

Abstract
βarrestin (βarr)-1 and -2 (βarrs) are universal G protein-coupled receptor adapter proteins that negatively regulate cardiac β–adrenergic receptor (βAR) function via βAR desensitization & downregulation. In addition, they mediate G protein-independent βAR signaling, which might be beneficial, e.g. antiapoptotic, for the heart. However, the specific role(s) of each βarr isoform in cardiac βAR dysfunction, the molecular hallmark of chronic heart failure (HF), remain unknown. Furthermore, adrenal βarr1 exacerbates HF by chronically enhancing adrenal production, and hence circulating levels of aldosterone and catecholamines. Herein, we sought to delineate specific roles of βarr1 in post-myocardial infarction (MI) HF by testing the effects of βarr1 genetic deletion on normal and post-MI cardiac function and morphology. We studied βarr1 knockout (βarr1KO) mice alongside wild type (WT) controls under normal conditions and after surgical MI. Normal (sham-operated) βarr1KO’s display enhanced βAR-dependent contractility and post-MI βarr1KO’s enhanced overall cardiac function (and βAR-dependent contractility) compared to WT’s. Post-MI βarr1KO’s also show increased survival, and decreased cardiac infarct size, apoptosis, and adverse remodeling, as well as circulating catecholamines & aldosterone, compared to post-MI WT’s. The underlying mechanisms are on one hand improved cardiac βAR signaling and function, as evidenced by increased βAR density and pro-contractile signaling, via reduced cardiac βAR desensitization due to cardiac βarr1 absence, and on the other hand decreased production leading to lower circulating levels of catecholamines & aldosterone due to adrenal βarr1 absence. Thus, βarr1, via both cardiac and adrenal effects, is detrimental for cardiac structure and function and significantly exacerbates post-MI HF.
Keywords: βarrestin1, β–adrenergic receptor desensitization, knockout, aldosterone, catecholamine, post-myocardial infarction heart failure
Despite recent advances in its prevention and management, heart disease, such as hypertension and post-myocardial infarction (MI) heart failure (HF), remains the leading cause of death in the western world and new treatments are needed.1 The molecular hallmark in chronic HF is cardiac β-adrenergic receptor (βAR) dysfunction due to increased receptor desensitization and downregulation.2–6 βARs belong to the superfamily of G protein-coupled receptors (GPCRs), which, upon agonist binding, activate the Gs protein-adenylyl cyclase (AC)-cyclic adenosine monophosphate (cAMP)-protein kinase A (PKA) signaling pathway leading to increased inotropy and chronotropy in the heart.2 In chronic HF though, there is a selective downregulation of cardiac β1ARs and significant functional desensitization (G protein uncoupling) of the remaining membrane β1ARs and β2ARs, which dramatically diminish cardiac adrenergic and inotropic reserves.3–6 These processes are mediated at the molecular level by the βarrestins (βarrs), which exist in two distinct isoforms (βarr1 and -2, also known as arrestin-2 and -3, respectively) in mammals, and are abundantly expressed in the heart.7–9 βarrs bind agonist-activated cardiac βARs, terminating their G protein-mediated signaling and targeting them for internalization, following receptor phosphorylation by GPCR kinase (GRK)-2 (mainly), the main cardiac GRK that is significantly elevated in HF.2,8–16 Consistent with their crucial role in cardiac βAR desensitization, βarr1 knockout (KO) mice display enhanced cardiac contractile responses to isoproterenol (a standard βAR full agonist) stimulation in vivo.17
In addition, the βAR-bound βarrs have been shown to elicit a second wave of G protein-independent signals, some of which might be beneficial for the failing heart, e.g. extracellular signal-regulated kinase (ERK) activation and epidermal growth factor receptor (EGFR) transactivation, both of which can promote cardiac cell survival, proliferation, etc.18–21 However, the actions of the two βarrs in vivo are rarely complementary; in fact, they might even oppose each other’s effects in certain tissues/organs, including in the cardiovascular system (e.g. vascular smooth muscle cells and cardiomyocytes).22,23 With regard to the heart, the exact role of each βarr in cardiac βAR desensitization or G protein-independent signaling is not presently known. The physiological hallmark of chronic HF is the sustained hyperactivity of several neurohormonal systems, in particular the sympathetic nervous (SNS) and the renin-angiotensin-aldosterone (RAAS) systems, accompanied by elevated circulating levels of the catecholamines (CAs) norepinephrine (NE) and epinephrine (Epi), and of aldosterone, respectively.9,14,15 All these circulating hormones exert detrimental effects on the failing heart, leading both to diminished function and to accelerated and aggravated adverse remodeling in chronic HF.24,25 The main source of these circulating hormones is the adrenal gland and we have shown in previous studies that βarr1, in particular, is a crucial mediator of adrenocortical aldosterone production,26,27 as well as of adrenomedullary CA production (acting in concert with adrenal GRK2),28–30 both normally and in post-MI HF in vivo.
Given that the specific roles of cardiac βarr1 in βAR (dys)function and signaling in HF have never been investigated and also the crucial role this protein plays in the adrenals in elevating the neurohormonal burden of the failing heart, we sought to investigate, in the present study, the effects of its genetic deletion on cardiac function, both normally and in post-MI HF. In order to study the (patho)physiological role(s) specifically of this βarr isoform in vivo, we took advantage of the available βarr1KO mouse model17 and studied these mice alongside age-matched wild type controls (of note, the double βarr1/2 KO mice are embryonic-lethal).9 We found that the absence of βarr1 from the heart and the adrenals leads to significantly elevated cardiac function in post-MI HF, accompanied by improved adverse remodeling, reduced neurohormonal activation and better cardiac βAR signaling and function.
Materials and Methods
Experimental animals and surgical procedures
The animals in this study were handled according to animal welfare regulations and protocols approved by the authors’ Institutional Review Boards. An expanded “Materials and Methods” section is available in the Supplementary Material online, which includes detailed methods on the following: animals and surgical procedures, echocardiography and hemodynamics, ELISA measurements of hormones and cytokines, infarct size measurements, TUNEL and Masson-Trichrome staining, real-time PCR, βAR density, cAMP and SERCA activity measurements, and western blotting.
Statistical analyses
Data are generally expressed as mean ± SEM. Unpaired 2-tailed Student’s t test and one- or two-way ANOVA with Bonferroni test were generally performed for statistical comparisons, unless otherwise indicated. For most 3-group statistical comparisons Dunnett’s test using SAS version 8.2 software was used, as well. For all tests, a p value of <0.05 was generally considered to be significant.
Results
Cardiac function of normal and post-MI βarr1KO mice
In order to investigate the impact on cardiac function of the genetic deletion exclusively of βarr1, we utilized the available global βarr1KO mouse model (Figure S1). These mice breed normally, without any gross abnormalities and present no overt cardiovascular or other phenotype.17 To induce HF, three-month-old male mice underwent surgical MI and were studied alongside age-matched male WT mice. We first examined the cardiac function parameters of these mice, both in sham and post-MI groups. Echocardiography revealed that, although similar between the sham groups (a finding consistent with the results of a previous study on these mice17), (basal) ejection fraction was severely diminished in WT mice at 4 weeks post-MI, as expected, but significantly higher in the post-MI βarr1KO’s at the same time point (Figure 1A & Table S1). Consistent with these findings, upon in vivo catheterization for hemodynamic measurements, post-MI βarr1KO mice also show significantly enhanced cardiac contractility, both basally and in response to isoproterenol stimulation, compared to post-MI WT mice (Figure 1B & Table S1), even though left ventricular end-systolic and end-diastolic pressures are similar between the two post-MI groups upon maximal isoproterenol challenge (Table S1). Notably, isoproterenol-induced contractility is elevated also in sham (normal) βarr1KO’s compared to sham WT’s (Figure 1B & Table S1), which is again consistent with the results of a previous study on these mice.17 Taken together, these findings strongly indicate that cardiac βarr1 is a major negative regulator of βAR-dependent contractility in vivo and that its absence leads to significant attenuation of cardiac dysfunction post-MI.
Figure 1.

(A) Ejection fraction (EF) % of sham-operated (sham) or of 4-week post-MI (MI) βarr1KO and WT mice. *, p<0.05, vs. either Sham; **, p<0.05, vs. WT MI; n=7 mice/group. (B) Basal and maximal dose (333 ng/kg) of isoproterenol (Max. Iso)-stimulated +dP/dtmax responses of these mice. *, p<0.05, vs. WT Sham-Max. Iso; #, p<0.05, vs. either Sham; **, p<0.05, vs. WT MI-Max. Iso; ^, p<0.05, vs. WT MI-Basal; n=5 mice/group.
Survival and neurohormonal status of post-MI βarr1KO mice
Next, we sought to further examine the phenotype of the post-MI βarr1KO mice. Kaplan-Meier survival curves indicated a markedly lower (overall) mortality of the post-MI βarr1KO’s compared to post-MI WT controls (p=0.012, Figure 2A). This was accompanied by significantly reduced elevations in the plasma circulating levels of the CAs NE and Epi in post-MI βarr1KO’s compared to post-MI WT’s (Figure 2B), indicative of reduced overall sympathetic activitation in post-MI HF βarr1KO mice. As for the other major cardiotoxic hormone, aldosterone, post-MI βarr1KO’s, remarkably, failed to exhibit any hyperaldosteronism (i.e. elevation of circulating aldosterone levels) whatsoever, in marked contrast to post-MI WT’s, which, as expected, display severe hyperaldosteronism (Figure 2C). This finding strongly corroborates the essential role of adrenal βarr1 in post-MI HF-associated hyperaldosteronism we have previously reported.26,27 Finally, and consistent with these effects on circulating levels of catecholamines and aldosterone, post-MI βarr1KO’s also display significantly lower mean arterial blood pressure (125 ± 4 mm Hg) than post-MI WT’s (141 ± 5 mm Hg) at 4 weeks post-MI (p<0.05, n=5).
Figure 2.

(A) Kaplan-Meier survival curves of the 4 groups of the study: sham-operated (Sham) and post-MI (MI) βarr1KO and WT mice. p=0.012 between MI WT and MI βarr1KO; n=15 mice/group for sham, 37 mice/group for MI mice. (B) Plasma circulating NE and Epi levels in sham-operated (Sham) or in 4-week post-MI (MI) βarr1KO (KO) and WT mice. *, p<0.05, vs. Sham-either genotype; ^, p<0.05, vs. WT MI; n=6 mice/group. (C) Serum aldosterone levels in these mice. *, p<0.05, vs. all other groups; n=6 mice/group.
Cardiac apoptosis, dilatation and infarct size in post-MI βarr1KO mice
The increased survival of the post-MI βarr1KO’s prompted us to investigate the cardiac apoptosis in these mice. Cardiac cellular apoptosis, as measured with the TUNEL assay in the hearts of the post-MI mice, was found significantly reduced in βarr1KO’s compared to WT mice at 24 hrs post-MI, which is consistent with the Kaplan-Meier survival results (Figure 3A). In addition, cardiac dilatation was also decreased in the βarr1KO’s compared to WT controls at 4 weeks post-MI (Figure 3B & Table S1), and, importantly, infarct size was also significantly reduced in the βarr1KO hearts compared to control WT hearts at the same post-MI time point (4 weeks) (Figure 3C & 3D). To exclude the possibility that differences in the extent of the initial injury inflicted by our surgical MI method upon the two lines might have been responsible for this result, infarct size at 24 hrs post-MI was also measured and found indistinguishable between the two groups (Figure S2A & S2B). Taken together, these results indicate that post-MI βarr1KO hearts have decreased apoptosis, dilatation, and infact size, which might underlie their favorable overall survival phenotype.
Figure 3.

(A) Apoptotic cell death at 24 hrs post-MI in βarr1KO (KO) and WT mice, as measured by TUNEL performed in the border zone of the infarct. Representative images of TUNEL-positive nuclei are shown (left), along with their quantitation (right). No difference in rate of apoptosis in the remote zone was found (data not shown). *, p<0.05, vs. either Sham; #, p<0.05, vs. WT MI; n=6 hearts/group. (B) Left Ventricular End Diastolic Diameter (LVEDD) of these mice at 4 weeks post-MI or post-sham operation. #, p<0.05, vs. either Sham; *, p<0.05, vs. WT MI; n=7 mice/group. (C–D) Infarct size at 4 weeks post-MI. Representative triphenyltetrazolium chloride (TTC)–stained cardiac cross-sections are shown in (C), and average left ventricular (LV) infarct size, expressed as % LV infarction and measured from 6 mice/group, is shown in (D). **, p<0.05, vs. WT MI.
Cardiac inflammation, fibrosis and adverse remodeling markers in post-MI βarr1KO mice
Next, we examined markers of post-MI cardiac inflammation and adverse remodeling in the two animal groups. Levels of all three major pro-inflammatory cytokines, tumor necrosis factor (TNF)-α (Figure 4A), interleukin (IL)-6 (Figure 4B), and IL-1β (Figure 4C), were found significantly decreased in post-MI βarr1KO hearts compared to control WT hearts at 4 weeks post-MI, indicating decreased overall cardiac inflammation in the post-MI βarr1KO mice. Moreover, cardiac fibrosis, as measured with Masson-Trichrome staining, was also markedly decreased in the post-MI βarr1KO mice compared to post-MI WT controls (Figure 4D & 4E). Finally, mRNA expression of all major adverse remodeling-associated biomarkers, i.e. collagen synthesis (Figure 4F), B-type natriuretic peptide (BNP) (Figure 4G), atrial natriuretic factor (ANF) (Figure S3A), and transforming growth factor (TGF)-β (Figure S3B), was significantly reduced in the post-MI βarr1KO hearts compared to post-MI WT hearts, strongly suggesting that adverse remodeling is markedly attenuated in post-MI βarr1KO hearts.
Figure 4.

(A–C) Levels of pro-inflammatory cytokines TNFα (A), IL-6 (B), and IL-1β (C), measured in serum of intra-cardiac blood from βarr1KO (KO) and WT mice at 4 weeks post-MI (MI) or post-sham operation (Sham). *, p<0.05, vs. all other groups; **, p<0.05, vs. WT Sham; n=5 mice/group. (D–E) Cardiac fibrosis staining in the 4-week post-MI mice. Blue denotes collagen fibers, red denotes muscle fibers, and black represents cell nuclei. Representative images are shown in (D) and quantification of the % fibrotic area in (E). *, p<0.05; n=5 hearts/group. (F–G) Heart mRNA levels of Collagen-1α1 (Col-1α1) (F) and of brain natriuretic peptide (BNP) (G) in these mice. #, p<0.05, vs. either Sham; *, p<0.05, vs. WT MI; n=6 hearts/group.
Cardiac βAR density, signaling and function in post-MI βarr1KO mice
In an effort to dissect the molecular mechanisms underlying the dramatic effect of the absence of βarr1 on cardiac contractile function, we also investigated several aspects of cardiac βAR signaling and function in the two post-MI groups. Consistent with the phenotypic and functional data, total βAR density in the post-MI βarr1KO hearts was significantly higher compared to post-MI WT hearts, which display marked βAR downregulation post-MI, as expected (Figure 5A). This attenuated βAR downregulation in post-MI βarr1KO hearts was accompanied by preserved total cAMP levels, again in sharp contrast with the WT hearts whose cAMP content is dramatically diminished post-MI (Figure 5B). Given that cAMP is the most critical mediator of cardiac βAR pro-contractile signaling,2,15 this result strongly indicates that the absence of cardiac βarr1 is sufficient to restore βAR-dependent contractility in post-MI HF. Another important component of the cardiac contractile machinery regulated by βARs is sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA)-2a.31 SERCA activity was also found significantly increased in post-MI βarr1KO hearts compared to post-MI WT hearts (Figure 5C), thus providing another line of evidence for the increased contractile function of post-MI βarr1KO hearts. Of note though, cardiac SERCA activity of βarr1KO’s appears elevated also in the normal (sham) groups (Figure 5C), indicating that cardiac βarr1 might affect the cardiac contractile machinery even under normal conditions (no HF). Finally, since cardiac βarrs, and in particular βarr2, have been shown to mediate EGFR transactivation from the β1AR upon the latter’s phosphorylation by GRK5, an effect that is considered anti-apoptotic in the heart,18 we also examined levels of EGFR transactivation in the post-MI hearts of βarr1KO’s and WT’s after acute stimulation with isoproterenol in the presence of ICI-118,551 (a β2AR-selective antagonist) in vivo (in order to activate the β1ARs only). Western blotting for phospho-Tyr845-EGFR (active EGFR) in cardiac extracts from these mice revealed significantly elevated EGFR transactivation in βarr1KO hearts compared to WT hearts at 24 hrs post-MI (and also in sham hearts) (Figure 5D & 5E), suggesting that cardiac βarr1 inhibits (rather than promotes) this β1AR-induced EGFR transactivation in the heart. Finally, in an effort to better dissect the relative contributions of cardiac vs. adrenal βarr1 on the observed phenotypes of the mice of the study, we overexpressed the known βarr1 inhibitor mini-gene βarr1ct27 specifically in the hearts of post-MI WT mice, in order to inhibit cardiac (only) βarr1 in vivo, and studied these hearts alongside control post-MI WT hearts receiving adenovirus encoding green fluorescent protein (GFP) at the time of MI. As shown in Figure S4, cardiac βarr1 inhibition with βarr1ct led to significant improvements in cardiac βAR density (Figure S4A) and cAMP accumulation (Figure S4B) at 4 weeks post-MI, indicating improved cardiac adrenergic and inotropic reserves, as expected; however, no significant changes in circulating catecholamines of the post-MI WT mice were observed with cardiac βarr1ct at 4 weeks post-MI (Figure S4C), indicating that catecholamine production/secretion is mediated by adrenal (rather than cardiac) βarr1.
Figure 5.

(A) βAR density in cardiac plasma membranes of sham-operated (Sham) or of 4-week post-MI (MI) βarr1KO (KO) and WT mice. *, p<0.05, vs. all other groups; n=5 hearts/group. (B) Steady-state total cAMP levels in cardiac homogenates purified from these mice. *, p<0.05 vs. all other groups; n=5 hearts/group. (C) Cardiac SERCA activity measured in membrane homogenates from the hearts of these mice. *, p<0.05, vs. WT Sham; **, p<0.05, vs. WT MI; n=5 hearts/group. (D–E) Cardiac EGFR transactivation in these mice. Representative western blots in total cardiac protein extracts for phospho-Tyr845-EGFR or total EGFR (loading control) are shown in (D) and their densitometric quantitation in (E). Relative EGFR transactivation was calculated by comparing the ratios of phospho-EGFR to total EGFR for each sample. *, p<0.05, vs. either WT; **, p<0.05, vs. KO Sham; n=5 hearts/group.
Discussion
βarr1 (and to a lesser extent βarr2) are abundantly expressed throughout the cardiovascular system7–9 and regulate the vast majority of cardiovascular GPCRs, including cardiac βARs, cardiac and adrenocortical angiotensin II (AngII) type 1 receptors (AT1Rs), adrenal and central SNS α2ARs, etc.9,32,33 Their importance in cardiovascular (and other systems’) biology is evidenced by the fact that the double βarr1/2 KO mouse is embryonic-lethal,32,33 Their actions on the heart have been attributed to both their functions as receptor desensitizers/internalizers and as G protein-independent signal transducers.9,32 However, and especially for their latter function, the exact in vivo roles of each cardiac βarr isoform in GPCR-dependent signal transduction are currently unknown. In the present study, we were able to delineate the cardiovascular phenotype of βarr1KO mice in post-MI HF, along with its associated underlying molecular signaling mechanisms. Specifically, we found that the absence of βarr1 from the heart leads to significantly better overall cardiac function in post-MI HF, dramatically increased cardiac βAR-dependent cardiac function both physiologically and in post-MI HF, and significantly attenuated adverse remodeling, apoptosis, inflammation and infarct size, leading to improved overall survival post-MI. At the same time, absence of βarr1 from the adrenal gland leads to a dramatically improved neurohormonal profile of the failing post-MI heart, with reduced circulating catecholamine and aldosterone levels. Finally, cardiac βAR signaling and function are dramatically elevated in the post-MI βarr1KO mice, translating into markedly improved cardiac adrenergic and inotropic reserves.
Thus, βarr1 appears to be the single most important negative regulator of βAR-dependent pro-contractile signaling and function, via the classical processes of desensitization and downregulation of the cardiomyocyte βAR content, and its absence results in dramatically elevated βAR-dependent contractility, both normally and after MI, and in almost restored functional βAR number and cAMP-mediated signaling post-MI, i.e. in dramatically improved β-adrenergic and inotropic reserves of the failing heart. Consistent with this, cardiac SERCA activity is also markedly elevated in post-MI βarr1KO’s compared to control WT mice. Of note however, SERCA activity is also significantly elevated in normal (sham) βarr1KO mice, suggesting that the effects of cardiac βarr1 on SERCA activity extend beyond (merely) βAR desensitization. Indeed, current investigations suggest that cardiac βarr2 directly interacts with SERCA2a, promoting its SUMOylation and increasing its activity,34 and that this is actually opposed by βarr1 (Lymperopoulos, A., unpublished data). On the other hand, βarr2 appears unable to compensate for the absence of βarr1 for any of these effects in vivo (and it is not compensatively upregulated, either; data not shown & Ref. 17). Taken together, these phenotypic findings strongly suggest that the βarr1 isoform is the salient βarr responsible for βAR desensitization and downregulation in the heart (Figure 6), processes whose dramatic elevation constitutes a molecular hallmark of chronic HF2,3,5. Therefore, prevention of the interaction of βarr1 with cardiac βARs, which is achieved (indirectly) via cardiac GRK2 inhibition with βARKct for instance,13,15,35 might be beneficial as a positive inotropic therapy for HF (Figure 6).
Figure 6.
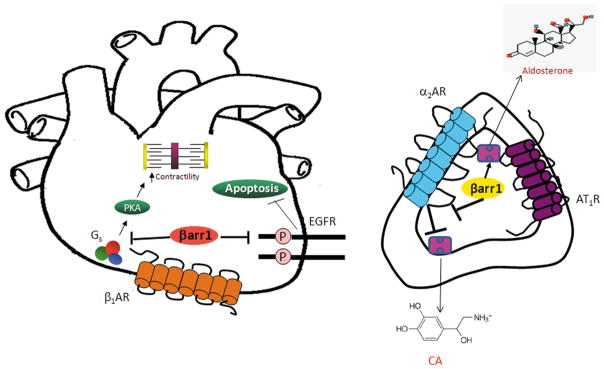
Schematic illustration of the cardiac (left) and adrenal (right) signaling mechanisms underlying the effects of βarr1 on cardiovascular function. Gs: stimulatory G protein; P: Phosphorylation; α2AR: alpha2-adrenergic receptor; AT1R: Angiotensin II type 1 receptor; CA: Catecholamine. See text for details and all other molecular acronym descriptions.
It is now well known and documented that βarrs, in addition to the original biological role ascribed to them as terminators of G protein-dependent signaling by GPCRs, can also serve as signal transducers in their own right independently of G proteins.32,33 Indeed, specifically in the heart, βarrs have been shown to mediate beneficial, anti-apoptotic and proliferation-promoting signals from the β1AR (upon its phosphorylation by GRK5 and not GRK2 though) and from the mechanical stretch-activated AT1R via EGFR transactivation and subsequent ERK phosphorylation/activation.18,36 In addition, they might even mediate pro-contractile signaling from the AT1R in cardiac myocytes, an effect ascribed to βarr2 and presumably inhibited by GRK2 and βarr1.23 We found that β1AR-induced EGFR transactivation is significantly elevated in βarr1KO hearts in vivo, both in normal (sham) conditions and (even more so) in post-MI HF, consistent with the apoptotic phenotype of the βarr1KO hearts in vivo. This strongly suggests that cardiac βarr2 normally mediates this beneficial, anti-apoptotic β1AR-dependent EGFR transactivation, whereas cardiac βarr1 opposes this signaling mechanism (Figure 6). Of course, studies in βarr2KO mice are warranted to further validate this scenario. Another important aspect of HF pathophysiology, in which the two βarr isoforms might exert differential or even opposing effects, is peri-infarct area neoangiogenesis;37 in fact, given the difference observed in infarct size at 4 weeks post-MI (Figure 3C–D), this is quite likely and certainly warrants further investigation in the future.
Finally, the neurohormonal profile of the post-MI βarr1KO mice confirms the essential roles of adrenal βarr1 in: a) chronically elevated CA secretion in post-MI HF (via chronic desensitization/downregulation of the sympatho-inhibitory α2ARs in the chromaffin cells of the adrenal medulla) (Figure 6), and b) post-MI HF-associated hyperaldosteronism (via increased AngII-dependent aldosterone synthesis and secretion in adrenocortical zona glomerulosa cells) (Figure 6), which we have uncovered over the past several years.26–30
Admittedly, the biggest limitation of the present study is the use of a global knockout model which precludes any safe conclusions on the relative contributions of adrenal vs. cardiac βarr1 on the observed phenotypic effects of the post-MI βarr1KO mice. Only development of tissue-specific KO mice can provide definitive answers to this important question. However, our data on post-MI WT mice having cardiac-specific blockade of βarr1 courtesy of cardiac-specific βarr1ct overexpression strongly suggest that cardiac βarr1 absence is responsible for the observed improvements in the adrenergic and inotropic reserves of the post-MI βarr1KO mice, while their vastly improved neurohormonal profile (i.e. lower circulating catecholamine levels and complete absence of hyperaldosteronism) is (mainly) due to βarr1 absence from the adrenal glands. Given the extensive cross-talk between the two organs (heart-adrenals) and that aldosterone and catecholamines can be produced also in the heart per se however,38,39 drawing of these conclusions warrants extreme caution. Nevertheless, all of the above findings are clinically important: we report for the first time that βarr1 is the βarr isoform mainly (if not exclusively) responsible for cardiac βAR desensitization and downregulation in vivo, a hallmark abnormality in chronic HF. In addition, βarr1 promotes elevation of the neurohormonal burden of the failing heart through its actions in the adrenal gland, and seems to oppose several beneficial signaling effects of its counterpart, βarr2, in the myocardium, such as EGFR transactivation and ERK signaling, which lead to inhibition of apoptosis and inflammation and to promotion of cardiac survival. Thus, βarr1 removal from both the heart and the adrenals results in significantly improved cardiac function, structural remodeling, β-adrenergic and inotropic reserves, reduced neurohormonal burden and increased cardiac and overall survival in post-MI HF. These findings strongly suggest that inhibition of βarr1 activity towards βARs in the heart and βarr1 blockade in the adrenals, either directly (with a pharmacological βarr1-specific inhibitor or via siRNA knockdown) or indirectly (e.g. via blockade of GRK2, which promotes association of βarr1 with cardiac βARs) might be a viable therapeutic strategy for chronic post-MI HF treatment, with the potential also of complementing or enhancing the benefits of β-blocker therapy, which is hampered by the adverse effect of negative inotropy.
Perspectives
The present study reports for the first time that cardiac βarr1 is the βarr isoform responsible for cardiac βAR desensitization and downregulation in vivo, a hallmark molecular abnormality in chronic HF. In addition, adrenal βarr1 promotes elevation of the neurohormonal burden of the failing heart by mediating catecholamine secretion and aldosterone synthesis and secretion in the adrenal medulla and cortex, respectively. Finally, βarr1 seems to oppose several beneficial signaling effects of its counterpart, βarr2, in the myocardium, such as EGFR transactivation and ERK signaling, which lead to inhibition of apoptosis and inflammation and to promotion of cardiac survival. Thus, βarr1 removal from both the heart and the adrenals results in significantly improved cardiac function, halted adverse remodeling, elevated β-adrenergic and inotropic reserves, reduced neurohormonal burden and increased cardiac and overall survival in post-MI HF. These findings strongly suggest that inhibition of βarr1 activity towards βARs in the heart and βarr1 blockade in the adrenals, either directly (with a pharmacological βarr1-specific inhibitor or via siRNA knockdown) or indirectly (e.g. via blockade of GRK2, which promotes association of βarr1 with receptors) might be a viable therapeutic strategy for chronic post-MI HF treatment.
Supplementary Material
Novelty and Significance.
What Is New?
βarr1 (rather than βarr2) is the βarr isoform responsible for βAR desensitization and downregulation in vivo in the heart, a hallmark molecular abnormality in chronic HF.
βarr1 in the adrenals is absolutely essential for post-MI hyperaldosteronism and catecholamine elevation in vivo, thereby heightening the neurohormonal burden of the failing heart.
Cardiac βarr1 also counters some beneficial effects of βarr2 in the myocardium, e.g. anti-apoptosis and suppression of inflammation.
What Is Relevant?
Adrenal βarr1 can promote hyperaldosteronism and sympathetic hyperactivity, two neurohormonal mechanisms that contribute significantly to the development of hypertension and HF.
Pharmacological or genetic βarr1 inhibition in vivo, at least in the adrenals and in the heart, might be a valid anti-hypertensive and anti-HF treatment strategy.
Summary.
Blockade of βarr1 activity in both the heart and the adrenal gland might be of therapeutic value for halting or even reversing post-MI HF progression.
Acknowledgments
We thank Dr. Yuhui Wen (Univ. of Miami, Miller School of Medicine) for excellent technical assistance with the in vivo experiments.
Sources of Funding
This work was supported in part by a Scientist Development Grant from the American Heart Association (AHA #09SDG2010138, National Center) and a Nova Southeastern University’s Health Professions Division (HPD) Research Grant (to A.L.), and NIH grants R37 HL061690, R01 HL085503, P01 HL075443 (Project 2) and P01 HL091799 (to W.J.K.).
Non-standard abbreviations and acronyms
- MI
myocardial infarction
- HF
heart failure
- AR
adrenergic receptor
- GPCR
G protein-coupled receptor
- AC
adenylyl cyclase
- PKA
protein kinase A
- GRK
GPCR kinase
- βarr
beta-arrestin
- KO
knockout
- EGFR
epidermal growth factor receptor
- ERK
extracellular signal-regulated kinase
- SNS
sympathetic nervous system
- CA
catecholamine
- NE
norepinephrine
- Epi
epinephrine
- WT
wild type
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
- TNFα
tumor necrosis factor alpha
- IL
interleukin
- cAMP
3′-5′ adenosine monophosphate (cyclic adenosine monophosphate)
- SERCA
sarco(endo)plasmic reticulum Ca2+-ATPase
- AngII
angiotensin II
- AT1R
AngII type 1 receptor
- siRNA
small interfering RNA
Footnotes
Disclosures
NONE.
The authors declare no relationships with industry or any other conflict of interest.
References
- 1.Tamargo J, López-Sendón J. Novel therapeutic targets for the treatment of heart failure. Nat Rev Drug Disc. 2011;10:536–555. doi: 10.1038/nrd3431. [DOI] [PubMed] [Google Scholar]
- 2.Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 3.Brodde OE. Beta-adrenoceptors in cardiac disease. Pharmacol Ther. 1993;60:405–430. doi: 10.1016/0163-7258(93)90030-h. [DOI] [PubMed] [Google Scholar]
- 4.Bristow MR, Ginsburg R, Umans V, Fowler M, Minobe W, Rasmussen R, Zera P, Menlove R, Shah P, Jamieson S. Beta 1- and beta 2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective beta 1-receptor down-regulation in heart failure. Circ Res. 1986;59:297–309. doi: 10.1161/01.res.59.3.297. [DOI] [PubMed] [Google Scholar]
- 5.Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, Billingham ME, Harrison DC, Stinson EB. Decreased catecholamine sensitivity and β-adrenegic receptor density in failing human hearts. N Engl J Med. 1982;307:205–211. doi: 10.1056/NEJM198207223070401. [DOI] [PubMed] [Google Scholar]
- 6.Port JD, Bristow MR. Altered beta-adrenergic receptor gene regulation and signaling in chronic heart failure. J Mol Cell Cardiol. 2001;33:887–905. doi: 10.1006/jmcc.2001.1358. [DOI] [PubMed] [Google Scholar]
- 7.Sterne-Marr R, Gurevich VV, Goldsmith P, Bodine RC, Sanders C, Donoso LA, Benovic JL. Polypeptide variants of β-arrestin and arrestin3. J Biol Chem. 1993;268:15640–15648. [PubMed] [Google Scholar]
- 8.Lymperopoulos A, Bathgate A. Pharmacogenomics of the heptahelical receptor regulators G-protein-coupled receptor kinases and arrestins: the known and the unknown. Pharmacogenomics. 2012;13:323–341. doi: 10.2217/pgs.11.178. [DOI] [PubMed] [Google Scholar]
- 9.Lymperopoulos A. Beta-arrestin biased agonism/antagonism at cardiovascular seven transmembrane-spanning receptors. Curr Pharm Des. 2012;18:192–198. doi: 10.2174/138161212799040475. [DOI] [PubMed] [Google Scholar]
- 10.Ungerer M, Böhm M, Elce JS, Erdmann E, Lohse MJ. Altered expression of beta-adrenergic receptor kinase and beta 1-adrenergic receptors in the failing human heart. Circulation. 1993;87:454–463. doi: 10.1161/01.cir.87.2.454. [DOI] [PubMed] [Google Scholar]
- 11.Rengo G, Lymperopoulos A, Koch WJ. Future G protein-coupled receptor targets for treatment of heart failure. Curr Treat Options Cardiovasc Med. 2009;11:328–338. doi: 10.1007/s11936-009-0033-5. [DOI] [PubMed] [Google Scholar]
- 12.Belmonte SL, Blaxall BC. G protein coupled receptor kinases as therapeutic targets in cardiovascular disease. Circ Res. 2011;109:309–319. doi: 10.1161/CIRCRESAHA.110.231233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rengo G, Lymperopoulos A, Leosco D, Koch WJ. GRK2 as a novel gene therapy target in heart failure. J Mol Cell Cardiol. 2011;50:785–792. doi: 10.1016/j.yjmcc.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lymperopoulos A, Rengo G, Koch WJ. Adrenal adrenoceptors in heart failure: fine-tuning cardiac stimulation. Trends Mol Med. 2007;13:503–511. doi: 10.1016/j.molmed.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 15.Lymperopoulos A, Rengo G, Koch WJ. GRK2 inhibition in heart failure: something old, something new. Curr Pharm Des. 2012;18:186–191. doi: 10.2174/138161212799040510. [DOI] [PubMed] [Google Scholar]
- 16.Penn RB, Pronin AN, Benovic JL. Regulation of G protein-coupled receptor kinases. Trends Cardiovasc Med. 2000;10:81–89. doi: 10.1016/s1050-1738(00)00053-0. [DOI] [PubMed] [Google Scholar]
- 17.Conner DA, Mathier MA, Mortensen RM, Christe M, Vatner SF, Seidman CE, Seidman JG. beta-Arrestin1 knockout mice appear normal but demonstrate altered cardiac responses to beta-adrenergic stimulation. Circ Res. 1997;81:1021–1026. doi: 10.1161/01.res.81.6.1021. [DOI] [PubMed] [Google Scholar]
- 18.Noma T, Lemaire A, Naga Prasad SV, Barki-Harrington L, Tilley DG, Chen J, Le Corvoisier P, Violin JD, Wei H, Lefkowitz RJ, Rockman HA. Beta-arrestin-mediated beta1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J Clin Invest. 2007;117:2445–2458. doi: 10.1172/JCI31901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tilley DG. G protein-dependent and G protein-independent signaling pathways and their impact on cardiac function. Circ Res. 2011;109:217–230. doi: 10.1161/CIRCRESAHA.110.231225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noor N, Patel CB, Rockman HA. βarrestin: a signaling molecule and potential therapeutic target for heart failure. J Mol Cell Cardiol. 2011;51:534–541. doi: 10.1016/j.yjmcc.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, Lefkowitz RJ. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc Natl Acad Sci USA. 2001;98:2449–2454. doi: 10.1073/pnas.041604898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim J, Zhang L, Peppel K, Wu JH, Zidar DA, Brian L, DeWire SM, Exum ST, Lefkowitz RJ, Freedman NJ. Beta-arrestins regulate atherosclerosis and neointimal hyperplasia by controlling smooth muscle cell proliferation and migration. Circ Res. 2008;103:70–79. doi: 10.1161/CIRCRESAHA.108.172338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rajagopal K, Whalen EJ, Violin JD, Stiber JA, Rosenberg PB, Premont RT, Coffman TM, Rockman HA, Lefkowitz RJ. Beta-arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes. Proc Natl Acad Sci USA. 2006;103:16284–16289. doi: 10.1073/pnas.0607583103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L. Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. CONSENSUS Trial Study Group. Circulation. 1990;82:1730–1736. doi: 10.1161/01.cir.82.5.1730. [DOI] [PubMed] [Google Scholar]
- 25.Weber KT. Aldosterone in congestive heart failure. N Engl J Med. 2001;345:1689–1697. doi: 10.1056/NEJMra000050. [DOI] [PubMed] [Google Scholar]
- 26.Lymperopoulos A, Rengo G, Zincarelli C, Kim J, Soltys S, Koch WJ. An adrenal beta-arrestin 1-mediated signaling pathway underlies angiotensin II-induced aldosterone production in vitro and in vivo. Proc Natl Acad Sci USA. 2009;106:5825–5830. doi: 10.1073/pnas.0811706106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lymperopoulos A, Rengo G, Zincarelli C, Kim J, Koch WJ. Adrenal beta-arrestin 1 inhibition in vivo attenuates post-myocardial infarction progression to heart failure and adverse remodeling via reduction of circulating aldosterone levels. J Am Coll Cardiol. 2011;57:356–365. doi: 10.1016/j.jacc.2010.08.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lymperopoulos A, Rengo G, Funakoshi H, Eckhart AD, Koch WJ. Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat Med. 2007;13:315–323. doi: 10.1038/nm1553. [DOI] [PubMed] [Google Scholar]
- 29.Lymperopoulos A, Rengo G, Zincarelli C, Soltys S, Koch WJ. Modulation of adrenal catecholamine secretion by in vivo gene transfer and manipulation of G protein-coupled receptor kinase-2 activity. Mol Ther. 2008;16:302–307. doi: 10.1038/sj.mt.6300371. [DOI] [PubMed] [Google Scholar]
- 30.Lymperopoulos A, Rengo G, Gao E, Ebert SN, Dorn GW, 2nd, Koch WJ. Reduction of sympathetic activity via adrenal-targeted GRK2 gene deletion attenuates heart failure progression and improves cardiac function after myocardial infarction. J Biol Chem. 2010;285:16378–16386. doi: 10.1074/jbc.M109.077859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 32.Luttrell LM, Gesty-Palmer D. Beyond desensitization: physiological relevance of arrestin-dependent signaling. Pharmacol Rev. 2010;62:305–330. doi: 10.1124/pr.109.002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 34.Kho C, Lee A, Jeong D, Oh JG, Chaanine AH, Kizana E, Park WJ, Hajjar RJ. SUMO1-dependent modulation of SERCA2a in heart failure. Nature. 2011;477:601–605. doi: 10.1038/nature10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rengo G, Lymperopoulos A, Zincarelli C, Donniacuo M, Soltys S, Rabinowitz JE, Koch WJ. Myocardial adeno-associated virus serotype 6-betaARKct gene therapy improves cardiac function and normalizes the neurohormonal axis in chronic heart failure. Circulation. 2009;119:89–98. doi: 10.1161/CIRCULATIONAHA.108.803999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rakesh K, Yoo B, Kim IM, Salazar N, Kim KS, Rockman HA. Beta-arrestin-biased agonism of the angiotensin receptor induced by mechanical stress. Sci Signal. 2010;3:ra46. doi: 10.1126/scisignal.2000769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Santulli G, Cipolletta E, Sorriento D, Del Giudice C, Anastasio A, Monaco S, Maione AS, Condorelli G, Puca A, Trimarco B, Illario M, Iaccarino G. CaMK4 Gene Deletion Induces Hypertension. J Am Heart Assoc. 2012;1:e001081. doi: 10.1161/JAHA.112.001081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Silvestre JS, Heymes C, Oubénaïssa A, Robert V, Aupetit-Faisant B, Carayon A, Swynghedauw B, Delcayre C. Activation of cardiac aldosterone production in rat myocardial infarction: effect of angiotensin II receptor blockade and role in cardiac fibrosis. Circulation. 1999;99:2694–2701. doi: 10.1161/01.cir.99.20.2694. [DOI] [PubMed] [Google Scholar]
- 39.Kuroko Y, Yamazaki T, Tokunaga N, Akiyama T, Kitagawa H, Ishino K, Sano S, Mori H. Cardiac epinephrine synthesis and ischemia-induced myocardial epinephrine release. Cardiovasc Res. 2007;74:438–444. doi: 10.1016/j.cardiores.2007.02.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.