

Abstract
The cyclooxygenase pathway is strongly implicated in breast cancer progression but the role of this pathway in the biology of breast cancer stem/progenitor cells has not been defined. Recent attention has focused on targeting the cyclooxygenase 2 (COX-2) pathway downstream of the COX-2 enzyme by blocking the activities of individual prostaglandin E (EP) receptors. Prostaglandin E receptor 4 (EP4) is widely expressed in primary invasive ductal carcinomas of the breast and antagonizing this receptor with small molecule inhibitors or shRNA directed to EP4 inhibits metastatic potential in both syngeneic and xenograft models. Breast cancer stem/progenitor cells are defined as a subpopulation of cells that drive tumor growth, metastasis, treatment resistance, and relapse. Mammosphere-forming breast cancer cells of human (MDA-MB-231, SKBR3) or murine (66.1, 410.4) origin of basal-type, Her-2 phenotype and/or with heightened metastatic capacity upregulate expression of both EP4 and COX-2 and are more tumorigenic compared to the bulk population. In contrast, luminal-type or non-metastatic counterparts (MCF7, 410, 67) do not increase COX-2 and EP4 expression in mammosphere culture. Treatment of mammosphere-forming cells with EP4 inhibitors (RQ-15986, AH23848, Frondoside A) or EP4 gene silencing, but not with a COX inhibitor (Indomethacin) reduces both mammosphere-forming capacity and the expression of phenotypic markers (CD44hi/CD24low, aldehyde dehydrogenase) of breast cancer stem cells. Finally, an orally delivered EP4 antagonist (RQ-08) reduces the tumor-initiating capacity and markedly inhibits both the size of tumors arising from transplantation of mammosphere-forming cells and phenotypic markers of stem cells in vivo. These studies support the continued investigation of EP4 as a potential therapeutic target and provide new insight regarding the role of EP4 in supporting a breast cancer stem cell/tumor-initiating phenotype.
Keywords: COX-2, Prostaglandin E, Prostaglandin E receptor EP4, Breast cancer, Stem cells, Tumor-initiating cells
Introduction
Elevated cyclooxygenase 2 (COX-2) expression is common in breast cancer and is associated with a worse prognosis [1, 2] but the role of COX-2 pathway members in the behavior of breast cancer stem cells has yet to be defined. The principle COX-2 product in tumors is prostaglandin E2 (PGE2) which mediates cellular responses by acting on a family of four G protein-coupled receptors (EP1–EP4). Prostaglandin E receptor 4 (EP4) is expressed in a wide range of epithelial malignancies [3–12] and pharmacologic blockade or genetic silencing of the EP4 receptor inhibits proliferation and migration in vitro and growth and metastasis in vivo [13–17]. These data provide evidence that EP4 and COX-2 are important to the behavior of the general population of tumor cells. Recently, mesenchymal stem cells were shown to create a supportive microenvironment for cancer stem cells by a PGE2-dependent mechanism [18]. We asked if the COX-2 pathway is supportive of breast cancer stem cell survival by examining the expression and function of COX-2 and EP4 in cells with a stem cell phenotype. We now show that both EP4 and COX-2 are highly induced on candidate tumor-initiating/stem cell populations and that EP4 antagonists reduce cancer stem cell properties in vitro and in vivo supporting the hypothesis that EP4 and/or COX-2 may represent novel targets expressed by the most high risk and resistant subpopulations.
Methods
Cell lines and mice
Murine mammary tumor lines 66.1 and 410.4 are highly tumorigenic and metastatic in syngeneic Balb/cByJ mice; lines 67 and 410 are poorly tumorigenic and non-metastatic in the same hosts. Human breast cancer cell lines MDA-MB-231, SKBR3, and MCF7 were obtained from ATCC. The EP4 antagonists AH23848 (Sigma Chem. Co, St. Louis, MO) and RQ-00015986, hereafter abbreviated RQ-15986 and RQ-08 (gifts of RaQualia Pharma, Inc., Ref. 19 as CJ-042794), Frondoside A (a gift of Coastside Bio Resources, Ref. 20), indomethacin (Sigma) were added to cell cultures to achieve final concentrations as indicated. Line 410.4 and 66.1 tumor cells were transfected with a plasmid expressing shRNA targeting the murine EP4 gene or control vector. For some studies, the targeting vector was from OpenBiosystems, Huntsville, AL [21]; for other studies an EP4shRNA was obtained from OriGene, Rockville, MD. MDA-MB-231-luc cells were a generous gift of Dr. Stuart Martin, UMB. Balb/cByJ female mice were purchased from the Jackson Laboratory (Bar Harbor, ME); Balb/c/SCID mice were purchased from Charles Rivers Laboratory (Wilmington, MA). Limiting dilution assays were carried out by injecting the indicated number of cells proximal to the inguinal mammary fat pad. Mice were euthanized on an individual basis when tumors measured 18 mm in largest diameter and lung surface tumor colonies were counted under a dissecting microscope. Tumor volume was calculated by the formula: (a × b 2) × 0.5236, where a = longest diameter and b = perpendicular diameter. Lung colonization was evaluated by injecting 1–2 × 105 viable tumor cells i.v. into the lateral tail vein of Balb/cByJ female mice. All mice were euthanized on day 18–22 post-transplantation or earlier if moribund. Lungs were examined for tumor colonies.
Live animal imaging
Balb/c/SCID mice were injected i.v. with 1 × 105 MDA-MB-231-luc cells treated with 3 μM RQ-15986 or DMSO. On the days indicated, sedated mice were injected with d-Luciferin (Caliper Life Sci., Hopkinton, MA) and whole-body bioluminescence determined by Xenogen system.
Immunohistochemistry
A tissue microarray was prepared and described previously [22]. Immunohistochemistry was carried out after antigen-retrieval using rabbit anti-human polyclonal EP4 antibody (Lifespan Biosciences, Seattle, WA) followed by secondary antibody (EnVision+dual link System, Dako).
qPCR
Quantitative PCR was conducted using SYBR green (Bio-Rad, Hercules, CA) and 400–700 ng cDNA per reaction. Relative expression levels for COX-2 and EP4 were normalized to GAPDH by the ∆∆C t method.
Metastasis PCR array
RNA was extracted from 66.1-vector and 66.1shEP4 cells and analyzed on a mouse tumor metastasis PCR array (Qiagen SABiosciences, Valencia, CA) per manufacturer’s protocol.
Western blotting
Total cellular protein was analyzed for COX-2, EP4, and beta-actin by immunoblotting; Cox-2 (Cayman Chemical, Invitrogen, Ann Arbor, MI), EP4 (Cayman), beta-actin (Sigma AC15), secondary RT for 1 h [KPL anti-mouse HRP, anti-rabbit HRP (Bio-Rad)] and visualized with ECL SuperSignal West Pico (Pierce Chem. Co., Rockford, IL).
ELISA
PGE2 levels were determined by PGE2 enzyme immunoassay (Cayman Chemical) and expressed as pg PGE2/μg total protein.
Mammosphere
Cells were grown in MammoCult medium (Stem Cell Technologies Inc., Vancouver, CA) supplemented with hydrocortisone, heparin, amphotericin B, and gentamicin (Sigma) and plated in ultra-low attachment plates (Corning Inc., Corning, NY). Three-dimensional spheroidal structures (mammospheres) resulting from the first plating in low-attachment conditions were designated MS-1; subsequent passage into secondary mammosphere cultures at days 7–12 were designated as MS-2 cultures.
Flow cytometry
Cells blocked with 1 % FBS and stained with antibodies recognizing CD44 (FITC-conjugated mouse antihuman CD44), CD24 (PE-conjugated mouse antihuman CD24) or appropriate isotype control (all from BD Biosciences, San Jose, CA). Aldehyde dehydrogenase was detected by Aldefluor kit (Stem Cell Technologies) used according to manufacturer’s protocol and analyzed by FACScan flow cytometry.
Biostatistics
In vivo data was analyzed by non-parametric Kruskal–Wallis test and pair-wise comparisons were carried out by exact two-sided Wilcoxon test. Tumor incidence data analyzed by generalized linear models approach. Data from in vitro studies analyzed by Student’s t test.
Results
EP4 is widely expressed in primary human breast cancer and targeting EP4 inhibits metastasis
We examined the expression of EP4 in 44 invasive ductal carcinomas of the breast by immunohistochemistry. EP4 expression was very low or absent in normal ducts (0, 1+, Fig. 1a), malignant epithelium was positive for cytoplasmic EP4 expression. On a scale of 0–3+ staining intensity, 21/44 (48 %) specimens had 1+ EP4 expression, 13/44 (29 %) were 2+ and 10/44 (23 %) were graded as 3+ in EP4 staining intensity. Nuclear staining was not observed.
Fig. 1.
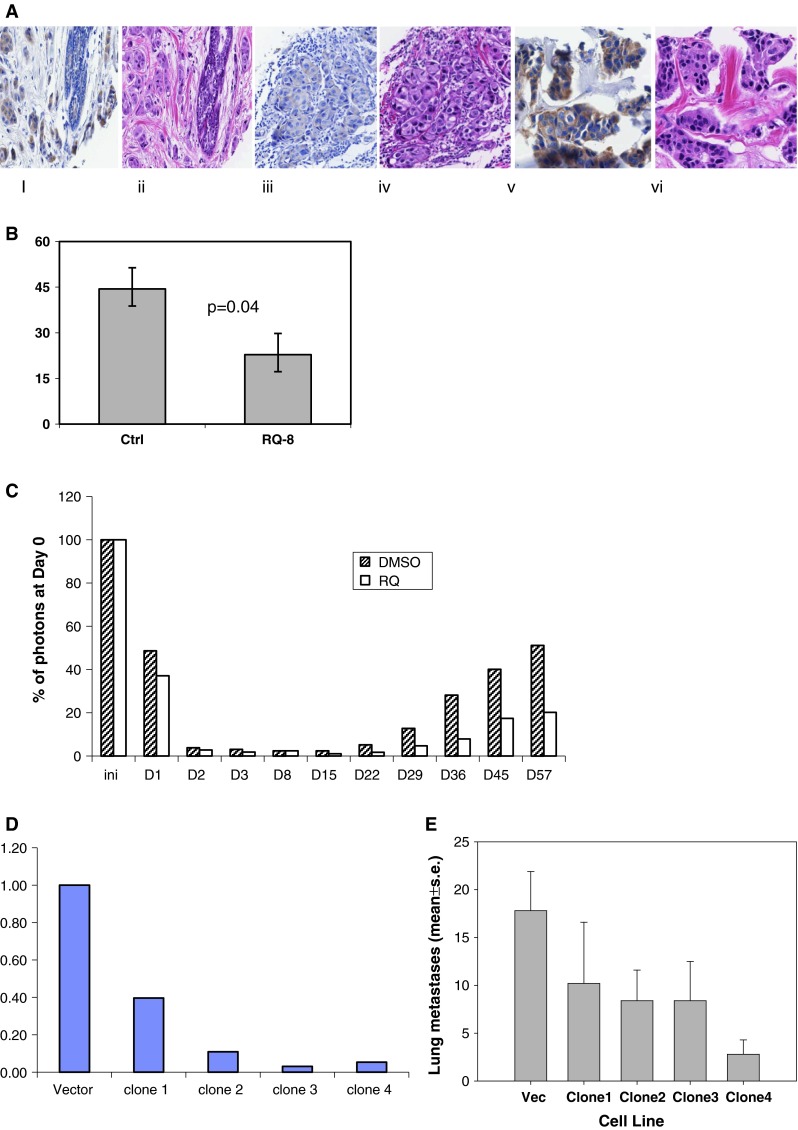
a A tissue microarray was prepared containing 44 invasive ductal carcinoma of the breast. EP4 and H&E by immunohistochemistry. (i) Benign lobule, EP4, 1+; (ii) H&E; (iii) invasive ductal carcinoma, EP4, 1+; (iv) H&E; (v) invasive ductal carcinoma, EP4, 3+; (vi) H&E. b Line 410.4 tumor cells injected proximal to the mammary fat pad of Balb/cByJ female mice treated with vehicle or RQ-08 (30 mg/kg/day). When tumors measured 18 mm in diameter, mice were euthanized and surface lung tumor colonies enumerated. Mean ± SE, P = 0.04. c MDA-MB-231-luciferase cells treated with RQ-15986 (3.0 μM/l) or DMSO vehicle and injected i.v. into groups of five Balb/SCID mice and live animal imaging carried out at 5 min and at the days indicated. Data expressed as percent photons detected relative to day 0. d Line 66.1 cells transfected with plasmid expressing shEP4 or vector; stable clones were derived and EP4 expression characterized by qPCR. e Cell lines from d injected i.v. into 5–10 Balb/cByJ female mice and surface lung tumor colonies quantified. Mean ± SE, P < 0.01
EP4 gene silencing or receptor inhibition with small molecule inhibitors block metastasis in a syngeneic murine breast cancer model [13, 20, 21, 23]. In this study, we confirmed, using a second tumor cell line and a different EP4 antagonist (RQ-08), that metastasis is inhibited by EP4 blockade. Line 410.4 tumor cells were implanted into syngeneic Balb/cByJ female mice and oral administration of RQ-08 (30 mg/kg × 28 days) was initiated on day +7. When tumors achieved an average diameter of 18 mm, mice were euthanized and metastatic disease was assessed. The growth of primary tumors was modestly inhibited by RQ-08 (not shown) but spontaneous metastasis to the lungs was reduced by 49 % (Fig. 1b, P = 0.04). Metastatic success of human MDA-MB-231-luc cells was also reduced by an EP4 antagonist (Fig. 1c). We studied cell-autonomous effects of EP4 antagonism on the tumor cell alone, by pre-treating tumor cells with RQ-15986 (3.0 μM/l) prior to i.v. injection into Balb/SCID mice. At day 1 after i.v. injection of tumor cells, less luciferase signal was detected when EP4 was antagonized. As the surviving tumor cell populations expanded with time, the difference between the two treatment groups became more pronounced. We also created multiple clones of 66.1 expressing EP4shRNA (Fig. 1d). Metastatic potential was reduced by 43, 53, 53, and 84 %, respectively, in comparison to mice injected with vector control cells (Fig. 1e). Thus, EP4 is widely expressed in breast cancer and either genetic or pharmacologic compromise of EP4 activity reduces metastatic potential.
Metastasis and stem cell-associated genes are downregulated in shEP4 cells
We employed a qPCR array of known metastasis-associated genes to compare gene expression patterns of 66.1-vector versus 66.1shEP4 cells. Table 1 shows genes that were downregulated by at least 1.5-fold in 66.1shEP4 cells compared to 66.1-vector cells that included Csf1, c-met, CXCL12, and CD44. Few genes were upregulated in the context of EP4 silencing, but included the metastasis-suppressor Nme4 (data not shown). The downregulation of Csf1, Timp2, and CD44 in 66.1shEP4 cells was confirmed by qPCR (Fig. 2a). While each of these gene expression changes may be important to the mechanism of metastasis inhibition, we focused our further studies on the reduction in CD44, a phenotypic marker of candidate breast cancer stem/tumor-initiating cells [24, 25].
Table 1.
Effect of EP4 reduced expression on metastasis-related genes
| Symbol | Fold down-regulation | Full name |
|---|---|---|
| Timp2 | −5.22 | Tissue inhibitor of metalloproteinase 2 |
| Csf1 | −4.29 | Colony stimulating factor 1 (macrophage) |
| Tcf20 | −4.06 | Transcription factor 20 |
| Ewsr1 | −4.01 | Ewing sarcoma breakpoint region 1 |
| Plaur | −3.89 | Plasminogen activator, urokinase receptor |
| Met | −3.53 | Met proto-oncogene |
| Cxcl12 | −3.47 | Chemokine (C-X-C motif) ligand 12 |
| Chd4 | −3.37 | Chromodomain helicase DNA binding protein 4 |
| Rorb | −2.92 | RAR-related orphan receptor beta |
| Ctbp1 | −2.88 | C-terminal binding protein 1 |
| CD82 | −2.69 | CD82 antigen |
| CD44 | −2.56 | CD44 antigen |
| Hras1 | −2.54 | Harvey rat sarcoma virus oncogene 1 |
| Col4a2 | −2.34 | Collagen, type IV, alpha 2 |
| Itga7 | −2.16 | Integrin alpha 7 |
| Rb1 | −2.15 | Retinoblastoma 1 |
| Mtss1 | −2.12 | Metastasis-suppressor 1 |
| Kras | −2.10 | V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog |
| Ctnna1 | −2.10 | Catenin (cadherin-associated protein), alpha 1 |
| Brms1 | −2.09 | Breast cancer metastasis-suppressor 1 |
| Smad4 | −2.04 | MAD homolog 4 (Drosophila) |
| Etv4 | −1.99 | Ets variant gene 4(E1A enhancer binding protein, E1AF) |
| Fat1 | −1.89 | FAT tumor suppressor homolog 1 (Drosophila) |
| Rpsa | −1.86 | Ribosomal protein SA |
| Pten | −1.82 | Phosphatase and tensin homolog |
| Mta1 | −1.74 | Metastasis-associated 1 |
| Htatip2 | −1.63 | HIV-1 tat interactive protein 2, homolog (human) |
| Trp53 | −1.57 | Transformation-related protein 53 |
RNA isolated from 66.1vector or 66.1shEP4 cells and analyzed on a mouse Metastasis array. Genes downregulated >1.5-fold in 66.1shEP4 cells relative to 66.1-vector cells are listed
Fig. 2.

a mRNA isolated from 66.1-vector and 66.1shEP4 cells and analyzed for expression of metastasis-related genes by metastasis pcr array. b 410.4 cells grown in standard culture or as mammospheres and on day 10 cells were harvested and 100 MS-1 or standard bulk culture cells injected into the mammary fat pad of Balb/cByJ female mice. Tumor growth monitored by caliper and expressed as tumor volume. Solid line = 100 MS-1 cells; dashed line = 100 bulk population cells
Tumor-initiating cells are identified in a syngeneic model of metastatic breast cancer
A true cancer stem cell should have heightened tumor-initiating capacity compared to the general population but a tumor-initiating cell has not been demonstrated in the murine cell lines under study. We cultured 410.4 cells in low-attachment conditions to demonstrate that mammospheres will form. These mammospheres can be dissociated and replated to form secondary and tertiary mammospheres (dns). Limiting dilution assays compared the tumorigenic properties of the general population of 410.4 cells versus mammosphere-forming (MS-1) 410.4 cells. Three of 10 mice injected with 100 of 410.4 bulk cells developed palpable tumors (dashed lines), whereas 8/9 mice injected with MS-1 cells (solid lines) developed progressively growing tumors (Fig. 3c). In an expanded limiting dilution assay using additional cell injection doses (Table 2), five of eight mice (62.5 %) injected with 100 MS-1 developed tumors and an average of 17 metastases per mouse; only 2/8 mice (25 %) injected with 100 cells of the bulk population developed tumors and an average of 0.5 metastases per animal, indicating heightened tumorigenic capacity of mammosphere-forming cells. At all cell doses, MS-1 cells were more tumorigenic than the same number of bulk population cells; the stem cell frequency was calculated as 1/145 MS-1 cells versus 1/526 for bulk cells (P < 0.00425). The average number of metastases was also higher in mice injected with MS-1 cells compared to an equal number of bulk population cells.
Fig. 3.

a, b MDA-MB-231 and MCF7 cells grown in standard culture conditions and analyzed for CD44 and CD24 expression by flow cytometry. Isotype control (left panel), double stain for CD44 and CD24 (right panel). c Aldefluor assay of MDA-MB-231 (left panel) and MCF7 (right panel) cells
Table 2.
Tumor-initiating capacity of mammosphere-forming cells
| Cell type | Tumor incidence | % Tumor incidence | Lung mets |
|---|---|---|---|
| 410.4 MS-1, 10 cells | 1/8 | 12.5 | 7 |
| 410.4 Bulk, 10 cells | 0/8 | 0 | 0 |
| 410.4 MS-1, 50 cells | 3/8 | 37.5 | 0.3 |
| 410.4 Bulk, 50 cells | 1/8 | 12.5 | 0 |
| 410.4 MS-1, 100 cells | 5/8 | 62.5 | 17 |
| 410.4 Bulk, 100 cells | 2/8 | 25 | 0.5 |
| 410.4 MS-1, 500 cells | 7/8 | 87.5 | 3.9 |
| 410.4 Bulk, 500 cells | 5/9 | 55.6 | 3.6 |
| 410.4 Bulk, 5 × 105 | 5/5 | 100 | 28.2 |
MS-1 are mammosphere-derived cells or bulk cells growing under attached culture conditions and injected at the indicated cell doses into the mammary fat pad of syngeneic Balb/cByJ female mice. Tumors palpated twice weekly and degree of metastatic disease determined at necropsy when tumors measured 18 mm in average diameter
EP4 and COX-2 are upregulated in mammospheres of aggressive breast cancer cells
Several laboratories have described the stem cell properties of human breast cancer cell lines [26–28] and we confirmed that MDA-MB-231 cells are highly enriched for a stem cell phenotype whereas this population is rare in MCF7 cells, which have a luminal gene signature. Consistent with the literature, more than 95 % of MDA-MB-231 cells are CD44hi/CD24low (Fig. 3a). In comparison, <1.0 % of MCF7 cells are CD44hi (Fig. 3b), however, these latter cells are highly positive for CD24 expression. The fraction of aldehydedehydrogenase (ALDH+) positive cells was highly variable from experiment to experiment; in one experiment, 5.4 % of MDA-MB-231 cells were ALDH+ (Fig. 3c); 0.23 % of MCF7 cells were ALDH+. Twenty-six percent of 410.4 cells are ALDH+; 57.4 % of 66.1 cells are positive for this marker (data not shown).
We compared EP4 and COX-2 expression levels in mammosphere-forming and bulk populations derived from human or murine breast cancer cell lines. MDA-MB-231 were employed as a model of breast cancer stem cells, MCF7 as a model of non-stem cells and SKBR3 represent the HER-2 phenotype. Metastatic murine cell lines 410.4 and 66.1 and non-metastatic cell lines 410 and 67 were employed for comparison. In both MDA-MB-231 and SKBR3 cells, EP4 mRNA levels were markedly increased in MS-1 versus bulk population cells (Fig. 4a) but EP4 mRNA was not elevated in MS-1-forming MCF7 cells. These patterns have been observed in three independent experiments. Like MDA-MB-231 and SKBR3 cells, murine 66.1 and 410.4 MS-1 cells expressed increased EP4 mRNA versus the bulk population (Fig. 4b), but EP4 was not increased in the comparatively benign 410 and 67 cells in either MS-1 or MS-2 cultures (Fig. 4c). Like EP4, COX-2 was increased in MS-1 versus bulk populations of MDA-MB-231 and SKBR3 cells (Fig. 4d) but slightly elevated in MCF7 MS-1 cultures. In murine cells, COX-2 was induced in MS-1 cultures of 66.1 and 410.4 (Fig. 4e), but not in 410 or 67 MS-1 cells (Fig. 4f). Consistent with the mRNA data, increased COX-2 protein was detected in MS-1 cells of MDA-MB-231 and SKBR3 grown in either DME or mammocult media; only a slight increase in COX-2 protein was detected in MCF-7 cells (Fig. 4g). Increased COX-2 in MS-1 cells resulted in more PGE2 detected in conditioned media of MDA-MB-231 and SKBR3, but not MCF7-derived MS-1 cells (Fig. 4h). Thus, in both human and mouse, EP4 and COX-2 are elevated in mammospheres produced by highly tumorigenic, basal-type cell lines. Luminal-type (MCF7) or less tumorigenic, non-metastatic (410, 67) cells do not elevate EP4 or COX-2 expression in MS-1 tumorspheres.
Fig. 4.

a MDA-MB-231, SKBR3, or MCF7 cells or b 66.1 or 410.4 or c 410 or 67 cells grown as MS-1, MS-2, or bulk cultures. mRNA harvested and analyzed for human or murine EP4 by qPCR. d–f The same cells analyzed for COX-2 mRNA expression. g Cell lysates of MDA-MB-231, SKBR3, or MCF7 cells grown as standard culture or in mammosphere (MS) assay and maintained in either DME media or mammocult (MC) media and immunoblotted for COX-2, EP4, or β-actin. h MDA-MB-231, SKBR3, or MCF7 cells grown as attached (open bar) or mammosphere (closed bar) in DME; conditioned media analyzed for PGE2 by ELISA and expressed as Mean ± SE, pg/μg protein
EP4 antagonists or EP4 gene silencing inhibits properties of breast cancer stem cells in vitro
We compared the growth properties of mammospheres derived from 410.4-vector cells to 410.4shEP4 cells (Fig. 5a). MS-1 mammospheres were disrupted and 5,000 cells from each MS-1 culture was replated and allowed to form secondary (MS-2) mammospheres. The average number of MS-2 spheres formed by 410.4-vector cells (5.6 ± 0.9) was reduced by 57 % in 410.4shEP4 mammospheres (2.4 ± 0.4 spheres). More marked effects of EP4 gene silencing were detected when the size of mammospheres, as indicated by the number of cells/sphere was considered. Mammospheres formed by 410.4-vector, 410.4shEP4, 410 or 67 cells were collected from each well and the total number of sphere-forming cells was determined (Fig. 5a). Five thousand MS-1 410.4-vector cells expanded into secondary tumorspheres to produce a total of 280,666 ± 6,992 cells; a 56-fold expansion in cell number from day 0. In contrast 410.4shEP4 cells expanded by 21-fold. Thus, when mammosphere-induced upregulation of EP4 was prevented by shEP4, mammosphere-forming ability (number and size) was compromised. Although line 410 and 67 cells were able to form small secondary mammospheres, the total average cell number was expanded by 12-fold and 3-fold, respectively. The reduced sphere-forming ability of 410 and 67 cells is consistent with the failure to upregulate EP4 or COX-2 in mammospheres of these cells.
Fig. 5.
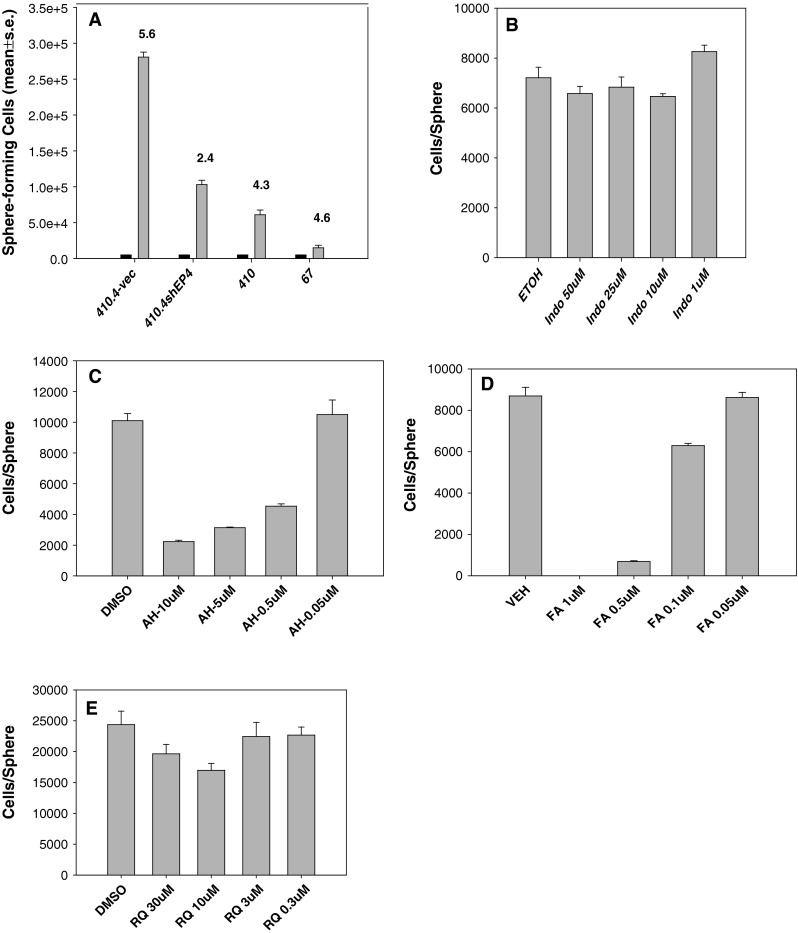
a 5 × 103 MS-1 cells of 410.4-vec, 410.4shEP4, 410 or 67 cells (black bars) were re-plated in secondary (MS-2) cultures and on day 10, sphere number and cellularity were determined for MS-2 cultures. Mean ± SE. b–e MDA-MB-231 cells placed in mammosphere culture and on day 2, treated with Indomethacin, AH23848, Frondoside A or RQ-15986 at the indicated concentrations. On day 10, sphere number and cellularity were determined
We determined the effect of EP4 antagonists or the COX-1/COX-2 inhibitor indomethacin on sphere-forming ability of MDA-MB-231 cells. In the presence of indomethacin (1.0–50 μM/l), neither the numbers of mammospheres nor the size of spheres was affected by COX inhibition (Fig. 5b); cell number was 90–115 % of that observed in vehicle-treated TS-1 cells. In contrast, three EP4 antagonists, AH23848, Frondoside A and RQ-15986 were all able to inhibit the size of mammospheres (Fig. 5c–e). In the presence of AH23848, mammospheres were actually increased in number, but individual spheres were much smaller as indicated by the dramatic reduction in cells/sphere. AH23848 at 10, 5, 0.5, or 0.05 μM/l reduced sphere cellularity by 78, 69, 55, and 2 %, respectively (Fig. 5c). In contrast, AH23848 did not affect the number of attached cells at any concentration examined (dns). While the number of total spheres was not affected by Frondoside A except at the highest concentration tested (1.0 μM/l), the sphere size was inhibited by 100, 92, 28, and 1 % in a dose-dependent manner (Fig. 5d). RQ-15986 (30, 10, 3, and 0.3 μM/l) inhibited sphere-forming cells by 20, 30, 8, or 7 % (Fig. 5e).
Taken together, these data indicate that tumor cells with heightened stem cell properties are more sensitive than the general population to EP4 antagonism.
EP4 antagonists inhibit the stem cell phenotype in vitro
To determine if EP4 inhibition would downregulate phenotypic markers of breast cancer stem cells, MDA-MB-231 cells were placed in mammosphere culture and on day 8, RQ-15986 (10 μM/l) was added. On day 10, cultures were analyzed for ALDH expression. In a typical experiment, RQ-15986 treatment reduced the proportion of ALDH+ MDA-MB-231 cells from 30.4 % to 16.7 % (Fig. 6a). RQ-15986, Frondoside A or AH23848 also modestly reduced the percentage of CD44-positive MS-1 cells. In the presence of PBS, 64 % of MDA-MB-231 (MS-1) cells were CD44-positive; Frondoside A (0.25 μM/l) reduced the fraction of CD44-positive cells to 54.3 %. In the presence of DMSO, 53.5 % of cells were CD44-positive; RQ-15986 (10 μM/l) or AH23848 (5 μM/l) reduced the CD44-positive fraction to 45.9 or 46.9 % of cells, respectively. CD44 expression was not changed in attached cells in the presence of EP4 antagonists (dns). CD24 is poorly expressed in MS-1 cells and the degree of expression was not changed by treatment with any EP4 antagonist (dns).
Fig. 6.

a MDA-MB-231 cells in mammosphere culture were treated with vehicle (left panel) or RQ-15986 (10 μM/l, right panel) on day 8 and 2 days later, Aldefluor positive cells were determined. b Five hundred or 50 of 410.4 MS-1 cells were injected into groups of 10 Balb/cByJ female mice. On day +7, RQ-08 or vehicle administered by gavage (30 mg/kg/day) and tumor incidence on day +46 is plotted. c Tumor volume for the same mice as in b that developed palpable lesions. Mean ± SE
EP4 antagonism inhibits tumor-initiating capacity in vivo
By limiting dilution assays, we assessed the ability of an EP4 antagonist to inhibit tumor-initiating cells in vivo. Balb/cByJ mice were transplanted with 500 or 50 of 410.4 MS-1 cells and, beginning on day +7, were treated with vehicle or RQ-08 for 28 days and tumor incidence and size were assayed. In two independent experiments, EP4 antagonism was able to significantly inhibit the tumor-forming capacity of small numbers of tumor cells as indicated by tumor incidence (Fig. 6b). When 500 cells were injected, tumor incidence was reduced from 90 % to 60 % by RQ-08 (P < 0.02); RQ-08 inhibited tumor-initiating capacity from 60 % tumor-positive mice to 20 % tumor-positive when 50 cells were injected (P < 0.058). The calculated stem cell frequency was reduced from 1/126 cells to 1/460 cells by RQ-08 (P < 0.018). While the average size of tumors is sometimes variable reflecting the different latency in individual mice, it is obvious that RQ-08 has a profound effect on the ability of palpable lesions to expand (Fig. 6c). In animals that developed palpable tumors in spite of RQ-08, the percentage of tumor cells that were CD44hi/CD24low was reduced from 84.2 % ± 1.3 (vehicle) to 74.1 + 1.3 % (P < 0.006).
Discussion
EP4 expressed on the malignant cell contributes to metastatic behavior in a cell-autonomous manner as demonstrated by reduced metastatic potential of tumor cells in which EP4 expression has been silenced or antagonized by small molecule inhibitors. Tumor-derived PGE2 also acts to suppress the anti-metastatic activities of NK cells by acting on EP4 expressed on the NK cell [23, 29, 30]. Other EP4-positive immune effector cells are also inhibited by PGE2 [31–33]. Thus, the function of multiple EP4 receptor-positive cells are affected directly by EP4 antagonism.
EP4 promotes multiple cancers [4–17] but little is known about the role of the COX-2 pathway in cancer stem cells. Mesenchymal stem cells are stimulated by IL-1 to produce PGE2 that creates a supportive microenvironment for cancer stem cells [18]. Stromal fibroblasts support the induction of MCF7-derived stem cells; in that model, PGE2 is not sufficient to directly act on the progenitor stem cells [34]; rather an indirect mechanism is proposed in which PGE2 acts indirectly through the cancer-associated fibroblast to induce a stem cell phenotype. The relevant EP receptor was not identified in that study.
Because mammary tumor cells have elevated endogenous COX-2 activity, we investigated the role of PGE2 and EP4 in an autocrine mechanism that would support breast cancer stem cells. We now show that a sub-population enriched in tumor-initiating cells upregulates both EP4 and COX-2. Upregulation of these genes was only observed in metastatic and/or basal-type human (MDA-MB-231, SKBR3) and murine (410.4, 66.1) cell lines but not in non-metastatic or luminal-type cells suggesting that tumor-initiating cells may contribute to the aggressive phenotype and may be relatively more sensitive than the non-stem cell population to direct inhibition by either EP4 antagonists or COX inhibitors.
The upregulation of EP4 expression in mammospheres is functionally linked with increased tumor-initiating cell capacity in vivo of mammosphere-forming cells. MS-1 cells were more tumorigenic than the bulk population with an increase in stem cell frequency. Importantly, tumorigenic potential of MS-1 cells was significantly reduced by an EP4 antagonist in vivo corresponding to a loss of CD44hi/CD24low cells. In the relatively few RQ-treated mice that developed palpable lesions, the tumors never expanded. This is in contrast to the modest inhibition of tumor size when bulk tumor cells are injected and is consistent with the increased expression of EP4 in MS-1 cells. Consistent with the studies in vivo, in comparison to 410.4-vector cells, 410.4shEP4 cells were less able to expand to form large mammospheres in culture. Interestingly, growth properties in vitro in conventional attached conditions are not different for EP4 high versus low cells supporting the hypothesis that EP4 has important functions in maintaining the stem/progenitor phenotype. 410.4-vector cells expanded by 56-fold during 10 days of culture under low-attachment conditions; the same number of 410.4shEP4 cells were only able to expand by 21-fold. Poorly tumorigenic 410 and 67 cell lines produced only small mammospheres. Likewise, the addition of three different EP4 antagonists to mammosphere-forming cultures were each able to inhibit mammosphere growth. Interestingly, the COX-1/COX-2 inhibitor indomethacin did not affect sphere size, indicating that it is not PGE2 synthesis per se, but more likely to be PGE2-mediated cell signaling that supports sphere formation. The fraction of CD44hi or ALDH1+ cells, both phenotypic markers of tumor-initiating cells, were reduced by each of three EP4 antagonists.
We did not observe appreciable upregulation of COX-2 in MCF7 tumorspheres consistent with the luminal properties of this cell line. Singh et al. detected a rare COX-2 highly positive cell in MS-1 cultures of MCF7; in these cells, the stem cell factor Oct4 was often co-expressed [35]. Those authors propose that the COX-2hi, Oct4+ is a breast cancer stem cell, consistent with the current report.
Taken together, these studies support the hypothesis that EP4-mediated activation can, in an autocrine or paracrine manner drive stem cell survival. Our studies show, for the first time, that a clinically relevant EP4 antagonist can inhibit breast cancer stem cells. Blocking EP4 inhibits breast cancer metastasis and tumorigenic capacity in vivo and stem cell properties including mammosphere-forming ability and phenotypic markers of breast cancer stem cells in vitro. EP4 antagonism may prove to be safer and more effective than global COX-2 inhibition. The demonstrated efficacy of EP4 antagonists in preclinical models of breast and other malignancies supports the continued evaluation of EP4 as a new therapeutic target in advanced malignancy.
Acknowledgments
We thank the Biostatistics, Genomics, Flow cytometry, and Tissue Biorepository Shared Services of the University of Maryland Greenebaum Cancer Center. We thank RaQualia Pharma Inc and Coastside Bio Resources for the gift of EP4 antagonists. These studies were supported by the United States Department of Health and Human Services and the United States Veterans Administration.
Competing interests
The authors declare no competing interests.
Ethical statement
All studies comply with current laws of the U.S.A. Mice were housed, cared for, and used in strict accordance with the U.S. Department of Agriculture regulations and the NIH Health Guide for the Care and Use of Laboratory Animals. All animal protocols were reviewed and approved by the University of Maryland Institutional Animal Care and Use Committee. The University of Maryland School of Medicine Animal Facility is fully accredited by the American Association for the Accreditation of Laboratory Animal Care.
Abbreviations
- ALDH
Aldehydedehydrogenase
- COX-2
Cyclooxygenase 2
- EP4
Prostaglandin E receptor 4
- PGE2
Prostaglandin E2
- MS
Mammosphere
Contributor Information
Namita Kundu, Email: nkundu@umaryland.edu.
Xinrong Ma, Email: Xma001@umaryland.edu.
Tyler Kochel, Email: tkoch001@umaryland.edu.
Olga Goloubeva, Email: ogoloubeva@som.umaryland.edu.
Paul Staats, Email: pstaats@umm.edu.
Keyata Thompson, Email: kethompson@som.umaryland.edu.
Stuart Martin, Email: ssmartin@som.umaryland.edu.
Jocelyn Reader, Email: jread001@umaryland.edu.
Yukinori Take, Email: yukinori.take@askat.co.jp.
Peter Collin, Email: pcollin48@gmail.com.
Amy Fulton, Phone: +1-410-7066479, Email: afulton@umaryland.edu.
References
- 1.Ristimaki A, Sivula A, Lundin J, Lundin M, Salminen T, Haglund C, et al. Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer. Cancer Res. 2002;62:632–635. [PubMed] [Google Scholar]
- 2.Kerikowske K, Molinaro AM, Gauthier ML, Berman HK, Waldman F, Bennington J, Sanchez H, et al. Biomarker expression and risk of subsequent tumors after initial ductal carcinoma in situ diagnosis. J Natl Cancer Inst. 2010;102:627–637. doi: 10.1093/jnci/djq101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reader JC, Holt D, Fulton AM. Prostaglandin E2 EP receptors as therapeutic targets in breast cancer. Cancer Metastasis Rev. 2011;30:449–463. doi: 10.1007/s10555-011-9303-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mutoh M, Watanabe K, Kitamura T, Shoji Y, Takahashi M, Kawamori T, et al. Involvement of prostaglandin E receptor subtype EP4 in colon carcinogenesis. Cancer Res. 2002;62:28–32. [PubMed] [Google Scholar]
- 5.Terada N, Shimizu Y, Kamba T, Inoue T, Maeno A, Kobayashi T, Nakamura E. Identification of EP4 as a potential target for the treatment of castration-resistant prostate cancer using a novel xenograft model. Cancer Res. 2010;70:1606–1615. doi: 10.1158/0008-5472.CAN-09-2984. [DOI] [PubMed] [Google Scholar]
- 6.Kim J, Lakshmikanthan V, Frilot N, Daaka Y. Prostaglandin E2 promotes lung cancer cell migration via EP4-B arrestin1-c-Src signalsome. Mol Cancer Res. 2010;8:569–577. doi: 10.1158/1541-7786.MCR-09-0511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y, Ritzenthaler J, Sun X, Roman J, Han S. Prostaglandin E2 stimulates human lung carcinoma cell growth through induction of integrin-linked kinase: the involvement of EP4 and Sp1. Cancer Res. 2009;69:896–904. doi: 10.1158/0008-5472.CAN-08-2677. [DOI] [PubMed] [Google Scholar]
- 8.Buchanan FG, Gorden DL, Matta P, Shi Q, Matrisian LM, DuBois RN. Role of β-arrestin 1 in the metastatic progression of colorectal cancer. Proc Natl Acad Sci USA. 2006;103:1492–1499. doi: 10.1073/pnas.0510562103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chell SD, Witherden IR, Dobson RR, Moorghen M, Herman AA, Qualtrough D, Williams AC, Paraskeva C. Increased EP4 receptor expression in colorectal cancer progression promotes cell growth and anchorage independence. Cancer Res. 2006;66:3106–3113. doi: 10.1158/0008-5472.CAN-05-3702. [DOI] [PubMed] [Google Scholar]
- 10.Rundhaug JE, Simper MS, Surh I, Fischer SM. The role of the EP receptors for prostaglandin E2 in skin and skin cancer. Cancer Metastasis Rev. 2011;30:465–480. doi: 10.1007/s10555-011-9317-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu J, Zhang Y, Frilot N, Kim JI, Kim W-J, Daaka Y. Prostaglandin E2 regulates renal cell carcinoma invasion through the EP4 receptor-Rap GTPase signal transduction pathway. J Biol Chem. 2011;286:33954–33962. doi: 10.1074/jbc.M110.187344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Subbaramaiah K, Hudis C, Chang S-H, Hla T, Dannenberg AJ. EP2 and EP4 receptors regulate aromatase expression in human adipocytes and breast cancer cells: evidence of a BRCA1 and p300 exchange. J Biol Chem. 2008;283:3433–3444. doi: 10.1074/jbc.M705409200. [DOI] [PubMed] [Google Scholar]
- 13.Ma X, Kundu N, Rifat S, Walser T, Fulton AM. Prostaglandin E receptor EP4 antagonism inhibits breast cancer metastasis. Cancer Res. 2006;66:2923–2927. doi: 10.1158/0008-5472.CAN-05-4348. [DOI] [PubMed] [Google Scholar]
- 14.Fulton AM, Ma X, Kundu N. Targeting prostaglandin E EP receptors to inhibit metastasis. Cancer Res. 2006;66:9794–9797. doi: 10.1158/0008-5472.CAN-06-2067. [DOI] [PubMed] [Google Scholar]
- 15.Timoshenko A, Guoziong X, Chakrabarti S, Lala P, Chakraborty C. Role of prostaglandin E2 receptors in migration of murine and human breast cancer cells. Exp Cell Res. 2003;289:265–274. doi: 10.1016/S0014-4827(03)00269-6. [DOI] [PubMed] [Google Scholar]
- 16.Robertson FM, Simeone AM, Mazumdar A, Shah AH, McMurray JS, Ghosh S, et al. Molecular and pharmacological blockade of the EP4 receptor selectively inhibits both proliferation and invasion of human inflammatory breast cancer cells. J Exp Ther Oncol. 2008;7:299–312. [PubMed] [Google Scholar]
- 17.Yang L, Huang Y, Porta R, Yanagisawa K, Gonzalez A, Segi E, et al. Host and direct antitumor effects and profound reduction in tumor metastasis with selective EP4 receptor antagonism. Cancer Res. 2006;66:9665–9672. doi: 10.1158/0008-5472.CAN-06-1271. [DOI] [PubMed] [Google Scholar]
- 18.Li JH, Reinhardt F, Herschman HR, Weinberg RA. Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin E2 signaling. Cancer Discov. 2012;2:840–855. doi: 10.1158/2159-8290.CD-12-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murase A, Taniguchi Y, Tonai-Kachi H, Nakao K, Takada J. In vitro pharmacological characterization of CJ-042794, a novel, potent, and selective prostaglandin EP(4) receptor antagonist. Life Sci. 2007;82:226–232. doi: 10.1016/j.lfs.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 20.Ma X, Kundu N, Collin PD, Goloubeva O, Fulton AM. Frondoside A inhibits breast cancer metastasis and antagonizes prostaglandin E receptors EP4 and EP2. Breast Cancer Res Treat. 2011;132:1001–1008. doi: 10.1007/s10549-011-1675-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kundu N, Ma X, Walser T, Goloubeva O, Ostrand-Rosenberg S, Fulton AM. Antagonism of the prostaglandin E receptor EP4 inhibits metastasis and enhances NK function. Breast Cancer Res Treat. 2009;117:235–242. doi: 10.1007/s10549-008-0180-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma X, Kundu N, Ioffe O, Goloubeva O, Konger R, Baquet C, Gimotty P, Fulton AM. Prostaglandin E receptor EP1 suppresses metastasis, is associated with better survival and may contribute to breast cancer disparities. Mol Cancer Res. 2010;8:1310–1318. doi: 10.1158/1541-7786.MCR-10-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma X, Holt D, Kundu N, Reader J, Goloubeva O, Take Y, Fulton AM. A prostaglandin E (PGE) receptor EP4 antagonist protects natural killer cells from PGE2-mediated immunosuppression and inhibits breast cancer metastasis. Oncoimmunology. 2013;2:e22647. doi: 10.4161/onci.22647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Al Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weng D, Penzner JH, Song B, Koido S, Calderwood SK, Gong J. Metastasis is an early event in mouse mammary carcinomas and is associated with cells bearing stem cell markers. Breast Cancer Res. 2012;14:R18. doi: 10.1186/bcr3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, Clouthier SG, Wicha MS. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci USA. 2012;109:2784–2789. doi: 10.1073/pnas.1018866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kenny PA, Lee GY, Myers CA, Neve RM, Semeiks JR, et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol Oncol. 2007;1:84–96. doi: 10.1016/j.molonc.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survival chemotherapy. Breast Cancer Res. 2008;10:R25. doi: 10.1186/bcr1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holt D, Ma X, Kundu N, Fulton AM. Prostaglandin E2 (PGE2) suppresses natural killer cell function through the PGE2 receptor EP4. Cancer Immunol Immunother. 2011;60:1577–1586. doi: 10.1007/s00262-011-1064-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holt DH, Ma X, Kundu N, Collin PD, Fulton AM. Modulation of host natural killer cell functions in breast cancer via prostaglandin E2 receptors EP2 and EP4. J Immunother. 2012;35:179–188. doi: 10.1097/CJI.0b013e318247a5e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boniface K, Bak-Jensen K, Li Y, Blumenschein W, McGeachy M, McClanahan T, et al. Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. J Exp Med. 2009;206:535–548. doi: 10.1084/jem.20082293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharma S, Yang S, Zhu L, Reckamp K, Gardner B, Baratelli F. Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of FOXP3 expression and CD4+ CD25+ T regulatory cell activities in lung cancer. Cancer Res. 2005;65:5211–5225. doi: 10.1158/0008-5472.CAN-05-0141. [DOI] [PubMed] [Google Scholar]
- 33.Yao C, Sakata D, Esaki Y, Li Y, Matsuoka T, Kuroiwa K, et al. Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nat Med. 2009;15:633–640. doi: 10.1038/nm.1968. [DOI] [PubMed] [Google Scholar]
- 34.Rudnick JA, Arendt LM, Klebba I, Hinds JW, Iyer V, Gupta PB, Naber SP, Kuperwasser C. Functional heterogeneity of breast fibroblasts is defined by a prostaglandin secretory phenotype that promotes expansion of cancer-stem like cells. PLoS One. 2011;6:e24605. doi: 10.1371/journal.pone.0024605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh B, Cook DR, Vincent L, Hall CS, Lucci A. Role of COX-2 in tumorospheres derived from a breast cancer cell line. J Surg Res. 2011;168:e39–e49. doi: 10.1016/j.jss.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]