, representing available genome sequences and reference strain EGD. This reference set included the intensely studied laboratory reference strains EGD-e, LO28, 10304S, F2365, and Scott A, as well as strains representing epidemic clones ECI, ECII, ECIII, and ECIa/ECIV.
Second, with the aim of evaluating the ability of PFGE and MvLST to discriminate within and among MLST-defined CCs, we selected 75 isolates (the clone diversity set) representing multiple isolates of the major MLST-defined CCs, i.e., CC1, CC2, CC6, CC7, CC8, and CC9 (9, 24). These isolates were selected from our previous global MLST study of L. monocytogenes (24) to represent geographically and temporally diverse isolates. They originated from the Institut Pasteur Listeria Collection (CLIP) and from Seeliger's Listeria Culture Collection (SLCC) (26).
Identification and serotyping.
Isolates were identified as L. monocytogenes by using API Listeria strips (bioMérieux, La Balme Les Grottes, France), and their serotype was determined by classical serotyping and PCR serogrouping methods (27). Genomic DNA used as the MLST or MvLST PCR template was extracted using the Wizard genomic DNA purification kit (Promega, Madison, WI).
MLST.
For the purposes of this study, MLST was performed as described previously (9). Novel alleles and profiles were incorporated into the international MLST database at www.pasteur.fr/mlst. MLST gene sequences from 16 publicly available genome sequences were extracted from the public sequence repositories.
MvLST.
In this study, eight virulence-associated genes of L. monocytogenes were sequenced for 111 strains, while for 16 other strains the gene sequences were extracted from the available genome sequences. Sequences for six of the MvLST genes (prfA, inlB, inlC, clpP, dal, and lisR) were obtained from the S. Knabel group's MvLST scheme (28). The two remaining genes, inlA and actA, were included because they were used to complement the six previous ones in a subsequent MvLST analysis (17). Note that we sequenced the entire length of the inlA gene (2,400 nucleotides [nt]), as we did in a previous study (9), whereas only 458 nt of this gene were sequenced by Chen et al. (17). For actA, because we experienced difficulties with PCR amplification when we used previously described primers, we sequenced 450 nt from the 3′ part of the gene, whereas the 582-nt template described by Chen et al. (17) corresponds to the 5′ region of the actA gene. PCR primers (Table 1) were based on those described by Chen et al. (17) or were designed in this study with eprimer3 (http://mobyle.pasteur.fr/cgi-bin/portal.py?#forms::eprimer3) based on the published genome sequence of Listeria monocytogenes strain EGD-e (NCBI accession number NC_003210.1). PCR amplification conditions were as follows: for the gene dal, 10 min at 95°C, followed by 30 cycles of 95°C for 30 s, 57.6°C for 30 s, and 72°C for 36 s, and a final extension for 5 min at 72°C. For prfA, inlB, inlC, clpP, and lisR, the conditions were 15 min at 95°C followed by 25 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min, with a final 7 min at 72°C. For actA, the conditions were 10 min at 95°C followed by 30 cycles of 95°C for 30 s, 54.7°C for 30 s, and 72°C for 36 s, and finally 5 min at 72°C. Finally, for inlA, the conditions were 5 min at 94°C followed by 35 cycles of 94°C for 30 s, 55.2°C for 30 s, and 72°C for 90 s, and a final 10 min at 72°C. The PCR products were purified by ultrafiltration (Millipore, France) and were sequenced on both strands by using BigDye v.1.1 chemistry on an ABI 3730XL sequencer (Applied BioSystems). As for MLST, each nucleotide was sequenced in both directions and validated by at least two independent chromatogram traces. MvLST gene sequences from the published genome were extracted from GenBank entries.
Table 1.
Primers used for MvLST
| Gene | Forward primer | Reverse primer | Location (EGD-e) | Size of PCR product (bp) | Size of template (bp) | Reference |
|---|---|---|---|---|---|---|
| actA | CGA CAT AAT ATT TGC AGC GAC A | GAA TCT AAG TCA CTT TCA GAA GCA T | 209,541–210,041 | 500 | 450 | This study |
| clpP | CCA ACA GTA ATT GAA CAA ACT AGC C | GAT CTG TAT CGC GAG CAA TG | 2,542,026–2,542,524 | 498 | 419 | Zhang et al. (28) |
| dal | GAA GGT ATC TAC ACG CAT TTT GC | GCC AAT TAT CGT TAC TTT TGA ACC | 925,391–925,909 | 518 | 428 | This study |
| inlB | CAT GGG AGA GTA ACC CAA CC | GCG GTA ACC CCT TTG TCA TA | 457,954–458,453 | 499 | 432 | Zhang et al. (28) |
| inlC | AAC CAT CTA CAT AAC TCC CAC | CGG GAA TGC AAT TTT TCA CTA | 1,860,322–1,860,822 | 500 | 457 | Zhang et al. (28) |
| lisR | CGG GGT AGA AGT TTG TCG TC | ACG CAT CAC ATA CCC TGT CC | 1,402,726–1,403,224 | 498 | 447 | Zhang et al. (28) |
| prfA | TGC GAT GCC ACT TGA ATA TC | AAC GGG ATA AAA CCA AAA CCA | 203,811–204,311 | 500 | 460 | Zhang et al. (28) |
| inlA | CGGATGCAGGAGAAAATCC | CTTTCACACTATCCTCTCC | 454,463–457,027 | 2,564 | 2,403 | Ragon et al. (9) |
PFGE.
Each isolate was typed by PFGE according to PulseNet standardized procedures, with AscI and ApaI restriction enzymes (29). Data analysis was performed using BioNumerics version 6.5 (Applied Maths, Sint-Martens-Latem, Belgium). ApaI and AscI PFGE types were defined as differing from other types by at least two bands for each individual enzyme.
Phylogenetic reconstructions.
Clonal complexes were defined based on MLST data as groups of allelic profiles sharing 6 out of 7 genes with at least one other member of the group (9). For phylogenetic analyses, gene sequences were concatenated independently for the MLST and MvLST schemes, and neighbor-joining trees were obtained using the BioNumerics program based on the concatenated sequences for the p-distance (i.e., the uncorrected percentage of nucleotide mismatches). Nucleotide polymorphism and summary statistics were calculated via Dnasp v5 (30). Minimum spanning trees were constructed using the BioNumerics program. Simpson's index and the adjusted Rand coefficient were computed using the tools on the website www.comparingpartitions.info (31).
Recombination and selection analyses.
We tested for recombination within phylogenetic lineages I and II for all loci independently, as well as for the concatenated MvLST and MLST loci, by using the LDhat v2.2 software (32). LDhat employs a coalescent-based method to estimate the population-scaled mutation rate (θ = 2Neμ) and recombination rate (ρ = 2Ner), where Ne is the effective population size, r is the rate at which recombination events separate adjacent nucleotides, and μ is the mutation rate per nucleotide. The ratio r/μ is calculated as ρ/(θ/L), where L is the gene length (sequence length). This r/μ ratio ranges from 0, which indicates full clonal reproduction, to ≫1, which is expected under free recombination. The significance of the evidence for recombination was tested using nonparametric, permutation-based tests implemented in LDhat.
For signatures of positive selection, flat ω(dN/dS) ratios (as generated with Dnasp) might fail to detect ongoing selection on a fraction of the molecule. We therefore tested the presence of both recombination and positive selection by using the OmegaMap program (33), which is able to disentangle the confounding effects of recombination and selection in a Bayesian framework. We chose a model with variable blocks of the ratio of nonsynonymous-to-synonymous rates, ω, and the population recombination rate, ρ. A first run was performed for all loci by using a block size set at 30 codons. A second run was performed using a block size set at 10 codons, for the loci where a posterior probability of selection greater than 0.80 was detected in the first run. For all runs, an inverse distribution of range of 0.01 to 100 was used for ω and ρ. For the other parameters, μ, κ, and ΦInDel, we used improper inverse distributions with starting values of 0.1, 3.0, and 0.1, respectively. All gene analyses were run with 100,000 iterations and 10 reorderings, with the first 8,000 iterations discarded as the burn-in period.
RESULTS AND DISCUSSION
Phylogenetic positions of outbreak and reference strains based on MLST.
To position reference strains, including isolates previously assigned to epidemic clones I to IV, within the diversity of L. monocytogenes, the sequences of internal portions of the seven MLST genes (3,288 nucleotides in total) were gathered from 14 available genome sequences, 34 strains of the ILSI reference collection analyzed previously (11), and 76 isolates of clones belonging to CC1, CC2, CC6, CC7, CC8, and CC9 (11). In addition, we sequenced the 7 MLST genes from strain EGD, which is used as a reference strain (34, 35) but has not been genotyped by MLST to our knowledge. For these 125 isolates or reference strains, alignment of the sequences of six genes revealed no insertion/deletion event, whereas the ldh sequence of strain EGD revealed a 2-codon insertion at alignment position 423. There were 20 to 61 variable nucleotide sites per gene, corresponding to 7.5% of variable sites on average (Table 2). This variation allowed distinction of 10 to 20 alleles per gene, resulting in 51 distinct sequence types (STs).
Table 2.
Polymorphisms of the MLST and MvLST genes, based on the 125 study isolatesa
| Gene group and name | Length | No. of alleles | No. of polymorphic sites | % polymorphic sites | Ks | Ka | Ka/Ks | π (%) |
|---|---|---|---|---|---|---|---|---|
| MvLST genes | ||||||||
| actA | 450 | 14 | 44 | 9.78 | 0.07 | 0.02 | 0.35 | 3.21 |
| clpP | 419 | 4 | 14 | 3.34 | 0.06 | 0.001 | 0.02 | 1.44 |
| dal | 428 | 14 | 54 | 12.62 | 0.15 | 0.002 | 0.01 | 3.88 |
| inlA | 2403 | 28 | 140 | 5.83 | 0.06 | 0.06 | 1.01 | 1.76 |
| inlB | 432 | 15 | 40 | 9.26 | 0.03 | 0.04 | 1.38 | 3.38 |
| inlC | 457 | 10 | 14 | 3.06 | 0.03 | 0.01 | 0.25 | 1.18 |
| lisR | 447 | 5 | 38 | 8.50 | 0.02 | 0 | 0 | 0.51 |
| prfA | 460 | 9 | 20 | 4.35 | 0.06 | 0.00008 | 0.001 | 1.37 |
| Concatenated | 5,496 | 40 | 364 | 6.62 | 0.06 | 0.03 | 0.53 | 1.96 |
| MLST genes | ||||||||
| abcZ | 537 | 14 | 37 | 6.89 | 0.08 | 0.001 | 0.01 | 1.96 |
| bglA | 399 | 15 | 21 | 5.26 | 0.04 | 0.0006 | 0.01 | 0.91 |
| cat | 486 | 17 | 26 | 5.35 | 0.08 | 0.002 | 0.03 | 1.93 |
| dapE | 462 | 19 | 40 | 8.66 | 0.11 | 0.01 | 0.08 | 3.27 |
| dat | 471 | 10 | 61 | 12.95 | 0.20 | 0.01 | 0.07 | 5.72 |
| ldh | 453 | 20 | 39 | 8.61 | 0.06 | 0.0008 | 0.01 | 1.37 |
| lhkA | 480 | 10 | 20 | 4.17 | 0.06 | 0.0026 | 0.04 | 1.55 |
| Concatenated | 3,294 | 51 | 244 | 7.41 | 0.09 | 0.004 | 0.05 | 2.41 |
Ks, number of synonymous changes per synonymous site; Ka, number of nonsynonymous changes per nonsynonymous site; π, nucleotide diversity.
As previously described (11), CC1 includes the reference strains of the 1981 Nova Scotia coleslaw outbreak, the 1983 to 1987 Switzerland Vacherin Mont d'Or outbreak, the 1986-1987 California Jalisco soft cheese outbreak, and the 1992 French pork tongue in jelly outbreak (Fig. 1). These strains were previously attributed to epidemic clone ECI (6). Clone CC2 comprised reference strains of the 1983 Massachusetts pasteurized milk outbreak (strain Scott A), the 1987-1988 United Kingdom and Ireland pâté outbreak, and the 1997 Italy gastroenteritis outbreak (Fig. 1). These strains have been attributed to ECIa (18), later renamed ECIV with a more restricted definition that excluded the 1983 Massachusetts milk outbreak (17). All these reference strains, including Scott A, belong to ST2, which is the central and most frequent genotype of CC2 (Fig. 1). These results showed that reference strains of ECI, ECII, and ECIa/ECIV fall in the MLST-defined CC1, CC6, and CC2 groups, respectively. CC3 included the reference strain of the 1994 Illinois chocolate milk outbreak. CC4 included the 1999-2000 France pork rillettes outbreak. CC6 included the 1998-1999 U.S. multistate hotdog outbreak and the 2002 U.S. multistate delicatessen turkey outbreak, previously attributed to ECII. Finally, the 2000-2001 North Carolina Mexican-style fresh cheese outbreak reference strain belongs to ST558 (Fig. 1). The unique genotype of this outbreak strain is consistent with its characterization as ST24 (12). Note that ST5 included the isolates of the recently described ECVI clone (4).
Fig 1.
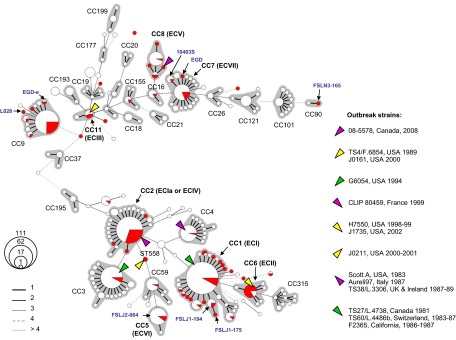
Minimum spanning tree of MLST data for 863 L. monocytogenes isolates. Each circle represents one ST, the size of which is related to the number of isolates with this ST, as indicated (111 represents ST1, the central ST of CC1). Red sectors represent the 125 isolates or reference strains included in this work, whereas white sectors represent previously published strains. Links between circles are represented according to the number of allelic mismatches between STs, as indicated. Gray zones surrounding groups of STs represent CC. Most CC numbers are indicated; those CCs that correspond to “epidemic clones” are written in bold with the corresponding EC number in parentheses. Laboratory reference strains are indicated in blue. STs of outbreak reference strains are indicated by colored triangles as shown on the right. Top, lineage II (starting with CC37); bottom, lineage I (starting with CC195).
Lineage II comprised reference strains of fewer outbreaks. ST11 included the 1988 Oklahoma turkey franks case and the two strains from the 2000 U.S. multistate sliced turkey deli meat outbreak, which was traced to the same food processing facility as the 1988 isolate. All three ST11 strains were previously attributed to ECIII (6). Note that whereas ECI, ECII, and ECIV have been implicated in multiple outbreaks (6, 8), this was not the case to our knowledge for ECIII. CC8 comprised the 2008 Canada ready-to-eat meat products outbreak, consistent with a previous report (3). Strains of this outbreak were recently proposed to represent ECV (19). Clone ECVII (involved in the 2011 U.S. cantaloupe outbreak) was shown to belong to CC7 (4).
The four widely used laboratory reference strains, EGD, EGD-e, LO28, and 10403S, belonged to two distant clonal complexes of lineage II, CC9 (EGD-e and LO28) and CC7 (10403S and EGD). These results indicated that despite their similar names, strains EGD and EGD-e appear to be phylogenetically distant (Fig. 1), excluding direct laboratory descent between them.
The genome reference strains of serotype 1/2b, FSL J2-064 and FSL N1-017, fell into major clonal groups CC5 and CC3, respectively, thus also representing important clonal groups of L. monocytogenes. In contrast, the three genome reference strains, FSL J1-194 (ST88), FSL J1-175 (ST87), and FSL N3-165 (ST90), represented rare STs not previously encountered, except for ST87, which was described for one food strain in Colombia (24).
To illustrate the position of the above-studied strains within the diversity of L. monocytogenes, a minimum spanning tree (MStree) that included all currently published MLST strains was constructed (Fig. 1). Whereas outbreak strains mostly fell into the central and most frequent STs of clonal complexes, the laboratory reference strains EGD, EGD-e, LO28, and 10403S represented variant STs that were not commonly found among clinical or food isolates (Fig. 1). Clearly, the diversity of L. monocytogenes is only partly represented by the laboratory reference strains that have been analyzed so far in studies of host-pathogen interactions, virulence, and other characteristics.
The distribution of MLST alleles showed that most CCs can be differentiated based on one or a few genes (Table 3). For example, allele lhkA-3 was uniquely observed for isolates of CC1, abcZ-1 was specific for CC2, and bglA-9 was shared by isolates of CC6. These CC-specific alleles allowed a rapid screening of isolates to identify those belonging to the CCs of interest. However, because recombination can occur, MLST based on the 7 gene sequences should be used for confirmation of selected isolates.
Table 3.
Distribution of MLST alleles within clones, based on the 125 study isolates
| Lineage | CC | No. of isolates | No. of distinct alleles (allele[s] found) for gene |
No. of STs | No. of STs without ldh | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| abcZ | bglA | cat | dapE | dat | ldh | lhkA | |||||
| I | 1 | 25 | 1 (3) | 2 (1; 56) | 3 (1; 20; 60) | 4 (1; 34; 42; 44) | 4 (3; 14; 18; 42) | 9 (1; 11; 12; 61; 90; 101; 103; 104; 105) | 1 (3) | 16 | 10 |
| 2 | 25 | 1 (1) | 2 (1; 38) | 1 (11) | 2 (3; 11) | 1 (2) | 1 (1) | 2 (5; 8) | 4 | 4 | |
| 3 | 5 | 1 (3) | 1 (4) | 1 (4) | 1 (4) | 1 (2) | 1 (1) | 1 (5) | 1 | 1 | |
| 6 | 15 | 1 (3) | 1 (9) | 1 (9) | 1 (3) | 1 (3) | 1 (1) | 1 (5) | 1 | 1 | |
| Others | 10 | 5 (1; 2; 11; 12; 34) | 4 (1; 2; 12; 39) | 3 (4; 11; 12) | 6 (1; 3; 14; 15; 16; 38) | 2 (2; 3) | 3 (1; 5; 39) | 5 (2; 3; 4; 5; 7) | 7 | 7 | |
| II | 7 | 6 | 1 (5) | 1 (8) | 1 (5) | 1 (7) | 1 (6) | 1 (2; 22; 38) | 1 (1) | 3 | 1 |
| 8 | 6 | 5 (5; 57) | 1 (6) | 1 (2) | 2 (9; 29) | 1 (5) | 3 (3; 89; 121) | 1 (1) | 5 | 3 | |
| 9 | 19 | 2 (6; 33) | 1 (5) | 1 (6) | 2 (4; 20) | 1 (1) | 3 (4; 57; 62) | 1 (1) | 5 | 3 | |
| Others | 14 | 5 (5; 6; 7; 21; 53) | 6 (5; 6; 10; 13; 27; 52) | 9 (2; 3; 8; 10; 16; 17; 24; 26; 39) | 6 (4; 6; 7; 8; 21; 67) | 5 (1; 5; 6; 13; 39) | 4 (2; 3; 24; 32) | 4 (1; 6; 14; 41) | 10 | 10 | |
Diversity of MvLST genes and comparison with MLST.
It has been suggested that the virulence-associated genes included in MvLST studies evolve faster that MLST genes, which code for proteins involved in central, housekeeping functions of the cell (28). To test this hypothesis, the 125 study isolates were sequenced at eight MvLST genes used for epidemic clone definition (4, 17, 19). The genetic variation recorded at MvLST genes (Table 2) represented 6.62% of nucleotide sites on average, ranging from 3.06% for inlC to 12.6% for the gene dal. This level of variation was therefore slightly lower than the amount observed for MLST genes (7.5%). The number of distinct alleles per gene varied from 4 alleles for gene clpP to 28 for gene inlA. When considering the eight MvLST genes, there were 364 nucleotide polymorphisms among the 5,496 sites that were sequenced, resulting in 40 distinct MvLSTs. Fifty-one distinct STs were found based on MLST. When excluding the atypically variable gene ldh from the MLST data (see below), 41 sequence types were distinguished, very similar to the number of MvLSTs. These results showed that MvLST does not provide more discrimination than MLST even when 8 MvLST genes are included, among which is the entire inlA gene sequence. In previous work, MvLST was found to be more discriminatory than MLST (28), but this suggestion was based on different MLST genes (prs, sigB, and recA) and only 14 strains.
Similarly, the variation of MvLSTs within the major MLST clones was limited (Table 4). No more than 4 distinct MvLSTs were observed within CCs. Besides, most isolates within a CC were identical at the eight MvLST genes, as the other MvLST types were represented by only one or few isolates. Remarkably, the homogeneity of MvLST gene sequences within CCs was observed across large geographical and time scales (see Table S1 in the supplemental material). For example, the most frequent MvLST type in CC1 was observed for isolates collected between 1963 and 2000 from 13 distinct countries in North America, Europe, Oceania, and Asia. These observations showed that MvLST gene sequences are stable through time and space, resulting in the same MvLST being observed for isolates that are considered unrelated from an epidemiological point of view. From a practical standpoint, the low variation of MvLST within single STs or CCs implies that adding the analysis of virulence genes to MLST does not improve the discriminatory power significantly: Simpson's index of discrimination was 0.946 (59 types; 95% confidence interval, 0.925 to 0.967) for MLST and MvLST combined, whereas it was 0.932 (0.908 to 0.956) for MLST and 0.898 (0.871 to 0.925) for MvLST.
Table 4.
MvLST allelic diversity within clones, based on the 125 study isolates
| Lineage | CC | No. of strains | No. of distinct alleles (allele[s] found) for gene |
No. of MVLSTs | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| actA | clpP | dal | inlA | inlB | inlC | lisR | prfA | ||||
| I | 1 | 25 | 2 (14; 15) | 1 (4) | 1 (5) | 1 (28) | 1 (3) | 2 (1; 4) | 1 (1) | 1 (3) | 3 |
| 2 | 25 | 2 (10; 13) | 1 (4) | 1 (5) | 1 (28) | 1 (4) | 1 (1) | 3 (1; 2; 5) | 1 (2) | 4 | |
| 3 | 5 | 1 (10) | 1 (4) | 1 (5) | 1 (22) | 1 (5) | 1 (4) | 1 (1) | 1 (2) | 1 | |
| 6 | 15 | 2 (10; 12) | 1 (4) | 1 (5) | 1 (29) | 1 (3) | 1 (4) | 1 (3) | 2 (2; 5) | 3 | |
| Others | 10 | 4 (9; 10; 11; 14) | 1 (4) | 1 (4; 5; 6; 7) | 6 (21; 23; 24; 25; 26; 27) | 4 (1; 2; 6; 7) | 3 (2; 3; 4) | 1 (3) | 4 (1; 2; 4; 6) | 7 | |
| II | 7 | 6 | 2 (6; 8) | 1 (2) | 2 (11; 13) | 2 (9; 10) | 2 (10; 13) | 2 (10; 11) | 1 (4) | 2 (7; 9) | 4 |
| 8 | 6 | 1 (4) | 1 (1) | 1 (8) | 2 (14; 15) | 2 (9; 12) | 1 (10) | 1 (4) | 1 (8) | 2 | |
| 9 | 19 | 1 (1) | 1 (2) | 2 (1; 2) | 6 (1; 2; 3; 4; 5; 6) | 1 (8) | 1 (5) | 1 (4) | 1 (8) | 3 | |
| Others | 14 | 7 (1; 2; 3; 4; 5; 6; 7) | 3 (1; 2; 3) | 7 (1; 3; 8; 9; 10; 12; 14) | 11 (4; 7; 8; 11; 12; 13; 16; 17; 18; 19; 20) | 6 (8; 9; 10; 11; 14; 15) | 6 (5; 6; 7; 8; 9; 10) | 2 (4; 6) | 1 (8) | 10 | |
InlA evolved by in-frame deletions as well as variation leading to truncated forms of internalin.
Internalin, encoded by inlA, plays a critical role in L. monocytogenes invasion of cultured epithelial cells (36), and it is a critical virulence determinant, mediating L. monocytogenes crossing of the intestinal and placental barriers (37, 38). Alignment of the MvLST gene sequences revealed that the gene inlA differed from the seven other genes by the presence of insertion-deletion events. First, all strains of CC6 had a deletion of 9 nucleotides corresponding to positions 2,212 to 2,220 in the inlA sequence of the other strains, which resulted in the predicted loss of 3 amino acids in the preanchor region of InlA. Second, ILSI strain FSL C1-122 had a 30-nt deletion in inlA (positions 2,141 to 2,170), which resulted in the predicted loss of 10 amino acids (723 to 732) in the preanchor region of InlA. These two in-frame changes are not expected to impair InlA function, as the functional domain of InlA is the LRR-IR region (39), and the CC6 and FSL C1-122 InlA variants harbor an intact anchor region (40). Third, three strains of CC9 had a G-to-A change at position 2,054 that resulted in a stop codon at amino acid position 694. Fourth, seven other strains of CC9, including LO28, had a 1-nt deletion in inlA at position 1635, resulting in the first premature stop codon described for inlA (41). Fifth, three other CC9 isolates had a G-to-A change at position 1,380 that resulted in a stop codon at position 460. Sixth, strain LM05-01099 had a G-to-T change at position 976 that resulted in a stop codon at position 326. Finally, strain CLIP 11308 had a 1-nt deletion at position 5 in gene inlA that resulted in a predicted stop codon at amino acid position 9. Functional, nontruncated InlA has been associated with the clinical origin of L. monocytogenes isolates (42), and premature stop codons in inlA leading to the secretion of a nonfunctional truncated protein have been associated with reduced pathogenicity (9, 41–43).
Patterns of recombination, selection, and phylogenetic relationships.
Homologous recombination and selection both influence the evolutionary rate and the diversity patterns of genes that they affect. To characterize the evolutionary forces that act on the diversification of clonal groups, we searched for evidence of recombination and selection in MLST and MvLST genes. Using LDhat, evidence for recombination was detected at five out of the 15 loci (see Table S2 in the supplemental material). Within lineage I, recombination was only detected at the inlB locus. Within lineage II, recombination was detected at the three MvLST genes actA, dal, and inlA. Among the MLST genes, recombination was detected only at gene dapE in lineages I and II. These results confirmed that recombination is infrequent in L. monocytogenes and indicated that MvLST genes are more affected by recombination than MLST genes.
To detect positive selection, the software OmegaMap (33) was used. This software separates the effects of recombination and positive selection, thus avoiding false positives for selection in the presence of recombination. By using a posterior probability threshold of 0.95, the results revealed positive selection within lineage I at MvLST genes actA and inlC, two major virulence factors, as well as at MLST loci dapE and ldh (see Fig. S1 in the supplemental material). No evidence for positive selection was found at the other loci within lineage I, including the recombining gene inlB. However, positive selection is highly likely to occur at gene inlA, which had a posterior probability of 0.94. No positive selection was detected within lineage II at any of the 15 genes. In this lineage, the highest posterior probability (0.84) was observed for the gene prfA, which encodes the master regulator of virulence genes. In the protein ActA, the positively selected amino acids within lineage I were at codons 124 and 130 (88 and 94 of our sequenced region) (see Fig. S1), located within the actin tail formation domain (44). Overall, these results indicate contrasting impacts of recombination and selection between lineages: while lineage II was more affected by recombination, consistent with previous findings (16), lineage I was more affected by positive selection.
The phylogenetic analyses of the concatenated gene sequences from the MLST gene set (Fig. 2, left) and the MvLST gene set (Fig. 2, right) gave highly similar results. As expected, the two major lineages of L. monocytogenes corresponded to the deepest branches and were clearly separated based on both gene sets. Based on MvLST genes, both lineages showed unequally long branches, consistent with a higher impact of recombination on MvLST genes. Nevertheless, both gene sets grouped strains into the same shallow branches, and MLST-defined CCs were recovered as monophyletic groups based on both gene sets (Fig. 2). We also classified isolates into MvLST clonal complexes by using their 8-digit allelic profile. These groupings were highly concordant with MLST clonal complexes, as illustrated by the adjusted Rand coefficients (0.988; 95% confidence interval, 0.976 to 1.000; 0.981 [0.964 to 1.000] when excluding the gene ldh [see below]). These results showed that groupings of L. monocytogenes isolates obtained based on either MLST or MvLST gene sets are nearly identical.
Fig 2.
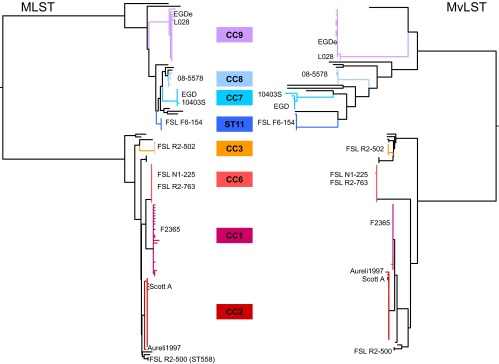
Compared phylogenies based on MLST (left) and MvLST (right) gene sequences of the 125 study isolates. Phylogenies were obtained based on concatenated gene sequences by using the neighbor-joining method based on uncorrected p-distances. Branches corresponding to clonal complexes are colored according to the central legend. The positions of reference strains are indicated.
Source-sink selection at the ldh gene and impact on CC definitions.
For most genes, all isolates of a given clonal complex shared the same allelic sequence (Table 3). However, gene ldh was a notable exception, as this gene was highly variable within CCs, resulting in a large proportion of STs differing from the central genotype of each CC solely by the gene ldh. We observed that almost all ldh nucleotide differences within CCs implied an amino acid change in the deduced protein sequence (see Table S1 in the supplemental material). Further, all amino acid changes were localized in the 3′ end (codon numbers 93 to 147) of the sequenced ldh region, consistent with the results obtained with OmegaMap (see Fig. S1 in the supplemental material). In addition, we observed the same amino acid substitutions in two pairs of isolates from different CCs: change Y95C was observed both in CC1 (ST252) and in CC8 (ST289), whereas change S120R was found in both CC1 (ST248) and CC7 (ST85). These evolutionary parallelisms represent strong evidence for positive selection on ldh and contribute to the atypically high diversity of this gene within CCs. Interestingly, the region from 93 to 137 within which changes were observed includes a variable flexible loop of enzyme lactate dehydrogenase, in which amino acid substitutions were associated with adaptation to cold temperatures in teleost fishes (45). In striking contrast to the observed pattern of intraclone variation, the amino acid sequence of lactate dehydrogenase was unchanged among the central genotype of most L. monocytogenes clonal complexes and was even conserved between lineages I and II. This absence of diversification at the amino acid level in the long term indicates strong purifying selection on ldh. Together, these results indicate that ldh evolved in a source-sink fashion, whereby variants were selected in the short term because they provided a selective advantage in a sink environment, but these variants were counterselected in the source environment, which sustains the population in the long run. Strikingly, the time since initial isolation of strains with amino acid substitutions in lactate dehydrogenase was significantly longer than for nonmutated isolates (P = 0.024, two-sided Mann-Whitney test). No substitution was observed in isolates collected after year 2000 (see Table S1). We hypothesize that nonsynonymous mutations in gene ldh could have been selected during long-term storage in the laboratory and do not represent naturally occurring variations.
The positive selection pattern at the 3′ end of the ldh sequence led us to evaluate the impact of this gene on the assignment of isolates to clonal complexes. We selected the 863 isolates and reference strains previously analyzed herein and in our previous studies (9, 11, 24, 46, 47). Classification of the isolates into CCs was performed using either the 7 MLST genes (Fig. 1) or the six genes after excluding ldh (MLST-6). For the 515 isolates of lineage I, there was a total correspondence between CCs obtained based on 7 or 6 genes. Similarly, in lineage III, there was no impact of exclusion of gene ldh on the grouping of the 25 isolates. However, when analyzing the 318 isolates of lineage II based on MLST-6, a few discrepancies were observed compared to MLST-7. First, CC8 and CC16 became merged into a single CC, as these two CCs differed by ldh (alleles ldh-3 and ldh-2, respectively). Similarly, CC11 (ldh-2) and CC19 (ldh-24) became merged based on MLST-6, and ST226 (ldh-96) became a member of CC177 (ldh-26). Finally, ST126 (ldh-65) became part of CC7 (ldh-2). Among the ldh alleles that affected CC membership, only ldh-65 had a nonsynonymous change (H121R) that could potentially have been selected. Therefore, the positive selection on the 3′ region of the ldh sequence potentially impacted the classification of only a single isolate (SLCC875, of ST126) out of the 863 isolates. The adjusted Rand coefficient between MLST-7 and MLST-6 was 0.992 (95% confidence interval, 0.979 to 1.000). We conclude that the use of gene ldh in MLST does not impact importantly the classification of isolates into clonal complexes.
Minimal age of clones and heterogeneity of PFGE results.
Although we believe it was not the intention of the author who coined the name “epidemic clone” (6), this denomination suggests that such groups have undergone recent emergence and have diffused within epidemiological time scales. To provide a minimal age of clones, we searched the public MLST database (as of 8 July 8 2013, 2,586 isolates) for the oldest isolate of the central ST of each major clone. ST1, ST2, ST3, ST4, ST5, ST6, ST7, ST8, and ST9 were isolated as early as 1936, 1954, 1955, 1965, 1992, 1990, 1927, 1991, and 1949, respectively. These dates are several decades older than the first described outbreak of the corresponding “epidemic clone,” and it is well possible that the central STs existed much earlier.
In order to estimate the diversity within clones, 110 isolates (all except the genome references) were analyzed by PFGE by using two enzymes separately. A total of 65 ApaI types, 48 AscI types, and 76 combined PFGE types were distinguished (see Fig. S2 in the supplemental material). PFGE patterns within an MLST-defined CC were more similar among themselves than with patterns of other clones. This was illustrated by cluster analysis of combined ApaI and AscI PFGE patterns (see Fig. S2), which grouped isolates of each clone within a single branch. One exception was the grouping of the two CC558 strains from the 2000 Mexican-style fresh cheese outbreak, which were clustered within the CC1 branch (see Fig. S2). As ST1 and ST558 are not closely related (differing by five out of seven MLST genes), this observation suggests an evolutionary convergence of the PFGE profiles of clones CC1 and CC558.
Although they clustered together, the PFGE patterns within a given clonal group showed a high level of heterogeneity (Table 5). First, the seven clonal groups represented by multiple isolates displayed 2 to 21 distinct PFGE types when we combined profiles obtained with ApaI and AscI enzymes. CC1 and CC2, which both comprised 25 analyzed isolates, were the most heterogeneous, with 21 and 17 distinct PFGE profiles, respectively. Furthermore, the most unrelated profiles within these CCs differed by more than 20 bands, considering both ApaI and AscI patterns (Table 5). These results showed that PFGE has a higher discriminatory power than MLST and that CCs exhibit high levels of PFGE diversity. As a matter of fact, isolates that differ by three or more PFGE bands are generally considered epidemiologically unrelated (48). Therefore, the PFGE diversity observed within L. monocytogenes clones confirmed the observation, based on date and country of origin, that most isolates that share the same or closely related MLST or MvLST are epidemiologically unrelated. As there is no evidence for epidemiological links between most outbreaks of L. monocytogenes caused by the same EC, the “epidemic clone” term is potentially misleading. Instead, these results favor the view that “epidemic clones” are evolutionarily successful lineages that are widely distributed and have caused multiple independent outbreaks due to their broad distribution (6, 8, 12, 24).
Table 5.
Pulsed-field gel electrophoresis diversity per clone
| Lineage | CC | No. of isolates | No. of AscI profiles (max. no. of different bands) | No. of ApaI profiles (max. no. of different bands) | No. of AscI + ApaI profiles |
|---|---|---|---|---|---|
| I | 1 | 25 | 11 (13) | 12 (9) | 21 |
| 2 | 25 | 7 (7) | 13 (15) | 17 | |
| 3 | 3 | 1 (0) | 2 (6) | 2 | |
| 6 | 15 | 2 (5) | 5 (10) | 6 | |
| Others | 6 | 5 (10) | 5 (12) | 5 | |
| II | 7 | 5 | 4 (5) | 4 (7) | 5 |
| 8 | 4 | 3 (8) | 2 (3) | 4 | |
| 9 | 20 | 6 (9) | 4 (16) | 7 | |
| Others | 11 | 9 (12) | 8 (13) | 9 |
It is possible that epidemic clones might in fact represent genetically distinct subgroups within MLST-defined CCs (24), even though the MLST and MvLST genes cannot discriminate such subgroups. Comparison of PFGE patterns showed that among isolates of CC1, the strains representative of the 1981 Nova Scotia coleslaw outbreak were clearly distinct from the representatives of the two other CC1 outbreaks (the 1987 Vacherin Mont d'Or and 1985 Jalisco outbreaks), which were more similar among each other. Within CC2, the 1987 United Kingdom and Ireland pâté outbreak and the 1983 Massachusetts milk outbreak strains were undistinguishable. Finally, within CC6, the strains from the 2002 multistate ready-to-eat meat outbreak and from the 1998 hot dog outbreak were distinguished by a conspicuous doublet when the AscI enzyme was used (see Fig. S2 in the supplemental material). These results showed that within a single CC, strains with distinct PFGE patterns were implicated in outbreaks. Clearly, the within-clone phylogenetic relationships need to be established more precisely, using the high resolution of whole-genome sequencing (49, 50), to determine whether they are structured into subgroups with distinct involvements in outbreaks.
Conclusions.
Research into the pathogenic or epidemiological differences among L. monocytogenes clonal groups would benefit from a widely accepted method to define clonal groups, as well as a reference system to name them. Serotyping has provided a widely shared language, but unfortunately this approach lacks discrimination and is unreliable as a classification system. Similarly, the recognition of epidemic clones as groups of similar isolates causing multiple outbreaks has been invaluable for research on L. monocytogenes epidemiology and biology. However, the concept of an epidemic clone was not designed to become a common nomenclature system for L. monocytogenes. First, it only includes clones involved in outbreaks, whereas most listeriosis cases are sporadic, thus an important fraction of L. monocytogenes diversity is excluded with this concept. Second, there is no consensual definition of an epidemic clone. For example, authors either accept gene sequence variation within clones (12) or they do not (4, 13, 17, 19, 20). In particular, ECIV to ECVII were defined based on complete identity at the six MvLST genes. Strict application of this definition led, for example, to the exclusion of strain Scott A, based on a single nucleotide change in lisR, from ECIV (17). As shown here, this strain is otherwise identical to other strains of CC2, ECIa, and ECIV at all other MvLST genes, as well as at MLST genes, suggesting that its exclusion from ECIV is too restrictive. An early definition of “clone” in a medical microbiology context was given by Orskov as genetically similar (but not identical) isolates from unrelated places and times that share many common features (51). The MLST-based definition of clones as CCs (21, 22) is in line with this definition, as it allows one isolate to belong to a CC even if it differs at one out of six genes from another member of the complex. The flexible definition of CCs thus accommodates random accumulation of variation within clones. In fact, MLST has initially been devised to provide a reliable way to recognize and define clonal groups (21, 22).
In this work, we have shown that reference isolates of distinct epidemic clones belong to distinct CCs. However, whereas all ECs have a corresponding group, based on MLST, only a small fraction of MLST diversity is covered by the EC nomenclature (Fig. 1). We demonstrated that MvLST genes evolve at a similar rate as MLST genes, implying that both approaches will delimit equivalent groups of isolates. Finally, we showed that CCs represent ancient, diverse, and globally distributed genetic groups. This result is in line with the view of CCs in many bacterial species (21, 22, 52).
Given the above, we regard EC denominations as a redundant but partial and less flexible nomenclature system for MLST groups. Given the close correspondence between MLST CCs and ECs, the identification methods developed previously for specific ECs (5, 15, 19, 53–56) should apply readily to the distinction of MLST clones. Unlike MvLST, MLST data are publicly available for comparison through a dedicated website, providing a continuously growing overview of L. monocytogenes diversity. The MLST database (http://www.pasteur.fr/mlst) currently contains 2,586 isolates and is being used by an increasing number of laboratories (4, 9, 11, 19, 23, 24, 46, 57–61) for studies of the epidemiology and population biology of this pathogen, thus providing de facto a common language for the denomination of clonal groups of L. monocytogenes.
Supplementary Material
ACKNOWLEDGMENTS
We thank Martin Wiedmann for providing the ILSI reference strain collection. Estimations of the minimal age of clonal complexes were improved thanks to deposition by Jana Haase and Mark Achtman of multilocus sequence typing data of ancient isolates into the Institut Pasteur MLST database. The National Reference Center for Listeria and Biology of Infection Unit acknowledges support by Institut Pasteur, Institut de Veille Sanitaire, Institut National de la Santé et de la Recherche Médicale, Fondation pour la Recherche Médicale, Ville de Paris, Fondation BNP Paribas, and LabEx IBEID.
This work was supported financially by Institut Pasteur, Institut de Veille Sanitaire, the Listress EU program, and the European Research Council.
Footnotes
Published ahead of print 4 September 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.01874-13.
REFERENCES
- 1.Denny J, McLauchlin J. 2008. Human Listeria monocytogenes infections in Europe: an opportunity for improved European surveillance. Euro Surveill. 13(13):pii=8082 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=8082 [PubMed] [Google Scholar]
- 2.Goulet V, Hedberg C, Le Monnier A, de Valk H. 2008. Increasing incidence of listeriosis in France and other European countries. Emerg. Infect. Dis. 14:734–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gilmour MW, Graham M, Van Domselaar G, Tyler S, Kent H, Trout-Yakel KM, Larios O, Allen V, Lee B, Nadon C. 2010. High-throughput genome sequencing of two Listeria monocytogenes clinical isolates during a large foodborne outbreak. BMC Genomics 11:120. 10.1186/1471-2164-11-120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lomonaco S, Verghese B, Gerner-Smidt P, Tarr C, Gladney L, Joseph L, Katz L, Turnsek M, Frace M, Chen Y, Brown E, Meinersmann R, Berrang M, Knabel S. 2013. Novel epidemic clones of Listeria monocytogenes, United States, 2011. Emerg. Infect. Dis. 19:147–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Evans MR, Swaminathan B, Graves LM, Altermann E, Klaenhammer TR, Fink RC, Kernodle S, Kathariou S. 2004. Genetic markers unique to Listeria monocytogenes serotype 4b differentiate epidemic clone II (hot dog outbreak strains) from other lineages. Appl. Environ. Microbiol. 70:2383–2390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kathariou S. 2002. Listeria monocytogenes virulence and pathogenicity, a food safety perspective. J. Food Prot. 65:1811–1829 [DOI] [PubMed] [Google Scholar]
- 7.den Bakker HC, Bundrant BN, Fortes ED, Orsi RH, Wiedmann M. 2010. A population genetics-based and phylogenetic approach to understanding the evolution of virulence in the genus Listeria. Appl. Environ. Microbiol. 76:6085–6100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piffaretti JC, Kressebuch H, Aeschbacher M, Bille J, Bannerman E, Musser JM, Selander RK, Rocourt J. 1989. Genetic characterization of clones of the bacterium Listeria monocytogenes causing epidemic disease. Proc. Natl. Acad. Sci. U. S. A. 86:3818–3822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ragon M, Wirth T, Hollandt F, Lavenir R, Lecuit M, Le Monnier A, Brisse S. 2008. A new perspective on Listeria monocytogenes evolution. PLoS Pathog. 4(9):e1000146. 10.1371/journal.ppat.1000146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wiedmann M, Bruce JL, Keating C, Johnson AE, McDonough PL, Batt CA. 1997. Ribotypes and virulence gene polymorphisms suggest three distinct Listeria monocytogenes lineages with differences in pathogenic potential. Infect. Immun. 65:2707–2716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chenal-Francisque V, Diancourt L, Cantinelli T, Passet V, Tran-Hykes C, Bracq-Dieye H, Leclercq A, Pourcel C, Lecuit M, Brisse S. 2013. Optimized multilocus variable-number tandem-repeat analysis assay and its complementarity with pulsed-field gel electrophoresis and multilocus sequence typing for Listeria monocytogenes clone identification and surveillance. J. Clin. Microbiol. 51:1868–1880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.den Bakker HC, Fortes ED, Wiedmann M. 2010. Multilocus sequence typing of outbreak-associated Listeria monocytogenes isolates to identify epidemic clones. Foodborne Pathog. Dis. 7:257–265 [DOI] [PubMed] [Google Scholar]
- 13.Lomonaco S, Chen Y, Knabel SJ. 2008. Analysis of additional virulence genes and virulence gene regions in Listeria monocytogenes confirms the epidemiologic relevance of multi-virulence-locus sequence typing. J. Food Prot. 71:2559–2566 [DOI] [PubMed] [Google Scholar]
- 14.Sperry KE, Kathariou S, Edwards JS, Wolf LA. 2008. Multiple-locus variable-number tandem-repeat analysis as a tool for subtyping Listeria monocytogenes strains. J. Clin. Microbiol. 46:1435–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ward TJ, Usgaard T, Evans P. 2010. A targeted multilocus genotyping assay for lineage, serogroup, and epidemic clone typing of Listeria monocytogenes. Appl. Environ. Microbiol. 76:6680–6684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.den Bakker HC, Didelot X, Fortes ED, Nightingale KK, Wiedmann M. 2008. Lineage specific recombination rates and microevolution in Listeria monocytogenes. BMC Evol. Biol. 8:277. 10.1186/1471-2148-8-277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, Zhang W, Knabel SJ. 2007. Multi-virulence-locus sequence typing identifies single nucleotide polymorphisms which differentiate epidemic clones and outbreak strains of Listeria monocytogenes. J. Clin. Microbiol. 45:835–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kathariou S. 2003. Foodborne outbreaks of listeriosis and epidemic-associated lineages of Listeria monocytogenes, p 243–256 In Torrence ME, Isaacson RE. (ed), Microbial food safety in animal agriculture. Iowa State University Press, Ames, IA [Google Scholar]
- 19.Knabel SJ, Reimer A, Verghese B, Lok M, Ziegler J, Farber J, Pagotto F, Graham M, Nadon CA, Gilmour MW. 2012. Sequence typing confirms that a predominant Listeria monocytogenes clone caused human listeriosis cases and outbreaks in Canada from 1988 to 2010. J. Clin. Microbiol. 50:1748–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Y, Zhang W, Knabel SJ. 2005. Multi-virulence-locus sequence typing clarifies epidemiology of recent listeriosis outbreaks in the United States. J. Clin. Microbiol. 43:5291–5294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feil EJ. 2004. Small change: keeping pace with microevolution. Nat. Rev. Microbiol. 2:483–495 [DOI] [PubMed] [Google Scholar]
- 22.Maiden MC. 2006. Multilocus sequence typing of bacteria. Annu. Rev. Microbiol. 60:561–588 [DOI] [PubMed] [Google Scholar]
- 23.Salcedo C, Arreaza L, Alcala B, de la Fuente L, Vazquez JA. 2003. Development of a multilocus sequence typing method for analysis of Listeria monocytogenes clones. J. Clin. Microbiol. 41:757–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chenal-Francisque V, Lopez J, Cantinelli T, Caro V, Tran C, Leclercq A, Lecuit M, Brisse S. 2011. Worldwide distribution of major clones of Listeria monocytogenes. Emerg. Infect. Dis. 17:1110–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fugett E, Fortes E, Nnoka C, Wiedmann M. 2006. International Life Sciences Institute North America Listeria monocytogenes strain collection: development of standard Listeria monocytogenes strain sets for research and validation studies. J. Food Prot. 69:2929–2938 [DOI] [PubMed] [Google Scholar]
- 26.Rocourt J, Seeliger HP. 1985. Distribution of species of the genus Listeria. Zentralbl. Bakteriol. Mikrobiol. Hyg. A 259:317–330 [PubMed] [Google Scholar]
- 27.Doumith M, Buchrieser C, Glaser P, Jacquet C, Martin P. 2004. Differentiation of the major Listeria monocytogenes serovars by multiplex PCR. J. Clin. Microbiol. 42:3819–3822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W, Jayarao BM, Knabel SJ. 2004. Multi-virulence-locus sequence typing of Listeria monocytogenes. Appl. Environ. Microbiol. 70:913–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Graves LM, Swaminathan B. 2001. PulseNet standardized protocol for subtyping Listeria monocytogenes by macrorestriction and pulsed-field gel electrophoresis. Int. J. Food Microbiol. 65:55–62 [DOI] [PubMed] [Google Scholar]
- 30.Librado P, Rozas J. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452 [DOI] [PubMed] [Google Scholar]
- 31.Carrico JA, Silva-Costa C, Melo-Cristino J, Pinto FR, de Lencastre H, Almeida JS, Ramirez M. 2006. Illustration of a common framework for relating multiple typing methods by application to macrolide-resistant Streptococcus pyogenes. J. Clin. Microbiol. 44:2524–2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McVean G, Awadalla P, Fearnhead P. 2002. A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics 160:1231–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson DJ, McVean G. 2006. Estimating diversifying selection and functional constraint in the presence of recombination. Genetics 172:1411–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuhn M, Kathariou S, Goebel W. 1988. Hemolysin supports survival but not entry of the intracellular bacterium Listeria monocytogenes. Infect. Immun. 56:79–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murray EGD, Webb RA, Swann MBR. 1926. A disease of rabbits characterised by a large mononuclear leucocytosis, caused by a hitherto undescribed bacillus Bacterium monocytogenes (n.sp.). J. Pathol. Bacteriol. 29:407–439 [Google Scholar]
- 36.Gaillard JL, Berche P, Frehel C, Gouin E, Cossart P. 1991. Entry of Listeria monocytogenes into cells is mediated by internalin, a repeat protein reminiscent of surface antigens from gram-positive cocci. Cell 65:1127–1141 [DOI] [PubMed] [Google Scholar]
- 37.Disson O, Grayo S, Huillet E, Nikitas G, Langa-Vives F, Dussurget O, Ragon M, Le Monnier A, Babinet C, Cossart P, Lecuit M. 2008. Conjugated action of two species-specific invasion proteins for fetoplacental listeriosis. Nature 455:1114–1118 [DOI] [PubMed] [Google Scholar]
- 38.Lecuit M, Vandormael-Pournin S, Lefort J, Huerre M, Gounon P, Dupuy C, Babinet C, Cossart P. 2001. A transgenic model for listeriosis: role of internalin in crossing the intestinal barrier. Science 292:1722–1725 [DOI] [PubMed] [Google Scholar]
- 39.Lecuit M, Ohayon H, Braun L, Mengaud J, Cossart P. 1997. Internalin of Listeria monocytogenes with an intact leucine-rich repeat region is sufficient to promote internalization. Infect. Immun. 65:5309–5319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lebrun M, Mengaud J, Ohayon H, Nato F, Cossart P. 1996. Internalin must be on the bacterial surface to mediate entry of Listeria monocytogenes into epithelial cells. Mol. Microbiol. 21:579–592 [DOI] [PubMed] [Google Scholar]
- 41.Jonquieres R, Bierne H, Mengaud J, Cossart P. 1998. The inlA gene of Listeria monocytogenes LO28 harbors a nonsense mutation resulting in release of internalin. Infect. Immun. 66:3420–3422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jacquet C, Doumith M, Gordon JI, Martin PM, Cossart P, Lecuit M. 2004. A molecular marker for evaluating the pathogenic potential of foodborne Listeria monocytogenes. J. Infect. Dis. 189:2094–2100 [DOI] [PubMed] [Google Scholar]
- 43.Nightingale KK, Ivy RA, Ho AJ, Fortes ED, Njaa BL, Peters RM, Wiedmann M. 2008. inlA premature stop codons are common among Listeria monocytogenes isolates from foods and yield virulence-attenuated strains that confer protection against fully virulent strains. Appl. Environ. Microbiol. 74:6570–6583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Travier L, Guadagnini S, Gouin E, Dufour A, Chenal-Francisque V, Cossart P, Olivo-Marin JC, Ghigo JM, Disson O, Lecuit M. 2013. ActA promotes Listeria monocytogenes aggregation, intestinal colonization and carriage. PLoS Pathog. 9(1):e1003131. 10.1371/journal.ppat.1003131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fields PA, Somero GN. 1998. Hot spots in cold adaptation: localized increases in conformational flexibility in lactate dehydrogenase A4 orthologs of Antarctic notothenioid fishes. Proc. Natl. Acad. Sci. U. S. A. 95:11476–11481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adgamov R, Zaytseva E, Thiberge JM, Brisse S, Ermolaeva S. 2012. Genetically related Listeria monocytogenes strains isolated from lethal human cases and wild animals, p 235–250 In Caliskan M. (ed), Genetic diversity in microorganisms. InTech, New York, NY [Google Scholar]
- 47.Leclercq A, Chenal-Francisque V, Dieye H, Cantinelli T, Drali R, Brisse S, Lecuit M. 2011. Characterization of the novel Listeria monocytogenes PCR serogrouping profile IVb-v1. Int. J. Food Microbiol. 147:74–77 [DOI] [PubMed] [Google Scholar]
- 48.Tenover FC, Arbeit RD, Goering RV, Mickelsen PA, Murray BE, Persing DH, Swaminathan B. 1995. Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. J. Clin. Microbiol. 33:2233–2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roetzer A, Diel R, Kohl TA, Ruckert C, Nubel U, Blom J, Wirth T, Jaenicke S, Schuback S, Rusch-Gerdes S, Supply P, Kalinowski J, Niemann S. 2013. Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: a longitudinal molecular epidemiological study. PLoS Med. 10(2):e1001387. 10.1371/journal.pmed.1001387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou Z, McCann A, Litrup E, Murphy R, Cormican M, Fanning S, Brown D, Guttman DS, Brisse S, Achtman M. 2013. Neutral genomic microevolution of a recently emerged pathogen, Salmonella enterica serovar Agona. PLoS Genet. 9(4):e1003471. 10.1371/journal.pgen.1003471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Orskov F, Orskov I. 1983. From the National Institutes of Health. Summary of a workshop on the clone concept in the epidemiology, taxonomy, and evolution of the enterobacteriaceae and other bacteria. J. Infect. Dis. 148:346–357 [DOI] [PubMed] [Google Scholar]
- 52.Achtman M. 2008. Evolution, population structure, and phylogeography of genetically monomorphic bacterial pathogens. Annu. Rev. Microbiol. 62:53–70 [DOI] [PubMed] [Google Scholar]
- 53.Chen Y, Knabel SJ. 2008. Prophages in Listeria monocytogenes contain single-nucleotide polymorphisms that differentiate outbreak clones within epidemic clones. J. Clin. Microbiol. 46:1478–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng Y, Kim JW, Lee S, Siletzky RM, Kathariou S. 2010. DNA probes for unambiguous identification of Listeria monocytogenes epidemic clone II strains. Appl. Environ. Microbiol. 76:3061–3068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ducey TF, Page B, Usgaard T, Borucki MK, Pupedis K, Ward TJ. 2007. A single-nucleotide-polymorphism-based multilocus genotyping assay for subtyping lineage I isolates of Listeria monocytogenes. Appl. Environ. Microbiol. 73:133–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yildirim S, Lin W, Hitchins AD, Jaykus LA, Altermann E, Klaenhammer TR, Kathariou S. 2004. Epidemic clone I-specific genetic markers in strains of Listeria monocytogenes serotype 4b from foods. Appl. Environ. Microbiol. 70:4158–4164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koopmans MM, Brouwer MC, Bijlsma MW, Bovenkerk S, Keijzers W, van der Ende A, van de Beek D. 2013. Listeria monocytogenes sequence type 6 and increased rate of unfavorable outcome in meningitis: epidemiologic cohort study. Clin. Infect. Dis. 57:247–253 [DOI] [PubMed] [Google Scholar]
- 58.Nilsson RE, Ross T, Bowman JP, Britz ML. 2013. MudPIT profiling reveals a link between anaerobic metabolism and the alkaline adaptive response of Listeria monocytogenes EGD-e. PLoS One 8(1):e54157. 10.1371/journal.pone.0054157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Parisi A, Latorre L, Normanno G, Miccolupo A, Fraccalvieri R, Lorusso V, Santagada G. 2010. Amplified fragment length polymorphism and multi-locus sequence typing for high-resolution genotyping of Listeria monocytogenes from foods and the environment. Food Microbiol. 27:101–108 [DOI] [PubMed] [Google Scholar]
- 60.Wang P, Yang H, Hu Y, Yuan F, Zhao G, Zhao Y, Chen Y. 2012. Characterization of Listeria monocytogenes isolates in import food products of China from 8 provinces between 2005 and 2007. J. Food Sci. 77:M212–M216 [DOI] [PubMed] [Google Scholar]
- 61.Wang Y, Zhao A, Zhu R, Lan R, Jin D, Cui Z, Li Z, Xu J, Ye C. 2012. Genetic diversity and molecular typing of Listeria monocytogenes in China. BMC Microbiol. 12:119. 10.1186/1471-2180-12-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.