

Abstract
Although de novo computational enzyme design has been shown to be feasible, the field is still in its infancy: the kinetic parameters of designed enzymes are still orders of magnitude lower than those of naturally occurring ones. Nonetheless, designed enzymes can be improved by directed evolution, as recently exemplified for the designed Kemp eliminase KE07. Random mutagenesis and screening resulted in variants with >200-fold higher catalytic efficiency, and provided insights about features missing in the designed enzyme. Here we describe the optimization of KE70, another designed Kemp eliminase. Amino acid substitutions predicted to improve catalysis in design calculations involving extensive backbone sampling were individually tested. Those proven beneficial were combinatorially incorporated into the originally designed KE70 along with random mutations, and the resulting libraries were screened for improved eliminase activity. Nine rounds of mutation and selection resulted in >400-fold improvement in the catalytic efficiency of the original KE70 design, reflected in both higher kcat and lower KM values, with the best variants exhibiting kcat/KM values of >5x104 s−1M−1. The optimized KE70 variants were characterized structurally and biochemically providing insights into the origins of the improvements in catalysis. Three primary contributions were identified: first, the reshaping of the active site cavity to achieve tighter substrate binding; second, the fine-tuning of the electrostatics around the catalytic His-Asp dyad; and third, stabilization of the active-site dyad in a conformation optimal for catalysis.
Keywords: computational protein design, directed evolution, enzymatic catalysis
Introduction
The advent of computational methods for predicting structure from sequence at atomic accuracy provides a new and powerful way of designing tailor-made active sites 1; 2; 3; 4. The computational design of enzymes depends on two factors: (i) the ability to design an active-site configuration that confers efficient catalysis; (ii) the ability to compute a sequence that confers the desired configuration. Both steps are currently performed with considerable success, but are far from optimal 5. Directed evolution, which requires no prior knowledge of structure-function, can be applied to improve computationally designed enzymes, and can therefore complement our limited design skills. However, the bottleneck of directed evolution is the very limited sequence space that can be covered by library screening.
We have recently described a series of computationally designed enzymes that catalyze an unnatural reaction dubbed the Kemp elimination 6. The Kemp elimination is a model reaction for proton transfer – a critical step in numerous enzymatic reactions. In this activated model system (in particular in the case of the 5-nitrobenzisoxazole substrate used here), a base-catalyzed proton transfer from carbon, concerted with the cleavage of nitrogen-oxygen bond, leads to the cyanophenol product (Scheme 1). The mechanism of the Kemp elimination has been extensively studied, and this reaction has been used as a probe for studying medium effects in catalysis 7; 8; 9. Several enzyme-like systems that catalyze this reaction have been explored, including catalytic antibodies 10; 11, synthetic polymers 12, and promiscuous catalysis by non-enzymatic proteins 13; 14. In all these protein catalysts, the reaction is catalyzed by a side-chain acting as a base within a hydrophobic active site. The alignment of the catalytic base relative to the substrate, medium effects that lead to the activation of the base catalysts, and charge-dispersing interactions that stabilize the negatively-charged transition state (TS), have all been shown to play a role 9; 15.
Scheme 1.
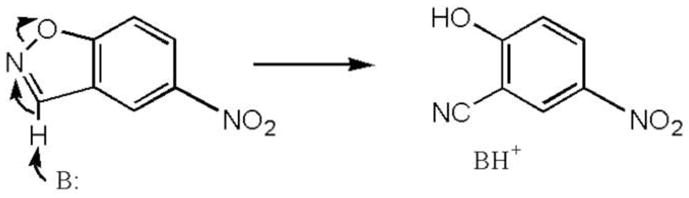
The computationally designed Kemp eliminases were accordingly designed to have a base for proton abstraction aligned with the substrate C-H bond in an otherwise apolar active site. Two catalytic bases were explored – a carboxylic acid side-chain (Glu, as, for example, in the KE07 design 6, or Asp), or a histidine polarized and positioned by an adjacent aspartic or glutamic acid (His-Glu/Asp dyad), as in the KE70 design described here. The locations of these catalytic residues relative to the transition state were optimized by quantum mechanical calculations. Other residues were included for maximal transition state stabilization, such as hydrogen-bond donor for the stabilization of the negative charge that develops on the phenolic oxygen, and aromatic residue(s) for substrate binding and delocalization of the TS’s negative charge. The RosettaMatch algorithm was used to search for constellations of protein backbones capable of localizing these catalytic residues based on a large set of natural proteins with known structures. The Kemp eliminase active-site residues were then installed within these natural scaffolds by replacing 12 to 20 residues of the natural protein 6.
Our previous report described the characterization and the directed evolution of KE07, one of the early designs 16. The structural and mechanistic data shed light on the properties of KE07 and the routes that led to its optimization. Critical contributions from the refinement of the electrostatics in the vicinity of the catalytic base, and of the active-site pocket in general, were identified. The mutations that accumulated in the directed evolution of KE07 led to a 200-fold increase in the kcat/KM, and could be classified into the following groups: a) Mutations improving binding of the substrate and/or transition state; b) Mutations optimizing the electrostatic environment and thus increasing the pKa of the catalytic base and its catalytic efficiency, c) Surface mutations most probably increasing protein solubility and stability.
This work describes the optimization of the KE70 design, which is one of the more advanced Kemp eliminase designs. KE70 is based on the TIM barrel scaffold of deoxyribose phosphate aldolase of E. Coli (PDB accession code 1JCL, Fig. 1a). Unlike the glutamate side-chain acting as a base in the KE07 design, the catalytic base in KE70 is a His17-Asp45 dyad located at the bottom of the active site. Other key residues include Tyr48 for π-stacking of the substrate and transition state, and Ser138 designed to serve as a hydrogen-bond donor in the stabilization of the phenolic oxygen (Fig. 1b). In addition, a number of hydrophobic residues (Ala19, Trp72, Ala103, Ile140, Val168, and Ile 202) were introduced to create a tight hydrophobic pocket for the substrate binding.
Fig. 1.
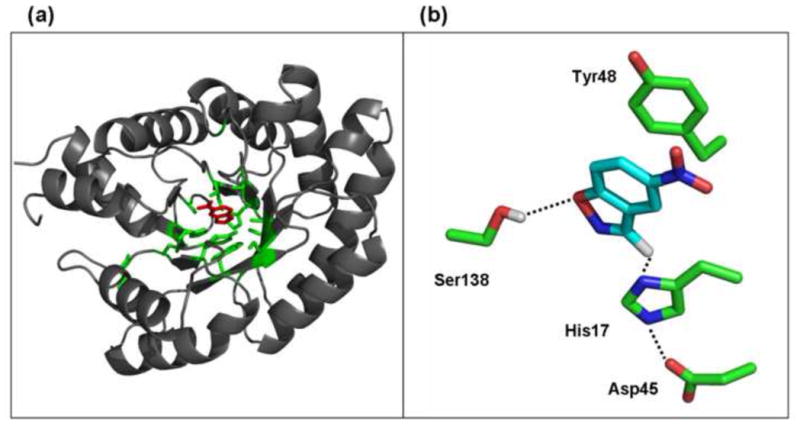
a. The KE70 design. Shown is the natural scaffold on which the design was based (PDB accession code 1JCL, grey), the modeled 5-nitrobenzisoxazole substrate (red), and the side chains of 16 residues replaced to form the designed Kemp eliminase active site (green). b. The key features of KE70’s active site. Shown are the 5-nitrobenzisoxazole substrate, the His17-Asp45 dyad, the H-bond donor (Ser138), and the stacking Tyr48 (Due to insertion of Ala after the initial Met to accommodate a restriction site for cloning the KE70 libraries, the numbering of the residues in this paper is n+1 relative to the numbering in the publication describing the original KE70 design 6,).
The catalytic proficiency of the KE70 design was nearly 10-fold higher than that of the KE07 design (kcat/KM ~80 vs. ~10 s−1M−1), and the rate acceleration is relatively high for a designed enzyme or enzyme mimic (kcat/kuncat~105). However, the catalytic efficiency of KE70 is still far below that of natural enzymes whose kcat/KM values are, on average, in the range of 105 s−1M−1, and can be as high as 108 s−1M−1 (Bar-Even et al., submitted for publication). We therefore opted for the optimization of KE70 with the ultimate goal of obtaining an enzyme with catalytic parameters comparable to the ones observed in natural enzymes. Whereas we used traditional random mutation based directed evolution to optimize KE07, for KE70, we included computationally guided optimization using several approaches that have not been applied in the original design. For example, to go beyond the fixed backbone approximation used in the original design calculations 6, backbone flexibility was incorporated in the computational optimization to search for mutations that mediate new backbone conformations conferring improved transition state binding. However, rather then remaking new designs and testing them (a process which is costly and laborious), the sequence changes suggested by the computational optimization were tested individually. Those that improved activity were introduced into the evolving KE70 variants in a combinatorial manner, to identify the most successful combinations of mutations. Overall, >400-fold improvement of KE70’s catalytic efficiency was obtained via extensive changes in the designed active site, complemented by random mutations enriched throughout the directed evolution process.
Results
Computational optimization of the KE70 design
In the original Kemp eliminase design calculations, the backbone was kept fixed for higher computational efficiency 6. However, small backbone movements can considerably increase the range of possible side-chain conformations. With the goal of achieving tighter transition state binding, we explored several approaches to incorporate backbone flexibility into the design calculations. We also explored alternative active site arrangements, loop geometries, and varying the electrostatic environment of the catalytic dyad residues. As the active site of KE70 resides within the β-barrel of the protein, drastic reshaping of the backbone was not allowed, as it would disrupt the hydrogen-bonding pattern between β-strands, which is critical for maintaining the TIM-barrel fold. The specific strategies we explored are described below.
Design category 1: Ensemble generation using backrub calculations
-
A straightforward way to go beyond the fixed backbone approximation is to generate an ensemble of slightly perturbed conformations within the neighborhood of a starting backbone structure. “Backrub” moves” 17, in which the coordinates of a residue pivot around an axis defined by adjacent residues, provide an effective means to densely sample local conformational space 18. The advantage of backrub moves is that the perturbations are purely local. Since the Cα-Cβ vector is perturbed, side-chains emanate from the perturbed backbones in a range of orientations so the variation in the positions of the terminal atoms can be greater than 1Å. Indeed, backrub ensembles were found to allow improved recapitulation of protein side-chain conformations following mutations18.
We therefore subjected the original KE70 design model to Monte Carlo based backrub sampling, focusing on moves in the vicinity of the active site and thus generating a large ensemble of models with slightly perturbed active site backbone geometries (Supplementary Fig. 1). For each of these, design calculations were carried out to identify amino acid sequences with particularly favorable transition state binding energies. These calculations identified a number of mutations and combinations of mutations, predicted to improve transition state stabilization: Ala19Thr/Ser, Tyr48Trp_Ser74Ala_Ser138Ala, Trp72Cys_Gly101Ala, Ser138Ala_His166Tyr, Arg70Ser, Tyr48Ala_Ser74Phe_Ala103Val, and Ser138Trp_His166Gly_Val168Ser. Since it is not certain that the backbone variation underlying each design could be realized by the predicted sequence changes, each mutant combination was tested before combining it with other sequence changes elsewhere in the protein. Experimental testing of the mutations indicated that activities of the double mutants Trp72Cys_Gly101Ala and Ser138Ala_His166Tyr were 2.6 and 1.9-fold higher than of the KE70 design, respectively. The Trp72Cys mutation is particularly interesting because it changes the packing environment of the catalytic His17 and may lead to a better positioning for proton abstraction of the substrate. Mutations of Trp72, Gly101, Ser138, and His166 were therefore introduced into the KE70 library at Round 3 of directed evolution (Table 1, Library #1).
To search more broadly for active site configurations that are related to but differ from the original KE70 design, we repeated the active site search using RosettaMatch 19 on the same scaffold (1JCL). In a subset of these calculations, we relaxed the requirement for a hydrogen bonding group to the negatively charged phenolic oxygen in the transition state, which resulted in a larger set of possible active site designs than in the earlier calculations. This followed the observation that both in KE07 and in the initial KE70 design, this hydrogen-bonding interaction did not promote higher rate accelerations. The catalytic geometry of each of the identified matches was optimized by gradient-based minimization and the remaining side chains of the active site were redesigned to maximize favorable interactions with the transition state. To further sample in the vicinity of the best solutions obtained at this stage, the lowest energy design variants were subjected to backrub Monte Carlo simulations and the side chains at the active site were redesigned in each of the resulting backbone conformations. Despite the additional sampling, the designs with the best catalytic dyad geometry and most favorable transition state binding geometry all retained the same catalytic dyad as KE70 (His17-Asp45), which appears to be the best solution in terms of catalytic geometry and transition state binding energy for this scaffold. Eight of these optimized variants were selected for construction and expression (Supplementary Tables 1 and 2). Perhaps because the structural variation introduced by the backrub sampling was actually not realized in most cases, only the KE113 design was as active as the original KE70 design. Mutations of KE113 relative to KE70, and certain variations of these mutations (denoted with a slash) (Ser74Ala/Gly, Phe77Tyr, Leu136Trp, Lys173Thr/Asn, Ala178Ser, Ala231Ser, Ala238Ser, Ser239Thr/Asn/His/Arg) were incorporated into the libraries at Rounds 5 and 6 of directed evolution (Table 1).
Table 1.
Summary of the directed evolution of KE70
| Round # | Random mutagenesis | Recombination | Mutation spiking by ISOR22 | Fold improvement measured with crude lysatesa |
|---|---|---|---|---|
| 1 | 2±1 random mutations per gene | - | - | ≤6-fold relative to designed KE70; best variant - R1 8/9C |
| 2 | - | Shuffling of the 15 best variants from Round 1 | - | ≤1.5-fold relative to R1 8/9C; best variant – R2 7/12F (see Table 2) |
| 3 | - | - | Shuffling of the 15 best variants from Round 2, with incorporation of designed mutations: Library #1 (Design categories 1a+2) – Met16Ile/Leu/Val/Phe, Leu18Ile/Leu/Val/Phe, Trp72Ser/Cys/His/Leu, Gly101Glu/Gln/Ala/Ser, Ser138Ala, His166Tyr/Asp/Asn/Ala/Ser. Library #2 (Design category 4a) – insertions after the residues Thr20 (Gly/Ser), Asn22 (Gly/Ser), Thr171 (Asn/Ala/Pro/Gly/Ser), Val204 (Ala/Pro/Gly/Ser), and Ser239 (Asn/Ala/Pro/Gly/Ser) | ≤3-fold relative to R2 7/12F; best variants – R3 2/6D (Library #1) and R3 9/3B (Library #2) |
| 4 | - | Shuffling of the 18 best variants from Round 3, both from Library #1 and Library #2 | - | ≤3-fold relative to R3 9/3B; best variants – R4 4/1B, R4 4/5B (see Table 2) |
| 5 | - | - | Shuffling of the 12 best variants from Round 4, with incorporation of designed mutations: Design category 1b - Ser74Ala/Gly, Phe77Tyr, Leu136Trp, Ala178Ser, Lys173Asn/Thr, Ala231Ser, Ala238Ser, Ser239Thr/Asn/His/Arg; Design category 4a - Ala21Asn/Gln/Arg, Asn22Gln/Arg, | ≤3-fold relative to R4 4/1B; best variant – R5 7/4A (see Table 2) |
| 6 | - | Shuffling of the 10 best variants from Round 5 | - | ≤1.2-fold relative to R5 7/4A; best variants – R6 6/10A, R6 4/8B (see Table 2) |
| 7 | 4 best variants from Round 5 and 4 variants from Round 6, 2±1 mutations per gene | - | - | ≤1.2-fold relative to R6 6/10A; best variant – R7 7/1C |
| 8 | - | - | Shuffling of the 14 best variants of Round 7, with the incorporation of designed mutations: Design category 2 - Met16Ala/Val, Leu18Ile/Val/Leu/Phe; Design category 4b - Ala238Met, Ser240Gly, Leu241Ala | ≤1.2-fold relative to R7 4/2E and 3/2B; best variants – R8 12/12B and R8 15/11E (see Table 2) |
| 9 | 22 best variants from Round 8, 2±1 mutations per gene | - | - | ≤1.2-fold relative to R8 12/12B and R8 15/11E |
The activity improvement measured in crude lysates is not corrected for protein expression, and is therefore only a preliminary measure for an increase in protein activity.
Design category 2: Beta strand perturbations
To fine-tune the positioning of the catalytic residues, we explored remodeling the conformation of the β-strands that support these residues, and the overall active site configuration, by systematically increasing and decreasing the size of the residues on the back side of the β-strands. The starting point for these computations was the double mutant Tyr48Phe_Phe77Tyr, suggested by the initial backrub calculations, which was ~1.7-fold more active than the KE70 design (the Tyr48Phe mutation was also identified in the first round of directed evolution, and the adjacent Phe77 was changed to the more hydrophilic tyrosine, since the hydroxyl group is solvent exposed). The side chains adjacent to His17 and Tyr48 (which interact with the side chains from the surrounding αhelices), were either elongated or shortened, to push the β-strand on which the side-chain resides into the active site, or pull it into the protein core, respectively. All the tested second-shell mutations around Phe48 (Ile47Val/Leu, Ile49Val/Leu) decreased the activity, indicating that the position of the aromatic side chain is likely to be optimal. In contrast, some of the mutations decreasing the side-chain size of residue 16 (Met16Ala/Val/Leu) or modifying the side-chain size of Leu18 (Leu18Val/Phe), were found to increase activity. Specifically, the mutations Met16Val and Leu18Phe increased the activity by ~2.9 and 2.6-fold, respectively. Mutations of Met16 and Leu18 were therefore introduced into the libraries of Round 3 and Round 8 (Table 1, Library #1), and the mutation Phe77Tyr was included at Round 5 (Table 1).
Design category 3: pKa modulation
To tune the pKa of the catalytic His17-Asp45 dyad, we mutated the adjacent positively charged residues to either neutral, polar, or negatively-charged amino acids, and adjacent hydrophobic/polar residues to negatively charged amino acids. Mutations Lys14Glu, Arg70Leu, Leu136Asp, Asn134Asp were tested, but with the exception of mutation Lys14Glu, which increased the activity ~1.4-fold, mutations which either removed a positive charge, or introduced a negative charge in the vicinity of the catalytic dyad, were found to slightly decrease the activity, or had no effect.
Design category 4: Loop redesign
Changes in loop length (insertions or deletions) are often associated with the evolution of new enzymatic activities in nature 20. We experimented with redesigning the active site loops to increase the catalytic efficiency of KE70.
Single amino acid insertions in four different loops were examined: loops formed by residues 20–27 (insertion of Gly/Ser after Thr20 and after Asn22); residues 168–180 (insertion of Asn/Ala/Pro/Gly/Ser after Thr171), residues 202–210 (insertion of Ala/Pro/Gly/Ser after Val204), and residues 238–241 (insertion of Asn/Ala/Pro/Gly/Ser after Ser239). These positions and the inserted amino acids were chosen with the aim of increasing the flexibility of the loop, so that either the newly introduced amino acid, or a polar residue within the existing loop sequence, could interact with the nitro group of the substrate. These insertions were incorporated into Library #2 of Round 3 of directed evolution (Table 1). In addition, mutations Ala21Asn/Gln/Arg and Asn22Gln/Arg were incorporated into the libraries of Rounds 5 and 6 of directed evolution (Table 1) to form additional potential interactions with the substrate’s nitro group.
A recently developed loop design protocol 21 was also applied, which computationally introduced a predefined hydrogen bond interaction to the nitro group of the substrate. The length of the loop from residue Phe236 through Leu242 (7 amino acids with the sequence FGASSLL) was varied between 5 and 8 amino acids while maintaining a hydrogen bond interaction between either a Ser or an Asn, to the nitro group of the substrate. Designs for each loop length were experimentally tested (Supplementary Table 3), and only the longest loop with the sequence FGMSAGAL was found to increase the activity. It increased the activity ~3.5-fold compared to the Tyr48Phe_Phe77Tyr mutant and combining it with the beta strand mutation Met16Val increased the activity ~9-fold. The FGMSAGAL loop was subsequently incorporated at Round 8 of directed evolution (Table 1).
Directed evolution of KE70
Table 1 summarizes the results of nine rounds of directed evolution starting from the designed KE70. The first two rounds employed random mutagenesis and improved the activity of KE70 by ~10-fold. Several first-shell residues were mutated, in particular, the designed stacking residue Tyr48 was changed into Phe, and Ala204 was changed into Val (Table 2, Figure 2a). These changes indicated that the KE70 design could be optimized by changes in the first-shell residues, which was not the case in the directed evolution of the KE07 design, where mutations in the 2nd and 3rd shells dominated, and only a single designed residue was mutated. The subsequent directed evolution rounds therefore included not only random mutagenesis, but also designed mutations identified as described in the previous section. At Round 3, two areas of the protein were optimized (Table 1). Library #1 included mutations in the beta strands at the bottom of the active site (Design categories 1a and 2; Fig. 2b). Library #2 included insertions in the loops at the top of the active site (Design category 4a; Fig. 2c). The designed mutations were spiked in a combinatorial manner by the ISOR method 22, so that each library variant contained on average 2 mutations out of the complete set of 22 mutations at 6 different positions for Library #1, and 18 insertions at 5 different positions for Library #2. Given the limited screening capacity (800–1600 variants per round), we aimed at libraries that carry only few designed mutations per gene. Therefore, designed mutations that could be readily introduced by random point mutations (e.g., Ala19Thr and Lys14Glu) were not incorporated by synthetic oligonucleotides. At Round 4, the improved variants from Library #1 and Library #2 were shuffled to combine the mutations in the loops and in the beta strands. At Rounds 5 and 6, further design optimizations were explored, based on design categories 1b and 4a (Table 1, Fig. 2d).
Table 2.
Summary of sequence, kinetic and structural data of representative KE70 variants.
| Variant | Kinetic parameters (5-nitro benzisoxazole)a, kcat, s−1 KM, mM kcat/KM. s−1M−1 (fold improvement in kcat/KM relative to the designed KE70) | Mutations incorporated by error-prone PCR (in bold – designed and active site residues)b | Beta strand optimization by backrub protocol (design Category #1a) | Insertions in loops (design Category #4a) | Additional design optimizations (Categories #1b and #4b) | pKa (kcat) | pKa (kcat/KM) | Structure: resolution and number of molecules in asymmetric unit (in brackets) |
|---|---|---|---|---|---|---|---|---|
| Designed KE70 | 0.14±0.01c 1.11±0.09 126±4 (1) |
- | - | - | - | 6.3±0.3 (basic) 6.6±0.3 (acidic) |
5.4±0.1 (basic) 7.4±0.1 (acidic) |
2.25Å (2) |
| R2 2/7E | 0.23±0.01 0.40±0.04 570±50 (4.5) |
Tyr48Phe, Asp212Glu | - | - | - | 2.15Å (2) | ||
| R2 3/5G | 0.229±0.004 0.21±0.03 1100±140 (8.7) |
Tyr48Phe, Val176Glu Ala204Val |
- | - | - | 2.02Å (2) | ||
| R2 7/12F | 0.316±0.005 0.237±0.006 1330±13 (10.5) |
Asp23Gly, Tyr48Phe Asp212Glu, His251Tyr |
- | - | - | 5.5±0.1 | 6.2±0.1 | 1.80Å (2) |
| R4 4/1B | 1.66±0.02 0.18±0.01 9240±560 (73) |
Lys29Asn, Thr43Asn Tyr48Phe, Ala204Val |
Trp72Cys Ser138Ala |
Ser20a Pro171a Ala239a |
- | |||
| R4 4/5B | 1.32±0.05 0.16±0.01 8000±320 (63) |
Lys29Asn, Thr43Asn Tyr48Phe, Ala204Val |
Trp72Cys Gly101Ser His166Asn |
Ser20a Ala204a Ser239a |
- | |||
| R4 8/5A | 0.79±0.02 0.18±0.01 4380±350 (35) |
Lys29Asn, Tyr48Phe Lys197Asn, Ala204Val |
Trp72Cys His166Tyr |
Ser20a Gly239a |
- | 5.9±0.1 | 5.9±0.2 | 1.40Å (1) |
| R5 7/4A | 5.38±0.55 0.14±0.01 37,800±1000 (300) |
Lys29Asn, Thr43Asn Tyr48Phe, Val176Glu Ala204Val |
Trp72Cys Gly101Ser Ser138Ala His166Asn |
Pro171a Ser239a |
Ala231Ser | 1.70Å (1) | ||
| R6 4/8B | 5.00±0.17 0.088±0.008 57,300±6140 (455) |
Lys29Asn, Thr43Asn Tyr48Phe, Lys197Asn, Thr198Ile, Ala204Val |
Trp72Cys Gly101Ser Ser138Ala His166Asn |
Ser20a Ala239a |
Ser74Gly Ala178Ser |
|||
| R6 6/10A | 5.26±0.45 0.096±0.012 54,800±5370 (435) |
Lys29Asn, Thr43Asn Tyr48Phe, Ala204Val |
Trp72Cys Gly101Ser Ser138Ala His166Asn |
Ser20a Ala239a |
- | 6.2±0.1 | 6.0±0.2 | 2.2Å (2) |
| R8 12/12B | 7.47±0.32 0.143±0.028 53,100±8180 (421) |
Lys29Asn,Thr43Asn Tyr48Phe, Thr75Ala Lys197Asn, Thr198Ile Ala204Val |
Trp72Cys Gly101Ser Ser138Ala His166Asn |
Ser20a Ala239a |
Ala178Ser | |||
| R8 15/11E | 5.30±0.18 0.150±0.015 34,900±2220 (277) |
Lys29Asn, Gln36Lys Thr43Asn, Tyr48Phe Ala204Val |
Trp72Cys Gly101Ser Ser138Ala His166Asn |
Ser20a Ala239a |
Ala178Ser Ala238Met |
The reported parameters are the average of at least three independent measurements, and error ranges reflect the differences between repeated determinations.
The numbering of the residues in this paper is n+1 relative to the numbering in the publication describing the original KE70 design 6, due to insertion of Ala after the initial Met, to accommodate a restriction site for cloning the KE70 libraries). Inserted loop residues are numbered as the residues after which they were inserted with the suffix “a”.
The deviation (≤1.6-fold) from the catalytic parameters previously reported for the KE70 design (kcat= 0.16±0.05s−1, KM=2.1±0.8mM, kcat/KM.=78±14s−1M−1)6 may stem from the differences in protein production, purification, and assay temperature in assays performed at different laboratories and times.
Fig. 2. Mutations in the evolved KE70 variants.
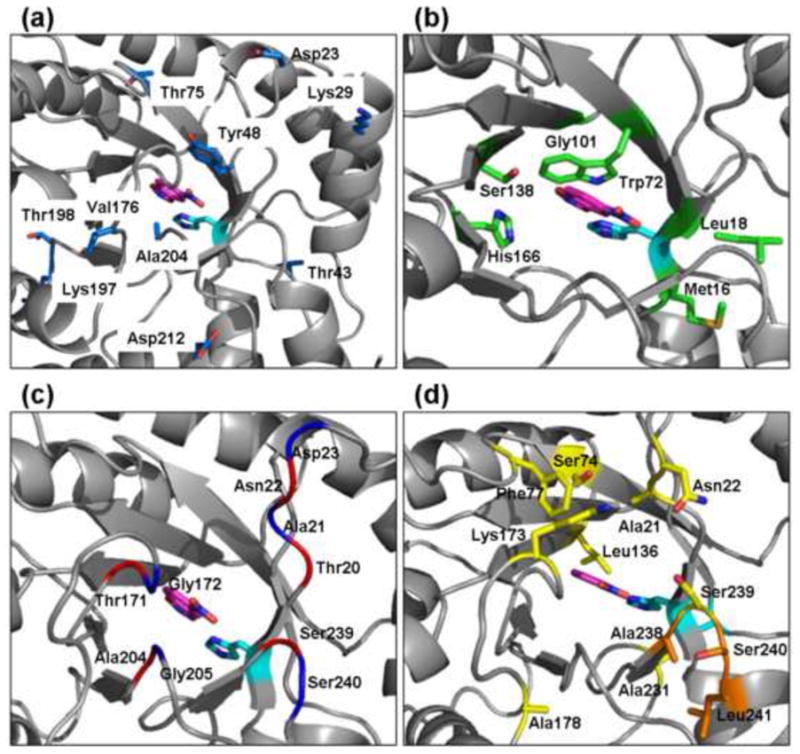
Shown is the model of KE70 design, with the side-chains that were mutated in the evolved variants. The catalytic His17 is shown in cyan, and the substrate is in pink. a. Residues in which random mutations accumulated during nine rounds of directed evolution (blue). b. Residues in the beta-strands at the bottom of the active site optimized by design categories 1 and 2 (green). c. Designed insertions in the loops above the active site (design category 4; noted in red are positions upstream to the insertions; in blue, positions downstream to insertions). d. Residues optimized at Round 5 (design category 5; yellow), and Round 8 (design category 4; orange).
Following another round of random mutagenesis (Round 7), the improved variants were shuffled again (Round 8). In parallel, at Round 8, additional mutations were explored by ISOR method based on design optimizations around the catalytic His17 and around insertions in the active site loops (Design categories 2 and 4b, Fig. 2d, Table 1). However, only mild improvements in the catalytic activity were obtained at Rounds 7 and 8. Round 9 of directed evolution, in which random mutagenesis was applied on the variants from Round 8, also failed to yield any additional improvement (Table 1). The improvement in the catalysis rate of KE70 therefore appeared to have reached a plateau.
The selected mutations and activity improvements
Only a subset of the designed mutations found individually to increase activity were incorporated in the final KE70 variants with the highest activities (Table 2). This suggests that an accurate calculation of the coupling between multiple simultaneous changes in the active site is challenging for current computational methods, and our combinatorial incorporation strategy is a pragmatic solution to this yet unsolved problem. Assuming that there was no bias in the incorporation method (as indicated by sequencing of the unselected libraries), the mutations that were not found in the selected KE70 variants, were probably not beneficial, or even deleterious in the context of other mutations that accumulated in the evolved variants. For example, mutations of the residues adjacent to the catalytic His17 (Met16 and Leu18, design category 2) were not incorporated although they were beneficial on their own and were introduced twice in the directed evolution process (at Round 3 and Round 8). In contrast, all the designed modifications of the beta strands at the bottom of the active site that increased the catalytic activity (Trp72Cys, Gly101Ser, Ser138Ala, and His166Asn) were ultimately incorporated. Insertions in the loops at the top of the active site were readily incorporated (Table 2, variants R4 4/1B and R4 4/5B), but after further rounds of optimization, only two insertions were fixated (Ser20a and Ala239a). Further design optimizations (design categories 1b and 4b) resulted in only small improvements of the catalytic activity, with the key mutations being Ser74Gly, Ala178Ser, Ala231Ser, and Ala238Met (Table 2, Fig. 2d).
The random mutations in the evolved KE70 variants occurred mostly in the active site (Table 2, Fig. 2a). Several of the designed residues, and residues adjacent to designed residues were mutated (Tyr48Phe, Thr43Asn, and Thr75Ala). Mutations in the loops adjacent to the active site, such as Asp23Gly, Val176Glu, Lys197Asn, and Thr198Ile, were as well associated with improvements in catalysis.
After six rounds of directed evolution, the catalytic proficiency of KE70 improved over 400-fold (Table 2). The turnover number (kcat) increased by >35-fold, from 0.14 s−1 to >5 s−1, and KM decreased by >10-fold, from ~1mM to <0.1mM. The evolved variants carried 10–14 mutations, of which approximately half came from design optimizations, and half from random mutagenesis (Table 2). Overall, the evolved variants differed from the natural enzyme that was used as a template (1JCL) by ≥19 positions. The catalytic machinery remained the same during the optimization process: mutating the catalytic His17 in the evolved variants from Round 4 and Round 6 decreased the activity >1000-fold, and mutating Asp45 caused ~5-fold decrease in activity (Supplementary Table 4).
Structural analysis of KE70 variants
Structures of the KE70 design and of six evolved mutants (R2 2/7E, R2 3/7G, R2 7/12F, R4 8/5A, R5 7/4A, and R6 6/10A) were solved at resolutions of 1.4–2.25Å. These structures provide insights into the mechanism and evolution of KE70. However, despite extensive efforts we could not obtain meaningful structures of KE70 variants with substrate or transition state analogues. Benzimidazole ligands did not exhibit any inhibition. The product of the reaction (5-nitro-2-cyanophenol) was used for co-crystallization and soaking experiments, but no electron density corresponding to it was observed. Benzotriazoles did inhibit some of the evolved variants (with Ki values in the range of 0.05–60 μM), and a structure of the KE70 variant R6 6/10A with 5-nitrobenzotriazole (Ki = 0.43±0.05 μM) was obtained at resolution of 2.2Å. The ligand was found to be located in the active site, and is aligned against the stacking Phe48. The key active site residues, such as the catalytic His17-Asp45 dyad and the stacking Phe48, did not change their conformations (Supplementary Fig. 2a), nor did other active site residues with the exception of Ser101 that binds the nitro group of the benzotriazole (Supplementary Fig. 2b). The nitro group of the ligand, however, is pointing towards His17, whereas the triazole nitrogens face the top of the active site (Supplementary Fig. 2a). Since both His17 and benzotriazole are protonated at the pH of crystallization (pH 4), this binding mode is somewhat expected and catalytically irrelevant. However, attempts to crystallize this complex at pH>8, namely with His17 in the catalytic deprotonated form, gave only poorly diffracting crystals.
The structure of KE70 variant R4 8/5A contained additional electron density in the active site, which was modeled as benzamidine (Supplementary Fig. 3a, b). However, benzamidine showed no inhibition of the eliminase activity (≤5mM benzamidine), and no binding was observed in isothermal calorimetry measurements (≤10mM). No inhibition of eliminase activity was observed also with molecules similar to benzamidine, such as benzoic acid, nitrobenzene and benzamide. In any case, as is the case with 5-nitrobenzotriazole, the presence of this unknown ligand did not cause any significant conformational changes in the active site. (Supplementary Fig. 3c).
Two structures of the original KE70 design were obtained, one with resolution of 2.25Å, and the other with resolution of 1.4Å. Both structures of KE70 design are overall similar to the computed model (Supplementary Table 5, Supplementary Fig. 4). However, in the structure of the KE70 design with 1.4Å resolution, in one of the monomers of the asymmetric unit (monomer A), there was a shift of one β-strand and of a helix adjacent to it (residues 229–238). The side chains of the residues on the shifted β-strand that point into the active site tunnel in the designed model were relocated into the protein core in the monomer A, and vice versa. In particular, the side-chain of Phe235 in the KE70 design structure is pointing into the active site, and may thus interfere with substrate binding (Fig. 3a, Supplementary Fig. 5). In the KE70 design structure with 2.25Å resolution, both monomers had a shifted β-strand. Interestingly, in the structures of the evolved variants, all the β-strands residues align perfectly with those in the KE70 model, and the side-chain of Phe235 is pointing into the protein core as in the computed model (Fig. 3b). Since the original design is active, the conformation observed in its crystal structure could be an artifact of crystallization, even though the shifted β-strand is not on the protein surface. In any event, mutations incorporated in the early rounds of directed evolution seem to have eliminated the shifted and presumably non-active conformation.
Fig. 3.
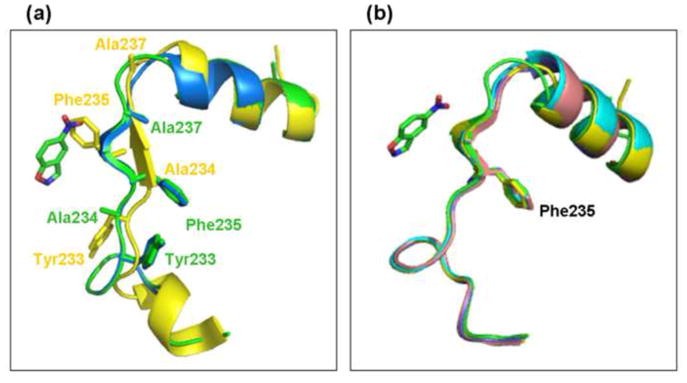
a. Strand register shift involving residues 229–238 in the crystal structure of KE70 design monomer A (yellow) relative to monomer B (blue) and to the KE70 design model (green). Shown is the region of residues 225–251 with key residues as sticks and the substrate in green. b. In the structures of the evolved KE70 variants, the region of the residues 229–238 aligns well with the KE70 model (green, KE70 model; yellow, R2 7/12F; cyan, R4 8/5A; pink, R5 7/4A; blue, R6 6/10A).
The largest deviations between the various structures are in the loops at the top of the barrel. This structural flexibility may explain why many insertions in the loops were tolerated. One of the loops (residues 21–27), is disordered and electron density is observed only in one of the structures (variant R2 7/12F). Some loop movements appear to cause shifts in the positions of certain helices, but the β-strands overlap in all structures (with the exception of the strand noted in Fig. 3). The position of the catalytic His17-Asp45 dyad is essentially the same in all structures, and the distances between His17 and Asp45 (3–4Å) vary by less than 1Å. However, as indicated by the MD simulations discussed below, these subtle changes may relate to the improved rates. Although the catalytic dyad remained largely unchanged, the shape of the active-site cavity changed significantly. Indeed, the active site tunnel of variant R6 6/10A, bearing two insertions in the loops, and a modified combination of amino acids at the bottom of the active site, is much deeper than in the original design. This is primarily caused by the mutation of Trp72 into the much smaller cysteine, which opened the central tunnel all the way to the other side of the protein (Fig. 4). As a result of the designed modifications at the bottom of the active site, the substrate is more tightly and firmly packed in the evolved variants, as indicated by the lower KM values and by the MD simulations discussed below.
Fig. 4.
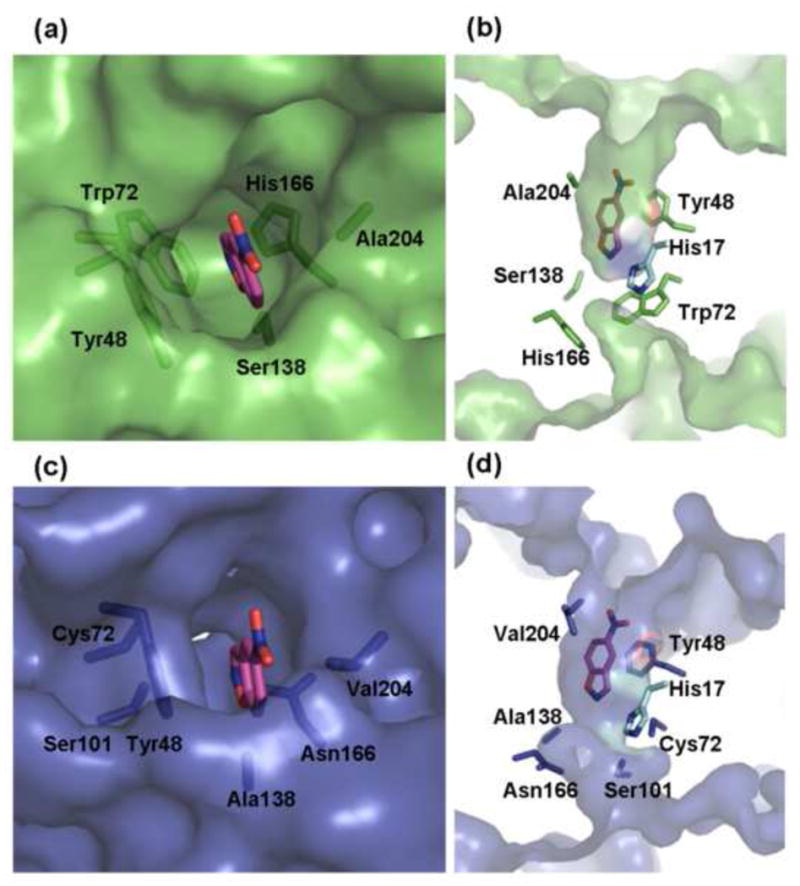
The active site cavity of the KE70 designed model ((a) - top view, (b) - crossection view), and of the evolved variant R6 6/10A ((c) - top view, (d) – crossection view). The substrate (magenta) was overlayed from the designed model. The active site cavity of variant R6 6/10A is deeper than that of the original KE70 design, primarily due to the mutation Trp72Cys at the bottom of the active site. The central tunnel is also opened all the way to the other side of the protein. In addition, the entrance to the active site is narrower in the evolved variant, primarily due to mutation Ala204Val.
Although the designed changes related to the catalytic dyad were not incorporated (design categories #2 and #3), there are significant changes in the residues around the His17-Asp45 dyad, and in the associated hydrogen bond networks. In the design model, Arg69 points away from the catalytic dyad, and the distance between Arg69 and Asp45 is 7.1Å (Fig. 5a). However, in the crystal structure of the KE70 design, Arg69 points towards the catalytic dyad, and the Arg69-Asp45 distance (2.8–3.1Å) corresponds to a salt bridge (Fig. 5b). The Arg69-His17 distance was also reduced from 10.5Å in the KE70 model to 3.1–4.0Å in the crystal structure. The proximity of Arg69 may lower the basicity of the dyad and therefore the catalytic potential of both Asp45 and His17. In the Round 2 variants, Arg69 and Asp45 are still within salt bridge formation, but Arg69 is detached from the catalytic His17 (>5.9Å). Interestingly, in the Round 2 7/12F variant, Arg69 adopts different conformations in the two monomers of the asymmetric unit. One conformer is similar to the computed model, and the other is similar to the crystal structure of the designed KE70 (Fig. 5c). In the Round 6 variant 6/10A, the mutation Gly101Ser caused Arg69 to move away from the catalytic dyad, and the distance between Arg69 and Asp45 increased to 6.2Å, probably rendering the His17-Asp45 dyad more basic and therefore more active (Fig. 5d).
Fig. 5.
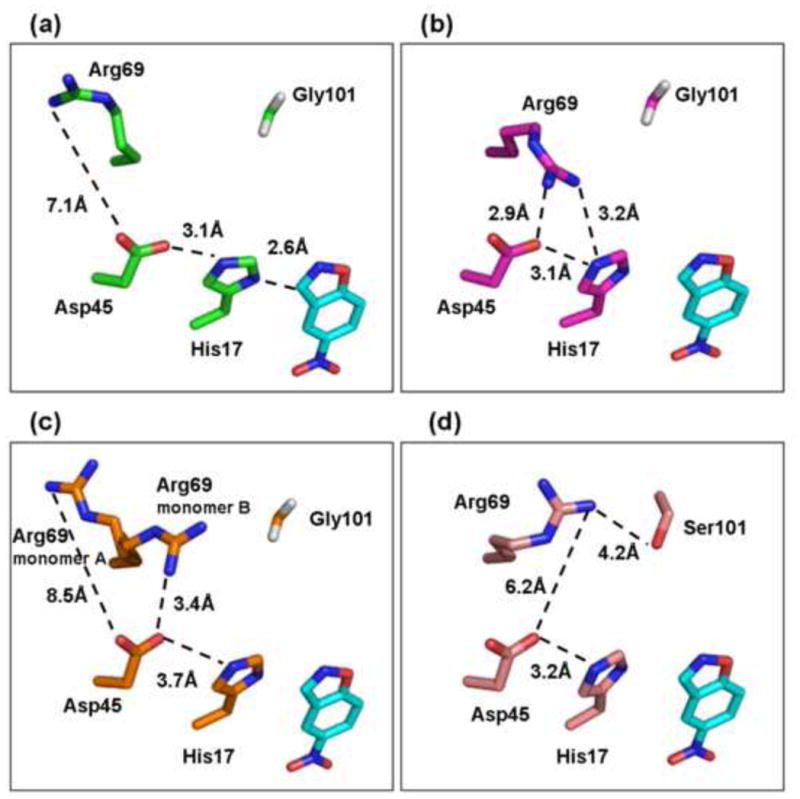
Refinement of the environment of the catalytic dyad. a. In the designed KE70 model, Arg69 is pointing away from the catalytic His17-Asp45 dyad. b. In the crystal structure of the KE70 design, Arg69 is pointing towards the catalytic dyad, and is within hydrogen-bond distance to both Asp45 and His17, thus reducing the basicity of His17. c. In the structure of the evolved variant from Round 2, 7/12F, two different conformations of Arg69 are observed in the two molecules in the asymmetric unit. One rotamer resembles the computed model (as in a), and the other aligns with Arg69 from the crystal structure of the KE70 design (as in b). d. In the evolved variant Round 6 6/10A, the mutation of Gly101 to serine appears to stabilize the rotamer of Arg69 that points away from the catalytic dyad. The substrate (cyan) was overlayed from the designed model.
pH-rate profiles of KE70 variants
The environment of the catalytic dyad was also probed by determining the pH-rate profiles of KE70 design and its evolved variants. The pH-rate profile of the KE70 design is bell-shape-like, although at pH>7.5 the rates deviate from a simple bell-shaped fit (Fig. 6). The basic pKa(kcat) of KE70 design is 6.3, and the basic pKa(kcat/KM) is much lower (5.3, Fig. 6, Table 2). The decrease in activity at higher pH suggests that in addition to His17, which is active in the basic form, there is another residue(s) with an acidic pKa in the range of 6–7 (pKa(kcat), −6.6, and pKa(kcat/KM), −7.4), whose deprotonation reduces the activity of the KE70 design. The origins of the decrease at higher pH is unclear, since both the catalytic residues His17 and Asp45 are supposed to be active in their deprotonated forms, but may be related to the strand register shift observed in the crystal structure of the original design (Fig. 3a). This is consistent with the absence of the acid shoulder in the pH-rate profiles of the evolved variants, in which the strand is correctly placed (Fig. 3b). The pH-rate profiles of the evolved variants, R2 7/12F, R4 8/5A, and R6 6/10A exhibit only one, basic shoulder, and could be therefore compared (Fig. 6). Consistently with their higher activities, the pKa(kcat) values of the evolved KE70 variants, indicating the pKa of the enzyme-substrate complexes, increased with the progress of directed evolution from 5.5 to 6.2 (Figure 6, Table 2). In contrast, the pKa(kcat/KM) values of the evolved KE70 variants, indicating the pKa of the free enzyme, are similar, and range between 5.9 and 6.2. The elevated pKa(kcat/KM) of variant R2 7/12F (6.2) is primarily due to a significant increase of the KM values with a decrease of pH below 6.5. This pattern suggests that changes in the environment of the catalytic dyad are primarily manifested at the level of the enzyme-substrate complex (changes in pKa(kcat) values), rather than the free enzyme (pKa(kcat/KM) values).
Fig. 6.
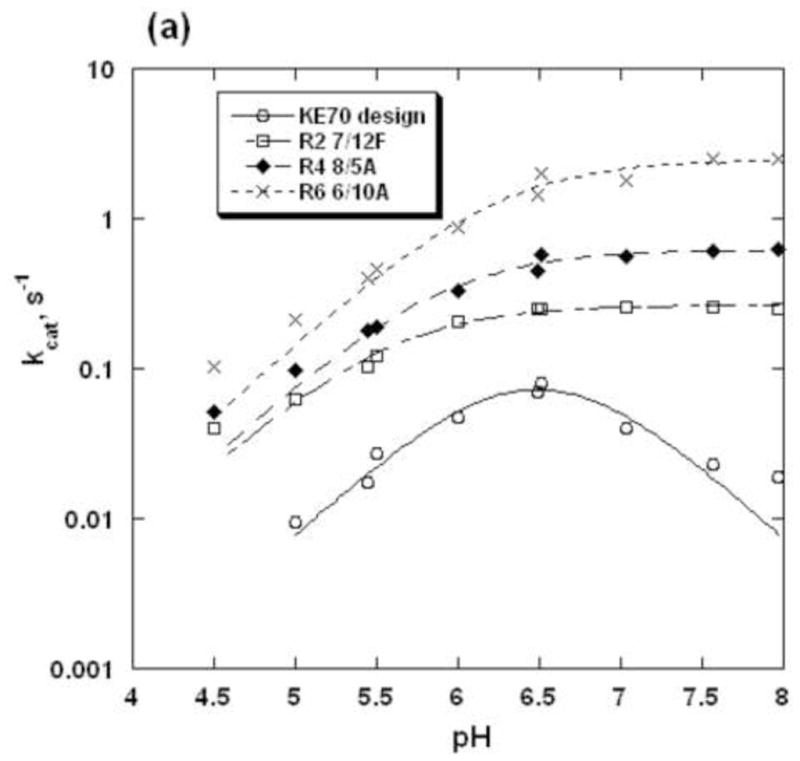
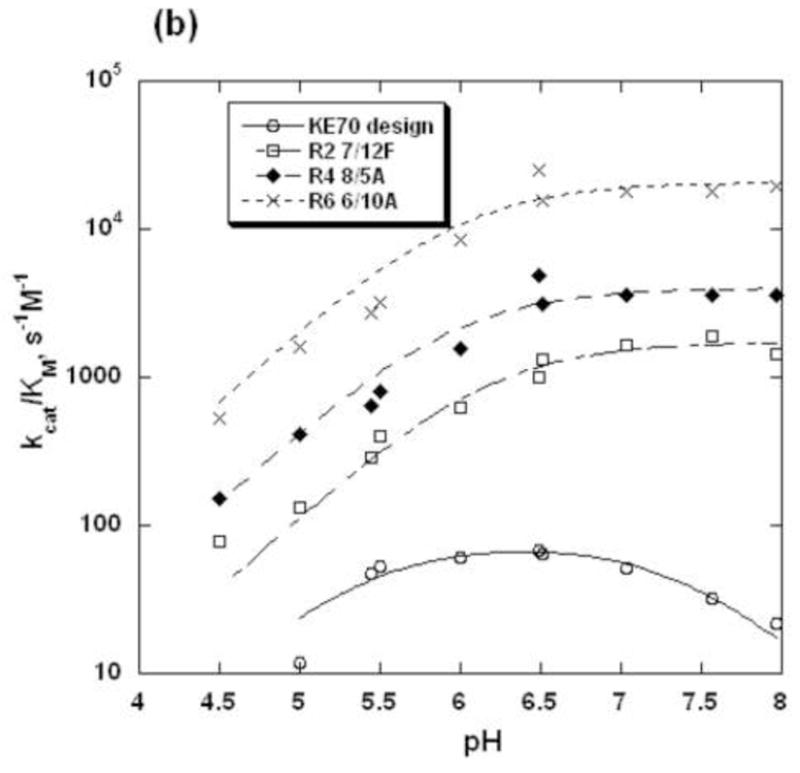
pH-rate profiles of the KE70 variants. a. kcat versus pH; b. kcat/KM versus pH. The resulting parameters are given in Table 2.
The increase in the pKa(kcat) values of the catalytic His17 during evolution (~0.7 units) is consistent with the changes in hydrogen-bonding of adjacent residues, and in particular with the repositioning of Arg69 (Fig. 5). These changes partly account for the increase in the kcat value of the evolved KE70 variants (>30-fold). However, as the catalytic efficiency (kcat/KM) has increased much more (>400-fold), other factors, such as optimization of substrate binding in the active site, and modification of the active site loops, also contributed the increase of catalytic power.
Molecular dynamics simulations
MD simulations were performed on the structures of the KE70 design and its evolved variants. We utilized a protocol that was previously established for the evaluation of general acid-base biocatalysts 23. Polar contacts, distances between the residues of catalytic dyad, distances between the catalytic residues and the corresponding atoms in the TS, root mean square displacements (RMSDs), and the atomic fluctuations (equivalent to b-factors) of the active site residues were monitored. For the purpose of the MD simulations, the active site of each structure was defined to consist of the residues His17, Ala19, Asp45, Tyr/Phe48, Trp/Cys72, Ala103, Ser/Ala138, Ile140, Val168, Ser170, and Ile202.
The MD-derived backbone RMSDs decreased as KE70 was evolved (Fig. 7a). Compared to the original KE70 design, the average atomic fluctuations of the active site and also their spread (as denoted by the standard deviation bars) appear lower in all the variants beyond the second round of directed evolution (Fig. 7b). This trend implies that key active site residues (His17, Ala19, and Asp45) that, in the original design, were more flexible than other active-site residues, rigidified as the design evolved (Supplementary Fig. 6, Supplementary Table 7). RMSD values quantify the degree to which the simulated structures deviate from a given reference (here the crystal structure), while the atomic fluctuations quantify the degree of flexibility (i.e., the variability between the runs of the same variant). As such, Fig. 7a–b and Supplementary Fig. 6 translate into the observation that each round of evolution rigidified the active site, giving rise to an increased level of pre-organization.
Fig. 7.
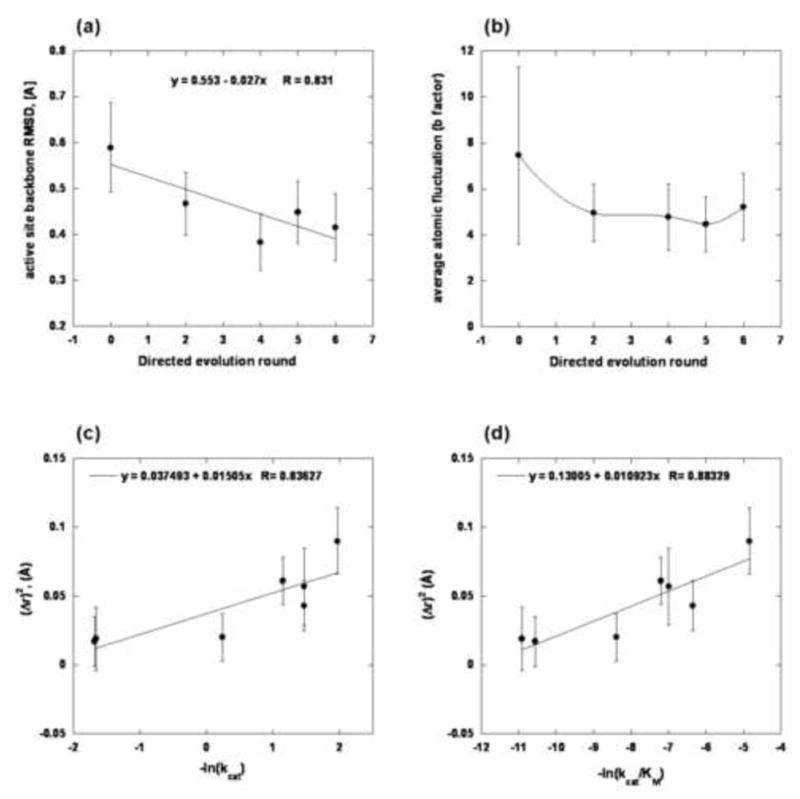
a. Active site backbone RMSD. The full width at half maximum (FWHM) of each RMSD distribution is represented by the individual error bars. b. Active site backbone b-factor versus the round of directed evolution. The error bars depict the spread (standard deviation) in atomic fluctuations across all active site residues. c. The square of the deviation (Δr)2 from the ideal hydrogen bond distance (1.8 Å) versus -ln(kcat). d. The square of the deviation (Δr)2 from the ideal hydrogen bond distance (1.8 Å) versus -ln(kcat/kM).
Fig. 7c and 7d plot (Δr)2 vs. -ln(kcat) and -ln(kcat/kM). Δr denotes the deviation of the measured His17-Asp45 distance (His-NεH – Oδ-Asp) from the ideal catalytic hydrogen bond distance (1.8Å)23. Assuming a simple harmonic model, the energetic cost of deviation from ideality is proportional to (Δr)2, and assuming a simple transition state model, ln(kcat) is proportional to the activation free energy. The linear relationship between (Δr)2 and ln(kcat) and ln(kcat/kM) suggests that the increased kcat of evolved variants results in part from tightening of the hydrogen bond between the catalytic dyad members, as the active site residues of the KE70 variants become more optimally placed and less mobile.
The distance between the catalytic base (His17) and the substrate proton which it abstracts, remained unchanged during the process of KE70 optimization, in the range of 2.6–2.7Å (Supplementary Table 6). This suggests that the distance between His17 and the subtracted proton in the KE70 design need not be further optimized.
Discussion
Our previous report described the optimization of the computationally designed Kemp eliminase KE07 by directed evolution 16. There are several significant differences between the KE70 and KE07 designs. Unlike the carboxylate catalytic base in the KE07 and other KE designs, in KE70, proton abstraction is mediated by a His-Asp dyad. The catalytic dyad feature is very abundant in enzymes, as it allows efficient positioning and activation of the His by the aspartic or glutamic acid residue 24. However, the catalytic dyad is also computationally challenging, as both the His and Asp residues, and the interaction between them have to be optimized. In addition, the pKa of histidine and its tautomeric state have to be regulated. The KE07 and KE70 designs also varied in their response to optimization by directed evolution. The KE07 design represented a deep energetic minimum. The design features and the designed residues remained essentially unchanged, and only underwent fine-tuning. Only one of the designed residues was mutated, and even this residue is a second-shell residue that does not directly interact with the substrate. The KE70 design turned out to be both more “evolvable” and re-designable. Several designed residues were spontaneously altered in the first rounds of directed evolution, including the substrate stacking residue (mutation Tyr48Phe). This change suggested that the designed active site could be computationally optimized, as proved to be the case.
Despite the significant changes in the optimized KE70 design, the catalytic dyad His17-Asp45 remained unmodified. Similarly, in the divergent evolution of families and superfamilies of enzymes, the basic mechanistic features and key active site residues remain unchanged, while the other parts of the active site, are modified to fit specific substrates and reactions 20. These results also highlight the strengths and weaknesses of current computational enzyme design methodology—the general outlines of the active site can be created in broad strokes, but the fine tuning of the active site electrostatics is very difficult to achieve only by computation.
Unlike the case of KE07, optimization of KE70 involved extensive computational design optimizations, rationally designed mutations, and directed evolution that combined the designed mutations with the random ones. In the case of KE07, it was shown that directed evolution, which requires no previous knowledge of structure-function, could be successfully applied to improve computationally designed enzymes, and can therefore bridge some of the gap created by our limited design skills 16. However, the bottleneck of directed evolution is the very limited sequence space that can be explored by error-prone mutagenesis (single-nucleotide changes, with strong biases against certain mutations), and by the medium-throughput screening. The screening throughput in 96-well plates using the colorimetric 5-nitrobenzisoxazole substrate is relatively low, as only ~1000 variants were screened per round. Structure-based computational design was therefore used not only to produce the starting point enzyme, but also to optimize it. Various combinations of mutations identified by the Rosetta algorithm were introduced into the evolved KE70 variants in a combinatorial manner. The incorporation of backbone flexibility yielded improved solutions compared to the original fixed backbone design, but only in a subset of cases—varying the backbone has associated perils (the protein may not fold, or the backbone shift may not be brought about by the sequence changes). To minimize the incorporation of mutations that are not beneficial, or even deleterious, the designed mutations were first experimentally validated. Those proven advantageous or neutral were then incorporated into the evolved KE70 variants in a combinatorial manner 22. This strategy allowed sampling the various combinations of mutations, while maintaining library sizes that are compatible with the screening capacity. Notably, many of the designed mutations involved changes in more than one nucleotide within the same codon (e.g. Ala21Asn/Gln/Arg, Ala238Met, and Ser239His) which are very unlikely to occur upon random mutagenesis. Likewise, insertions are not a likely outcome of error-prone mutagenesis.
Whilst several of the computationally designed mutations have been incorporated into the evolving KE70 variants, it remains unclear which design strategy may yield the best designs. The procedures applied here followed a ‘trail and error’ approach, rather than a systematic exploration of various approached. Nonetheless, the approach of including computationally designed mutations in a ‘spiking’ manner enabled us to explore a relatively large number of possibilities, and ‘hedge the bets’ by including mutations that may actually reduce rather than increase activity.
Changes in the designed residues of KE70, complemented by random mutations introduced by directed evolution, led to >400-fold improvement of KE70’s catalytic efficiency. Substrate binding was improved (up to 12-fold decrease in KM), as well as the turnover number (up to 53-fold increase in kcat). The most efficient evolved KE70 variants exhibit kcat/KM values of ~5x104 s−1M−1, which brings them closer to natural enzymes. Nonetheless, in view of the high reactivity of 5-nitrobenzisoxazole, it is clear that the catalytic powers of the evolved KE70s are far below those of natural enzymes. The kcat/kuncat values of the evolved KE70 variants are ~5x106, which compares very favorably with the other enzyme mimics 25. For comparison, the kcat/KM of the evolved KE7 is 2.6x103 s−1M−1, and the kcat/KM of the catalytic antibody 34E4, which also catalyzed Kemp elimination of 5-nitrobenzisoxazole, is 5.45x103 s−1M−1, and its kcat/kuncat is 2.1x104 10.
The improvement in the catalytic activity of the KE70 variants has three main origins:
(1) Modification of the active-site electrostatics
Whilst the His17-Asp45 dyad remained unchanged, the electrostatics surrounding it were modified, in particular the unfavorable bonds between Arg69 and the catalytic dyad (especially Asp45), that likely quenches the dyad’s basicity, were broken (Fig. 5). Modification of the electrostatic environment was also a key feature in the optimization of the KE07 design, where an unfavorable bond between the catalytic base (Glu101) and Lys222 was broken, thus leading to higher basicity and activity of the catalytic base 16. While electrostatic effects play a crucial role in the catalysis of Kemp elimination (and perhaps most other reactions) 26, they are not well modeled by the computational design methodology. The different rotamers adopted by Arg69 (Fig. 5c) exemplify the difficulty in modeling of the charged residues at protein surface. The different rotamers seem to be very close in energy, as indicated by their co-existence in the two molecules within the same asymmetric unit (Fig. 5c). A similar situation was observed in the analysis of the KE07 design and its evolved variants 16. Improvements in computation of electrostatic interactions, which enable prediction of the energetics of hydrogen bonding networks would clearly contribute to the improvement of computational design methodology.
(2) Reshaping the active site cavity
The active site cavity of the designed KE70 was completely reshaped, resulting in a deeper pocket with a narrower entrance (Fig. 4). This led to improved substrate binding (indicated by lower KM values).
(3) Mobility of the active site residues
The MD simulations suggest that the optimization of KE70 design led to increased pre-organization of the catalytic dyad and restricted mobility of most active site residues, and particularly of residues His17 and Asp45, in a way that could enhance the catalytic activity. The simulations further suggest that the increased kcat and kcat/KM of the evolved variants correlate with the formation of a more favorable geometry in the catalytic His-Asp dyad as a shorter hydrogen bond between these residues is achieved. This is in line with the primal requirement emphasized early on by Jencks 27, that the catalytic residues and the substrate need to be precisely and tightly positioned, to optimize the interactions essential for catalysis 28.
In summary, computational and evolutionary optimization methods were combined to yield improved Kemp eliminases. Computational algorithms can both generate the initial designs, which can then serve as starting points for directed evolution, and assist in the optimization of these designs by identifying beneficial amino acid substitutions for testing29. Further improvements in the computational methods may include better modeling of the electrostatic networks between charged residues, including additional parameters in the calculations (e.g. pKa of the catalytic base), and MD simulations of the top-ranked designs to assess the relevant parameters, such as bond lengths and residue fluctuations. Future work will be aimed at generating de novo designed and optimized catalysts not only for model reactions with activated substrates, such as 5-nitrobenzisoxazole, but also for more challenging reactions of the type catalyzed by the natural enzymes.
Materials and methods
Cloning
The synthetic gene encoding the designed KE70 protein was purchased from Codon Devices, Inc., and the gene was cloned into his-tag expression vector pET29b (Novagen) using NdeI and XhoI restriction sites. In the libraries, the NdeI site was replaced by NcoI, which involved the addition of an alanine residue at the N-terminus, just after the initial methionine.
Backrub protocol
TS placement was carried out as previously described 19 on the rigid backbone scaffold (PDB code 1JCL) using the RosettaMatch algorithm. In addition to the previously described theozymes used, theozymes lacking a hydrogen-bonding group to the phenoxide’s oxygen (Ser, Tyr, or Lys in previous theozymes) were allowed. For each of the matches a gradient-based minimization was used to optimize the catalytic geometrical constraints. Residues surrounding the TS were then redesigned to maximize the stability of the active site conformation. Five hundred of the lowest-scoring designed variants were then subjected to a backrub simulation, as previously described 18 to generate 5 variants for each starting design. In general, the level of observed divergence of the loop regions was small (Supplementary Table 1), but the resulting subtle changes in Cα-Cβ bond vectors changed the allowed spectrum of designed residues. Each of the backrub variants was then redesigned as before, using both the standard Rosetta vdw function and a soft vdw function for a total of 10 redesigns per starting model. All models (flexible and fixed-backboned) were subjected to both energetic and geometric filters (cavities, exposed surface area, number of hydrogen bonds). The mutations of the 8 variants that were finally selected for expression are listed in Supplementary Table 2.
Library making and screening
Several mutagenesis methods were used to create genetic diversity in KE70 genes. Error-prone PCR with mutazyme (Genemorph PCR mutagenesis kit, Stratagene) was used for random mutagenesis, and at certain rounds (Table 1), the genes were shuffled as previously described 16. Selected positions were mutated by incorporation of spiking oligonucleotides during the assembly PCR as described 22. After mutagenesis and/or shuffling, the KE70 genes were re-cloned into the original pET29b plasmid, and the ligated DNA was transformed into E. coli DH5α cells. Typically, 104–105 tranformants were obtained, and the plasmids encoding the libraries were extracted. It should be noted that random mutagenesis and gene shuffling are not exclusive, that is, during random mutagenesis, the genes are shuffled, and the shuffling procedure incorporated a certain level of random mutations created by polymerase errors (~1 mutation per gene, on average). The libraries were screened with 5-nitrobenzisoxazole as previously described 16.
Enzymatic characterization
Variants subjected to detailed analysis were produced and purified as described 16. The enzymes were stored at 4°C in storage buffer (25 mM HEPES pH 7.25 with 100 mM NaCl, 5% glycerol, and 0.02% sodium azide). For the kinetic characterization, the reactions were started by adding 150 μl of substrate (final concentration − 0.13–1.05 mM, or 0.065–0.525mM, in 25 mM HEPES pH 7.25 with 100 mM NaCl), to 50μl of enzyme in storage buffer (various concentrations were used for different variants). 5-nitrobenzisoxazole was used from 0.1M stock in acetonitrile. The co-solvent percentage was 1.5 %, and glycerol percentage was 1.25% in all the reaction mixtures. Storage buffer without protein was used for the background reaction. Product formation was monitored spectrophotometrically at 380 nm (Power HT microtiter scanning spectrophotometer), in 200 μl reaction volumes, using 96-well plates. The reported results are the average of at least three independent measurements, and the error ranges between the repeated experiments were <20%.
pH-rate profiles
kcat and KM values were determined with 5-nitrobenzisoxazole at pH 4.5–8.0 for KE70 variants Round 2 7/12F, Round 4 8/5A, and Round 6 6/10A, and at pH 5.0–8.0 for the KE70 design. Initial velocities (v0) were determined at eight different substrate concentrations (0.13–1.05 mM). The buffers used (at 50 mM) were: Citrate (pH 5.0–5.5), MES (pH 5.5–6.5), and Bis-tris propane (pH 6.5–8.0) at 50 mM. At pH 5.5 and 6.5, the activity was measured with both relevant buffers to eliminate the buffer effects. Measurements at pH>8 were precluded due to substrate decomposition and the low activity of the KE70 design. The ionic strength was adjusted to a total of 0.1M with NaCl. The enzyme stocks were kept in 10 mM HEPES pH 7.25 containing 100 mM NaCl and 5 % glycerol.
Data analysis
Kinetic parameters (kcat, KM, kcat/KM) were obtained by fitting the data to the Michaelis-Menten equation [v0=kcat[E]0[S]0/([S]0+KM)], using the program Kaleidagraph 5.0. When working with low substrate concentrations, data were fitted to the linear regime of the Michaelis-Menten model [v0= [S]0[E]0kcat/KM], and kcat/KM was deduced from the slope. All the data presented are the averages of at least three independent experiments with standard deviations. The pH-rate data (kcat and kcat/KM values for each pH value ((kcat)H and (kcat/KM)H) were fitted using the equations (kcat)H=(kcat)maxx10−pKa/(10−pH+10−pKa) and (kcat/KM)H=( kcat/KM)maxx10−pKa/(10−pH+10−pKa); where (kcat)max and (kcat/KM)max are the plateau values of kcat and kcat/KM, and pKa is the apparent pKa value for the acidic group. The pH-rate data of the KE70 design were fitted to a “bell-shaped” model using the equations (kcat)H=(kcat)max/[(10−pH/10−pKa1)+(10−pKa2/10−pH)+1] and (kcat/KM)H=(kcat/KM)max/[(10−pH/10−pKa1)+(10−pKa2/10−pH)+1]; where (kcat)max and (kcat/KM)max are the plateau values of kcat and kcat/KM, and pKa1 and pKa2 are the apparent pKa values for the acidic and the basic groups, respectively.
MD simulations
The crystal structures of KE70 and its evolved variants R2 2/7E, R2 3/5G, R2 7/12F, R4 8/5A, R5 7/4A, and R6 6/10A were used as starting geometries. Co-crystallized water molecules were removed. The substrate was docked into each active site with AutoDock4 30. Substrate parameters were generated within the antechamber module of AMBER 10 31 using the general AMBER force field, with partial charges set to fit the electrostatic potential generated at HF/6-31G* by RESP 32. The charges were calculated according to the Merz-Singh-Kollman scheme 33; 34 using Gaussian 03 35. The electron density of the D-loop could not be resolved for all crystal structures. Here, the geometry of the KE70wt chain-B D-loop (Ala18 through Thr25) was used and grafted onto affected structures prior to MD.
The structures were immersed in a truncated octahedral box with a 10 Å buffer of TIP3P water molecules 36. The systems were neutralized by addition of explicit counter ions. All subsequent calculations were done using the widely tested Stony Brook modification of the Amber 99 force field 37. A two stage geometry optimization approach was utilized, initially minimizing the positions of water molecules and ions, followed by an unrestrained minimization of all atoms. The systems were heated gently in six 50K increments, using 50ps steps from 0 to 300K at constant volume periodic boundary conditions. Harmonic restraints of 30 kcal/mol were applied to the solute, and the Langevin equilibration scheme was used to control and equalize the temperature. The time step was kept at 1 fs during the heating stages, allowing potential inhomogeneities to self-adjust. Each system was then equilibrated for 2 ns with a 2 fs timestep at a constant pressure of 1 atm. Water molecules were triangulated with the SHAKE algorithm such that the angle between the hydrogen atoms is kept fixed. A 20 ns production MD simulation was performed for each of the systems (with and without the substrate bound to the active site) using pmemd 38. Geometries and velocities were saved every 100 steps (0.2 ps) which resulted in a total of 100,000 frames from each production run. Long-range electrostatic effects were modeled using the particle-mesh-Ewald method 39. Post-MD data-extraction and analysis was performed using the ptraj module of AMBER 10 and the statistical analysis software OriginPro8 40.
Crystallization, data collection, and refinement
For crystallization, the designed KE70 and its evolved variants were purified by Ni-NTA HiTrap chelating HP column (Amersham), followed by gel filtration (HiLoad 16/60 Superdex™, Amersham).
Some of the crystals of KE70 evolved variants and the design were obtained by the microbatch method under oil by using the Oryx6 robot (Douglas Instruments Ltd., East Garston, Hungerford, Berkshire, U.K.) and others by the sitting drop vapor diffusion method using the Mosquito robot (TTP LabTech Inc, Cambridge, MA, USA) (Table 3). The protein concentration used for crystallization of all variants was 20–25 mg/ml. Diffraction data were integrated, scaled, and reduced using the HKL2000 program package 41. The structure of R2 2/7E KE70 variant was solved by molecular replacement using the program PHASER 42, by using the refined structure of deoxyribose phosphate aldolase of E. Coli (PDB accession code 1JCL) as a model. The structures of the remaining KE70 variants were solved using the R2 2/7E KE70 variant structure as a model. All the steps of atomic refinement were carried out with the program CCP4/Refmac5 43. The models were built to σa-weighted, 2Fobs – Fcalc, and Fobs – Fcalc maps using the program COOT 44. The coordinates of all variants have been deposited to the Protein Data Bank and their accession codes are listed in Table 3. The KE70 models of the design and variants were evaluated with the program PROCHECK 45.
Table 3.
Summary of crystallization, data collection and refinement of the KE70 variants structures.
| Variant | KE70 design | KE70 design | KE70 R2 2/7E |
KE70 R2 7/12F |
KE70 R2 3/5G |
KE70 R4 8/5A |
KE70 R5 7/4A |
KE70 R6 6/10A |
KE70 R6 6/10A with 5-nitro benzisoxazole |
|---|---|---|---|---|---|---|---|---|---|
| Crystal parameters | |||||||||
| Crystallization conditions | 0.2M NaF 0.1M BisTris Propane pH=7.5 20% PEG 3350 |
0.2M NaF 0.1M BisTris Propane pH=7.5 20% PEG 3350 |
0.2M NH4Cl 0.1M MES pH=6 20% PEG 6000 |
0.1M NaCitrate pH=5.5 20% PEG 3000 |
0.1M PCB pH=5 20% PEG 1500 |
0.2M NH4NO3 20% PEG 3350 pH=6.3 |
0.2M (NH4)2SO4 0.2M NaAc pH=4.6 30% PEG MME 2000 3% Tri-methyl amine N-oxide |
0.2M MgCl2 0.1M NaAc pH=5 20% PEG 6000 |
0.1M NaMalonate, Imidazole and Boric acid pH=4 25% PEG 1500 |
| Crystallization method | sitting drop vapor diffusion | sitting drop vapor diffusion | sitting drop vapor diffusion | microbatch under oil | microbatch under oil | sitting vapor diffusion | microbatch under oil | sitting drop vapor diffusion | sitting drop vapor diffusion |
| Temperature (ºC) | 19 | 19 | 19 | 19 | 4 | 19 | 4 | 19 | 19 |
| Space Group | P21 | P21 | P212121 | P212121 | C2 | P43212 | C2 | P21 | P21 |
| Cell Dimensions | a=62.22 Å b=53.29 Å c=81.84 Å α=90.00º β=110.19º γ=90.00º |
a=62.59 Å b=53.51 Å c=81.07 Å α=90.00º β=110.34º γ=90.00º |
a=52.39 Å b=82.87 Å c=127.96 Å α=90.00º β=90.00º γ=90.00º |
a=52.13 Å b=83.33 Å c=129.20 Å α=90.00º β=90.00º γ=90.00º |
a=99.01 Å b=96.23 Å c=71.27 Å α=90.00º β=125.88º γ=90.00º |
a=52.82 Å b=52.82 Å c=153.81 Å α=90.00º β=90.00º γ=90.00º |
a=90.52 Å b=64.72 Å c=49.03 Å α=90.00º β=104.69º γ=90.00º |
a=49.10 Å b=41.22 Å c=139.44 Å α=90.00º β=94.96º γ=90.00º |
a=48.84 Å b=41.33 Å c=138.81 Å α=90.00º β=95.35º γ=90.00º |
| # of copies in the asymmetric unit | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 2 | 2 |
| PDB Accession Code | 3NPU | 3NPV | 3NPW | 3NPX | 3NQ2 | 3NQ8 | 3NQV | 3NRO | 3Q2D |
| Data Collection | |||||||||
| Data Collected | In-house source | ESRF beam ID14-4 | In-house source | ESRF beam ID14-4 | In-house source | ESRF beam ID14-4 | In-house source | In-house source | In-house source |
| Wavelength (λ) | 1.54178 | 0.9765 | 1.54178 | 0.975 | 1.54178 | 0.939 | 1.54178 | 1.54178 | 1.54178 |
| Resolution range (Å) | 50.00-2.25 | 50.00-1.48 | 50.00-2.15 | 50.00-1.80 | 50.00-2.02 | 50.00-1.40 | 50.00-1.70 | 50-2.20 | 50.00-2.20 |
| Last resolution shell (Å) | 2.33-2.25 | 1.51-1.48 | 2.23-2.15 | 1.86-1.80 | 2.09-2.02 | 1.45-1.42 | 1.73-1.70 | 2.24-2.20 | 2.24-2.20 |
| # of observations | 154,480 | 751,597 | 220,501 | 506,485 | 177,142 | 881,041 | 150,631 | 109,838 | 493,378 |
| # of unique reflections | 23,599 | 80,055 | 31,353 | 53,243 | 34,934 | 43,027 | 28,862 | 27,694 | 27,722 |
| Completeness (%)a | 97.8 (83.8) | 98.9 (98.6) | 99.9 (98.9) | 99.6 (99.1) | 97.9 (95.3) | 97.9(100.0) | 95.8 (92.4) | 95.4 (95.7) | 96.4 (79.9) |
| Redundancy | 6.5 (4.9) | 5.4 (5.3) | 7.0 (5.5) | 9.5 (8.4) | 5.1 (4.4) | 20.5 (23.1) | 5.2 (3.5) | 4.0 (4.0) | 4.6 (3.9) |
| <I>/<σ (I) >a | 22.3 (4.9) | 24.6 (6.6) | 24.2 (2.9) | 28.7 (5.8) | 19.6 (3.1) | 57.28 (12.5) | 14.5 (2.6) | 15.5 (3.0) | 15.1 (2.5) |
| Rmergeb on I (%)a | 8.1 (21.7) | 5.2 (24.2) | 8.1 (39.4) | 9.1 (31.0) | 7.0 (32.0) | 5.8 (24.3) | 13.1 (37.0) | 9.8 (49.6) | 9.3 (34.2) |
| Refinement and model statistics | |||||||||
| Total number of reflections | 22,380 | 78,893 | 29,720 | 50,467 | 33,171 | 40,785 | 27,408 | 26,268 | 26,309 |
| Number of reflections in the test set | 1,292 | 4,150 | 1,574 | 2,703 | 1,757 | 2,155 | 1,451 | 1,416 | 1,403 |
| Water molecules | 40 | 263 | 83 | 228 | 159 | 166 | 124 | 67 | 35 |
| Mean B value of protein (Å2) | 30.2 | 17.6 | 28.9 | 19.0 | 19.3 | 14.6 | 26.1 | 31.4 | 28.6 |
| Mean B value of water (Å2) | 24.2 | 25.8 | 25.9 | 24.6 | 19.3 | 22.7 | 30.7 | 26.7 | 20.0 |
| Rcryst=(%)c | 18.9 | 14.3 | 21.96 | 19.69 | 20.54 | 17.4 | 20.5 | 20.7 | 21.81 |
| Rfree=(%)d | 24.1 | 17.01 | 25.8 | 22.31 | 25.6 | 19.0 | 23.4 | 26.8 | 27.12 |
| Rmsd bond length (Å) | 0.020 | 0.020 | 0.019 | 0.011 | 0.010 | 0.007 | 0.012 | 0.026 | 0.026 |
| Rmsd bond angles (º) | 1.73 | 2.02 | 1.63 | 1.26 | 1.35 | 1.13 | 1.44 | 2.08 | 2.01 |
| Stereochemical parameters | |||||||||
| Ramachandran Plot: | |||||||||
| Residues in most favored regions (%) | 95.2 | 95.7 | 95.6 | 95.4 | 94.5 | 95.4 | 94.0 | 94.2 | 94.0 |
| Residues in additional allowed regions (%) | 4.1 | 3.9 | 4.0 | 3.6 | 5.1 | 4.2 | 5.6 | 5.3 | 5.5 |
| Residues in generously allowed regions (%) | 0.0 | 0.0 | 0.0 | 0.5 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Residues in disallowed regions (%) | 0.7 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
In prentices is last resolution shell.
Rmerge= Σ|I - <I>|/Σ I, where I denotes the observed intensities.
Rcryst= Σ||Fo|-|Fc||/Σ|Fo|, where Fo denotes the observed structure factor amplitude and Fc the structure factor calculated from the model.
Rfree is for 5% of randomly chosen reflections excluded from the refinement.
Supplementary Material
Acknowledgments
Financial support by the EU network BioModularH2, the Defense Advances Research Projects Agency (DARPA), the Research Grant from Meil de Botton Aynsley, and the Adams Fellowship (Israel Academy of Science) to O.K., are gratefully acknowledged. D.S.T. is the incumbent of the Nella and Leo Benoziyo Professorial Chair.
Abbreviations
- TS
transition state
- WT
wild-type
- PDB
protein data bank
Footnotes
Accession numbers.
Coordinates and structure factors for the structures of KE70 design and variants have been deposited in the Protein Data Bank with the following accession numbers: 3NPU (KE70 design at 2.25Å resolution), 3NPV (KE70 design at 1.4Å resolution), 3NPW (R2 2/7E), 3NPX (R2 7/12F), 3NQ2 (R2 3/5G), 3NQ8 (R4 8/5A), 3NQV (R5 7/4A), 3NR0 (R6 6/10A), and 3Q2D (R6 6/10A with 5-nitrobenzotriazole).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Das R, Baker D. Macromolecular modeling with Rosetta. Annu Rev Biochem. 2008;77:363–82. doi: 10.1146/annurev.biochem.77.062906.171838. [DOI] [PubMed] [Google Scholar]
- 2.Rohl CA, Strauss CE, Misura KM, Baker D. Protein structure prediction using Rosetta. Methods Enzymol. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- 3.Jiang L, Althoff EA, Clemente FR, Doyle L, Rothlisberger D, Zanghellini A, Gallaher JL, Betker JL, Tanaka F, Barbas CF, 3rd, Hilvert D, Houk KN, Stoddard BL, Baker D. De novo computational design of retro-aldol enzymes. Science. 2008;319:1387–91. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mandell DJ, Kortemme T. Computer-aided design of functional protein interactions. Nat Chem Biol. 2009;5:797–807. doi: 10.1038/nchembio.251. [DOI] [PubMed] [Google Scholar]
- 5.Baker D. An exciting but challenging road ahead for computational enzyme design. Protein Sci. 2010;19:1817–9. doi: 10.1002/pro.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rothlisberger D, Khersonsky O, Wollacott AM, Jiang L, DeChancie J, Betker J, Gallaher JL, Althoff EA, Zanghellini A, Dym O, Albeck S, Houk KN, Tawfik DS, Baker D. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453:190–5. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 7.Casey ML, Kemp DS, Paul KC, Cox DD. The physical organic chemistry of benzisoxazoles. I. The mechanism of the base-catalyzed decomposition of benzisoxazoles. J Org Chem. 1973;38:2294–2301. [Google Scholar]
- 8.Kemp DS, Cox DD, Paul KG. The physical organic chemistry of benzisoxazoles. IV. The origins and catalytic nature of the solvent rate acceleration for the decarboxylation of 3-carboxybenzisoxazoles. J Amer Chem Soc. 1975;97:7312–7318. [Google Scholar]
- 9.Hollfelder F, Kirby AJ, Tawfik DS. On the magnitude and specificity of medium effects in enzyme-like catalysts for proton transfer. J Org Chem. 2001;66:5866–74. doi: 10.1021/jo015723v. [DOI] [PubMed] [Google Scholar]
- 10.Thorn SN, Daniels RG, Auditor MT, Hilvert D. Large rate accelerations in antibody catalysis by strategic use of haptenic charge. Nature. 1995;373:228–30. doi: 10.1038/373228a0. [DOI] [PubMed] [Google Scholar]
- 11.Debler EW, Ito S, Seebeck FP, Heine A, Hilvert D, Wilson IA. Structural origins of efficient proton abstraction from carbon by a catalytic antibody. Proc Natl Acad Sci U S A. 2005;102:4984–9. doi: 10.1073/pnas.0409207102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hollfelder F, Tawfik D. Efficient catalysis of proton transfer by synzymes. J Amer Chem Soc. 1997;119:9578–9579. [Google Scholar]
- 13.Hollfelder F, Kirby AJ, Tawfik DS. Off-the-shelf proteins that rival tailor-made antibodies as catalysts. Nature. 1996;383:60–2. doi: 10.1038/383060a0. [DOI] [PubMed] [Google Scholar]
- 14.Hollfelder F, Kirby AJ, Tawfik DS, Kikuchi K, Hilvert D. Characterization of proton-transfer catalysis by serum albumins. J Amer Chem Soc. 2000;122:1022–1029. [Google Scholar]
- 15.Hu Y, Houk KN, Kikuchi K, Hotta K, Hilvert D. Nonspecific medium effects versus specific group positioning in the antibody and albumin catalysis of thebase-promoted ring-opening reactions of benzisoxazoles. J Amer Chem Soc. 2004;126:8197–8205. doi: 10.1021/ja0490727. [DOI] [PubMed] [Google Scholar]
- 16.Khersonsky O, Rothlisberger D, Dym O, Albeck S, Jackson CJ, Baker D, Tawfik DS. Evolutionary optimization of computationally designed enzymes: Kemp eliminases of the KE07 series. J Mol Biol. 2010;396:1025–42. doi: 10.1016/j.jmb.2009.12.031. [DOI] [PubMed] [Google Scholar]
- 17.Davis IW, Arendall WB, 3rd, Richardson DC, Richardson JS. The backrub motion: how protein backbone shrugs when a sidechain dances. Structure. 2006;14:265–74. doi: 10.1016/j.str.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 18.Smith CA, Kortemme T. Backrub-like backbone simulation recapitulates natural protein conformational variability and improves mutant side-chain prediction. J Mol Biol. 2008;380:742–56. doi: 10.1016/j.jmb.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zanghellini A, Jiang L, Wollacott AM, Cheng G, Meiler J, Althoff EA, Rothlisberger D, Baker D. New algorithms and an in silico benchmark for computational enzyme design. Protein Sci. 2006;15:2785–94. doi: 10.1110/ps.062353106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glasner ME, Gerlt JA, Babbitt PC. Evolution of enzyme superfamilies. Curr Opin Chem Biol. 2006;10:492–7. doi: 10.1016/j.cbpa.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 21.Murphy PM, Bolduc JM, Gallaher JL, Stoddard BL, Baker D. Alteration of enzyme specificity by computational loop remodeling and design. Proc Natl Acad Sci U S A. 2009;106:9215–20. doi: 10.1073/pnas.0811070106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herman A, Tawfik DS. Incorporating Synthetic Oligonucleotides via Gene Reassembly (ISOR): a versatile tool for generating targeted libraries. Protein Eng Des Sel. 2007;20:219–26. doi: 10.1093/protein/gzm014. [DOI] [PubMed] [Google Scholar]
- 23.Kiss GDR, Baker D, Houk KN. Protein Science in press. [Google Scholar]
- 24.Bartlett GJ, Porter CT, Borkakoti N, Thornton JM. Analysis of catalytic residues in enzyme active sites. J Mol Biol. 2002;324:105–21. doi: 10.1016/s0022-2836(02)01036-7. [DOI] [PubMed] [Google Scholar]
- 25.Kirby AJ. Enzyme mechanisms, models, and mimics. Angew Chem Int Ed Engl. 1996;35:707–724. [Google Scholar]
- 26.Roca M, Liu H, Messer B, Warshel A. On the relationship between thermal stability and catalytic power of enzymes. Biochemistry. 2007;46:15076–88. doi: 10.1021/bi701732a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jencks WP. Binding energy, specificity, and enzymic catalysis: the circe effect. Adv Enzymol Relat Areas Mol Biol. 1975;43:219–410. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- 28.Warshel A. Electrostatic origin of the catalytic power of enzymes and the role of preorganized active sites. J Biol Chem. 1998;273:27035–8. doi: 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]
- 29.Mena MA, Treynor TP, Mayo SL, Daugherty PS. Blue fluorescent proteins with enhanced brightness and photostability from a structurally targeted library. Nat Biotechnol. 2006;24:1569–71. doi: 10.1038/nbt1264. [DOI] [PubMed] [Google Scholar]
- 30.Huey R, Morris GM, Olson AJ, Goodsell DS. A semiempirical free energy force field with charge-based desolvation. J Comput Chem. 2007;28:1145–52. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 31.Case DA, Darden TA, Cheathem TE, et al. AMBER10. University of California; San Francisco: 2008. [Google Scholar]
- 32.Bayly CI, Cieplak P, Cornell WD, Kollman PA. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints For Determining Atom-Centered Charges: The RESP Model. J Phys Chem. 1993;97:10269–10280. [Google Scholar]
- 33.Besler BH, Merz KM, Kollman PA. Atomic Charges Derived from Semiempirical Methods. J Comput Chem. 1990;11:431–439. [Google Scholar]
- 34.Singh UC, Kollman PA. An Approach to Computing Electrostatic Charges for Molecules. J Comput Chem. 1984;5:129–145. [Google Scholar]
- 35.Frisch MJ, et al. Gaussian 03, Revision C.02. Gaussian, Inc; Wallingford CT: 2004. [Google Scholar]
- 36.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926. [Google Scholar]
- 37.Wang JM, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules. J Comput Chem. 2000;21:1049–1074. [Google Scholar]
- 38.Duke RE, Pedersen L. PMEMD. University of North Carolina; Chapel Hill: 2003. [Google Scholar]
- 39.Darden TA, York D, Pedersen L. Particle mesh Ewald an Nlog(n) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 40.OriginLab. Origin. Northampton, MA: [Google Scholar]
- 41.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 42.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr. 2004;60:432–8. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 43.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 44.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 45.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.