

Abstract
The Protein Model Portal (PMP) has been developed to foster effective use of 3D molecular models in biomedical research by providing convenient and comprehensive access to structural information for proteins. Both experimental structures and theoretical models for a given protein can be searched simultaneously and analyzed for structural variability. By providing a comprehensive view on structural information, PMP offers the opportunity to apply consistent assessment and validation criteria to the complete set of structural models available for proteins. PMP is an open project so that new methods developed by the community can contribute to PMP, for example, new modeling servers for creating homology models and model quality estimation servers for model validation. The accuracy of participating modeling servers is continuously evaluated by the Continuous Automated Model EvaluatiOn (CAMEO) project. The PMP offers a unique interface to visualize structural coverage of a protein combining both theoretical models and experimental structures, allowing straightforward assessment of the model quality and hence their utility. The portal is updated regularly and actively developed to include latest methods in the field of computational structural biology.
Database URL: http://www.proteinmodelportal.org
Introduction
Three-dimensional protein structures are crucial for developing a detailed understanding of the many functions of proteins occurring in nature. Thanks to the efforts of the structural biology community the number of available experimental structures has grown considerably. In recent years, structural genomics efforts (1–3) have contributed to the field by establishing high-throughput structure determination approaches and determining the structures of many unique proteins. Despite these achievements, the number of protein sequences in current databases remains orders of magnitude larger than the number of experimentally solved protein structures. Homology (or comparative) modeling methods are currently the most accurate approaches (especially for larger proteins and protein complexes) for obtaining all-atom models of proteins (4–6) and bridging this knowledge gap. These methods make use of experimental protein structures (‘templates’) to build models for evolutionarily related proteins (‘targets’). Experimental structural biology and homology modeling thereby complement each other in the exploration of the protein structure space and as a result structural coverage to a large extent is now available for the proteomes of many model organisms such as Escherichia coli (7) or Thermotoga maritima (8).
The Worldwide Protein Data Bank (wwPDB) (9, 10) is the single archive for experimental information on macromolecular structures; however, theoretical models cannot be deposited in the PDB (11), and the Protein Model Portal (PMP) has hence been developed to unify access to homology models built by well-established modeling methods. PMP is one of the modules of the Nature Protein Structure Initiative (PSI) Structural Biology Knowledgebase (SBKB) (3). The goal of the portal is firstly to give access to the combined structural coverage leveraged from homology models and experimental protein structures, while transparently communicating the accuracy of the models to the user. This is a crucial aspect for aiding the user in determining the utility of a given model for a particular biological application.
The quality of individual protein structure models may vary substantially and thus model validation has been recognized to play a central role in the process of predicting the structure of a target protein. Potential inaccuracies of models arise from structural divergence between the template and the target during evolution, but also from the modeling process itself, e.g. erroneous alignment(s) of the target sequence to the template(s), or incorrect modeling of loops or amino acid side-chain conformations. The combination of different sources of errors limits the overall quality of a model and thereby the utility to address specific scientific questions (11). High-quality models can be used in a similar way as experimental structures, while models of lower quality can still be valuable for applications requiring lower resolution information, e.g. designing mutagenesis experiments to elucidate the function of a protein.
In this article, we first illustrate how PMP provides structural information for proteins by combining experimental structures in the PDB and theoretical models from various modeling resources, and explain how PMP communicates model quality information. We describe the interactive servers, which allow users to trigger modeling of a protein sequence and show how to submit existing models to quality estimation servers. We then introduce a unique feature of PMP—the analysis of structural variability of experimental structures and models. Finally, we briefly describe the Continuous Automated Model EvaluatiOn (CAMEO) project, aiming at continuously evaluating the accuracy and reliability of protein structure prediction methods in a fully automated manner.
Materials and Methods
Portal content and architecture
PMP relies on models provided by several research groups such as Center for Structures of Proteins in Membranes (CSMP) (12), North Eastern Structural Genomics Center (NESG) (13), New York Structural Genomics Research Consortium (NYSGRC) (14), ModBase (15), SWISS-MODEL Repository (16) and GPCRDB (17) (Table 1). For each model at the partner sites, PMP stores metadata describing the templates(s) used, the sequence identities to these templates, the range of the target sequence modeled and URLs to the providers’ model information pages and model coordinates. In this way, PMP effectively overcomes the fact that the models are provided in various formats at different sites. PMP uses cryptographic MD5 hashes of raw UniProt (31) sequences to identify a specific protein sequence and to allow unambiguous lookup of models in its database of metadata. Thus one of the main obstacles in the past impeding the efficient retrieval of model information has been remedied: the distinct ways of accessing all models available for a given protein using incompatible accession code systems. Because all model data sources are mapped into a common UniProt reference system, additional queries such as PFAM domain annotation are possible, even if the underlying model resource does not offer this functionality.
Table 1.
Servers and data providers currently available in PMP, listed by type of service
| Resource | Features/comments |
|---|---|
| Modeling servers | |
| HHPred | Homology detection and protein-structure prediction by HMM-HMM comparison (18). |
| I-TASSER | Service for protein structure prediction. 3D models are built based on fold recognition and threading techniques (19). |
| ModWeb | Comparative protein structure modeling server (15) based on Modeller (20). |
| M4T | Comparative modeling server using a combination of multiple templates and iterative optimization of alternative alignments (21). |
| SWISS-MODEL Workspace | Integrated web-based homology modeling expert system (22, 23). |
| Quality estimation servers | |
| ModEval | ModEval (24) reports quality scores such as predicted RMSD and native overlap, along with scores based on statistical potentials (DOPE) (25), GA341 (26) assessing the reliability of a model. |
| ModFOLD3 | ModFOLD V3.0 is comparing multiple models for the global and local assessment of models (27, 28). |
| QMEAN | Server for protein model quality estimation based on four statistical potentials terms combining geometrical and interaction scores with two terms for agreement between calculated and predicted secondary structure (29). |
| Protein Model Providers | |
| PSI-Structural-Genomics Centers | CSMP (12) (30),NESG (13), NYSGRC (14)a |
| ModBase | Database of annotated comparative protein structure models built by ModWeb (15). |
| SWISS-MODEL Repository | Database of annotated three-dimensional comparative protein structure models (16) generated by the fully automated homology-modeling pipeline SWISS-MODEL (22) for a selection of model organisms. |
| GPCRDB | Information System for G protein coupledreceptors (GPCR) (17) |
aModels for the targets of the structural genomic centers are provided via ModBase, except for the NESG, which uses a different modeling pipeline.
PMP is updated regularly following the UniProt release cycle and metadata updates by model database providers.
Access to the Portal
Interactive queries of PMP are possible using the main search field allowing queries by free text, or by specifying various biological databases identifiers (UniProt, PDB, RefSeq). The conversion of PDB identifiers to UniProt Accession codes is implemented via Structure integration with function, taxonomy and sequence (SIFTS) (32). The mapping of UniProt codes to other database accession codes is derived from iProClass [a resource provided by Protein Information Resource (PIR), current release 4.12] (33). User queries in the main search field on the portal home page will be automatically interpreted as text /ID queries for search strings of <30 characters, otherwise the query will be interpreted as a protein sequence search. The free text searches are based on the Apache search platform Solr using the Lucene search library. Currently the content of the UniProt fields Description (‘DE’), Gene name (‘GN’), Keyword (‘KW’) and Organism Species (‘OS’) is queried.
From the ‘Advanced Search Page’, additional searches (e.g. gene loci or Entrez/GI accession codes) are supported.
For programmatic access, PMP provides a DAS service (34) and allows convenient RESTful queries, which are e.g. used by the RCSB PDB website to show protein residue ranges for which PMP can provide additional structure information based on theoretical protein models.
Interactive modeling and quality estimation servers
PMP provides an interface to several protein structure prediction servers to interactively start the computation of theoretical models for a protein of interest as well as a submission interface to servers to estimate the accuracy of a predicted protein model. The submission to modeling servers currently supports HHPredB (18), I-TASSER (19), ModWeb (15), M4T (21) and SWISS-MODEL Workspace (22, 23) (Table 1). For estimating the quality of models, currently ModEVAL (24), ModFOLD3 (27) and QMEAN (35) are accessible through the portal (Table 1).
The structural variability analysis
Structural variability among a set of experimental structures and/or models is determined with a superposition-free Cα-distance–based approach.
First, for each model m, where m = 1,..,n, an all-against-all distance matrix Am is generated from the Cα atoms. A column i in one of these matrices contains distances dm,ij of the Cα atom i to all other Cα atoms j where j = 1,..,L for a protein of length L. Second, the standard deviation sij of the distances is calculated following formula (1) for each pair of Cα atoms. To focus the analysis on local accuracy, the influence of long-range distances is weighted down. To achieve this, the Euclidean distance from the mean 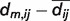 is weighted with an exponential term analogous to the Holm/Sander approach (36). The element sij is then stored in the matrix S, representing the variability within the selected set of models. A graph of S is shown on the ‘Structure Comparison Results’ page of PMP (Figure 1, panel III). Regions in blue correspond to low, whereas regions in red represent high variability. The exponential weighting term has been parameterized in a set of single-domain proteins such that 1.4 Å deviation in a structure-based superposition is visually detectable in the graph.
is weighted with an exponential term analogous to the Holm/Sander approach (36). The element sij is then stored in the matrix S, representing the variability within the selected set of models. A graph of S is shown on the ‘Structure Comparison Results’ page of PMP (Figure 1, panel III). Regions in blue correspond to low, whereas regions in red represent high variability. The exponential weighting term has been parameterized in a set of single-domain proteins such that 1.4 Å deviation in a structure-based superposition is visually detectable in the graph.
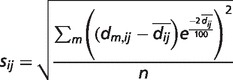 |
(1) |
Figure 1.

Structural variability analysis of the models for the oxygen sensor protein DosP across a region spanning a GGDEF domain and an EAL domain. Panel I indicates the five models (light blue bars) computed for the N-terminal part of the DosP protein (depicted as a red bar). The models selected for the structural variability analysis are shown with a light-brown background color. Pfam domain information is displayed by gray bars, followed by a short description of the target protein and a link to the corresponding entry in the UniProt database for protein functional information. For each of the five analyzed models, Panel II illustrates regions of the models that deviate more from the ensemble in a local (per residue) deviation plot. Most of the variability in this example is observed around residues 430–550. Panel III shows the underlying variability matrix S. Panel IV displays a structural superposition to visualize structural variability among the five selected models. This picture shows the two structural and functional domains GGDEF (with the superposed models #2, #4 and #5 and partially model #3—in blue, red, orange and green colors, respectively) and EAL [with the superposed models #1 (black) and #3 (green)].
To identify regions of high variability between individual models, the per-residue deviation is shown in a second plot. Here, the mean 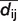 is calculated from each of the cells containing
is calculated from each of the cells containing 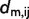 in column i and row j of each of the n matrices of type A. Subsequently, for each model m, the absolute difference
in column i and row j of each of the n matrices of type A. Subsequently, for each model m, the absolute difference  between
between 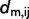 and the mean
and the mean 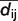 are computed and averaged over the number of residues L of the model [formula (2), Figure 1, panel II].
are computed and averaged over the number of residues L of the model [formula (2), Figure 1, panel II].
| (2) |
Results
The query interface
From the main PMP entry site, it is possible to search for experimental structures or theoretical models for a target protein, using amino acid sequence, free text (e.g. ‘oxygen sensor protein’) or biological database accession codes (e.g. UniProt, RefSeq, PDB, …) in the same query box without the need for the user to specify the input format. Amino acid sequence queries (both fragments and entire protein sequences) are first matched to corresponding UniProt database entries, and sequence similarity searches are performed if no direct match can be identified. It is also possible to search by specifying a PDB identification code (and optionally also chain name) to identify other experimental structures and models for the same protein sequence.
The results summary page
The PMP displays the structural coverage (experimental structures deposited in the PDB up to 90% sequence identity to the target protein and homology models) available for a given protein in a summary page (Figure 2). It features a visual representation as well as a tabular list (Figure 2) of available structures and models for the protein of interest. Information about available experimental structures of the query protein are also provided as well as biological (38) and domain architecture annotations (39) (Figure 2). For computational models, interactive mouseovers outline information about the model providers (panel I, Figure 2), data about the template used (panel II, Figure 2) and a first rough estimate (in the form of a traffic light) of the expected model quality based on sequence similarity. A graphical visualization is used to facilitate the interpretation of how difficult the modeling task was expected to be using categories labels ‘A–C’ (panel III, Figure 2). ‘A’ (red area of the graph) indicates a difficult modeling task, requiring close inspection of the results, whereas ‘C’ (green area of the graph) indicates a rather straightforward modeling task.
Figure 2.

PMP results summary page for the oxygen sensor protein (UniProt Accession Code P76129). Experimentally determined structures for the protein (i.e. PDB entries with sequence identity >90%) are shown in green, models in shades of blue, the darker the higher the sequence identity of the template to the target protein. Pfam domain annotations are depicted in gray, to the right of each domain is a direct link to InterPro providing more information about the different domains. Apart from the graphical representation of structural coverage, detailed lists of experimental structures (not shown) and models are presented. As example, a model from NESG, which was built on the template 3ICL chain A, is highlighted in orange (mouseover text of corresponding model bar is shown in panel I). More details of the template used is given in panel II, where also the template deposition date, a preview image of the structure and the experimental method are given. Expected quality of the model based on the sequence identity between aligned target and template sequences is illustrated in panel III, where a vertical red bar marks the sequence identity of 27% to the template and the expected model accuracy based on the work of Chothia and Lesk (37).
The model details page
The model details page provides additional information about each model such as template information, an interactive Jmol (40) based 3D molecular viewer, a first indication of model quality and a structure-based target-template alignment. Details about the template used in the modeling procedure are provided. Should the model have not been updated for more than three months, a warning is displayed together with a link to the submission interface for the interactive modeling servers registered with PMP (for more details see ‘Modeling servers and Quality estimation servers’ and Table 1 for the list of providers). The user has therefore the possibility to interactively trigger the computation of a new comparative model for the protein of interest based on the latest up-to-date template information. The model details page allows for an interactive graphical display of the protein to inspect the structural details in 3D. The quality of the target protein-template alignment present in the details page is a first estimate for the expected accuracy of the model (Figure 2 panel III). Models produced based on sequence alignments with a low sequence identity are not generally expected to be sufficiently accurate to be used in structure-based drug design/ligand docking applications or to study the molecular mechanisms of proteins. Still, often these models are useful for molecular replacement phasing in X-ray crystallography or in combination with other experimental constraints in integrative modeling approaches. Hence, to facilitate the identification of regions of the model expected to be of high quality (i.e. highly conserved sequence alignment with no or few insertions and deletions), the structurally derived target-template alignment is shown in colors, highlighting regions of conservation between the modeled protein and the structure template (41). Finally, each model can be submitted directly to the quality estimation servers registered in PMP, allowing the user to apply state-of-the art quality estimation prediction algorithms to the obtained structural model of the protein of interest.
Modeling and quality estimation servers
Model accuracy critically depends on the accessibility of suitable template structures. The databases containing precompiled models are based on the best template structures available at the time the model was computed and usually focus their efforts on a limited number of target proteins for efficiency reasons. Hence, model and template verification dates are clearly displayed on the model detail pages in PMP. While a precomputed model in PMP represents a ‘minimal structure information’ baseline for a given protein sequence, improved models may be accessible using more recently released structures in the PDB. Therefore, PMP provides an interface to several established modeling servers to interactively initiate a new template selection and modeling process for a target protein.
PMP features convenient submission interfaces to both modeling and model quality estimation servers available both in the PMP menu and from each model detail page. While in the first case the user needs to supply the sequence to be modeled or the structure coordinates of the model to be validated, in the second case, PMP conveniently fills out this information. The first option can also be used to submit private structure files in PDB format to quality estimation servers. The results produced by the respective servers are sent directly to the email address provided during submission. Table 1 in ‘Materials and Methods’ summarizes modeling and quality estimation servers currently participating to the portal. PMP is an open system, and new servers are continuously included on request from the community.
Analysis of structural variability
Experimental structures for a given protein may show substantial structural variability due to domain motions, mobile loops, different functional states or induced fit upon ligand binding. The analysis of the ensemble of models is an effective way of distinguishing structurally conserved regions in a protein family from more variable regions. Besides these native effects, additional variability is added in the case of theoretical models by differences in algorithmic approaches, the choice of template(s) and modeling errors. Regions with large variability within an ensemble of theoretical models often reflect segments, which cannot be predicted with high confidence, e.g. due to the variation in alternative template structures, flexible loop regions or unaligned regions (indels) in the target-template alignment (42). For these reasons, PMP provides an interface to analyze the variability within an ensemble of experimental structures and models for a protein, which is built based on the computational structural biology framework OpenStructure (43). In the results overview page of PMP, the structure comparison tool (Figure 1 panel I) can be used to compare an overlapping subset of structures and models. Local deviation plots indicate for each amino acid residue the divergence from the ensemble (Figure 1, panel II) for each model or structure. Alternative visualizations of the structural variability information include ‘distance variance maps’ (Figure 1, panel III) or the visualization of structural superpositions using an interactive Jmol applet (40) (Figure 1, panel IV).
Continuous Automated Model EvaluatiOn
CAMEO is a new independent project that continuously evaluates the accuracy and reliability of protein structure prediction methods in a fully automated manner. The service was originally created for monitoring the performance and reliability of those modeling servers registered with PMP. CAMEO currently assesses predictions in two categories (3D protein structure modeling and ligand binding site residue predictions). The project is open to everyone and has been used by several method developer groups to benchmark and monitor their servers and new developments.
Within each CAMEO assessment category, registered servers are provided with the prereleased sequences (‘targets’) of those structures that are to be released next by the PDB. The participating servers then have 4 days to model the sequence using their in-house methods and return the predictions by email until the PDB releases the structures. CAMEO then compares the predictions to the experimentally determined structures. The assessment results are presented on a public Web site, which allows the modeling community to quickly understand how a particular method performed in comparison with others for a given target. Owing to the large number of experimental structures released, the targets within CAMEO represent a wide range of challenges in protein modeling. All raw data (models and scores) are publicly available for download in compressed weekly archives, allowing for more detailed investigation.
CAMEO supports both the developers of structure prediction servers as well as the users of theoretical models: For users of models, the retrospective evaluation of prediction accuracy allows to select the best-performing tools for the modeling task at hand. Because the accuracy requirements for different scientific applications vary (11), CAMEO offers a variety of scores assessing different aspects of a prediction (coverage, local accuracy, completeness, responsiveness, etc.). For the developers of predictions methods, CAMEO provides continuous real-time data on the performance of new approaches. Thus, direct comparison on the same targets at the same point in time with other state-of-the-art techniques helps debugging and benchmarking of new developments. The large number of evaluated prediction targets provides a statistically significant measure of the algorithmic improvements. Because CAMEO evaluates blind prediction accuracy on prereleased target proteins, CAMEO data is well suited to demonstrate a method’s performance in publications by means of an independent direct benchmark.
CAMEO is an open platform, applying assessment criteria established by the protein structure prediction community, and implementing new assessment categories on demand.
CAMEO URL: http://www.cameo3d.org/
Discussion and Conclusion
The PMP has been developed to foster effective use of molecular models in biomedical research by providing convenient and comprehensive access to structural information for a specific protein. For the first time, both experimental structures and theoretical models for a given protein can be searched simultaneously and analyzed for structural variability. Each model’s quality is indicated, and outdated models are flagged to be directly resubmitted via the portal own interface to the registered modeling servers. PMP can be queried using amino acid sequences, free text or various database accession codes, and the results are presented in an intuitive graphical way. The available structural information (experimental or theoretical) is visualized and complemented with functional and domain annotation for a protein of interest. For each precomputed model, PMP displays technical information such as the date of creation and date of verification, the sequence identity of the template and the expected model accuracy based on the evolutionary distance between the target and the template. However, with the ever growing number of new protein sequences, it is no longer feasible to maintain precomputed models for all proteins, and as a consequence, most model resource providers have decided to focus on subsets of the data, e.g. selected model organisms or specific protein families. The PMP submission interfaces to interactive modeling engines therefore are expected to become more relevant in the future.
Based on the results of CAMEO continuously assessing modeling servers, PMP will soon be in a position to provide guidance on which modeling approach might be most suitable for a given protein sequence and modeling task by extrapolating the retrospective assessment data from CAMEO. To educate first-time or occasional users of models, PMP further maintains a set of ‘modelling 101’ resources, which are constantly updated and extended.
All current efforts are dedicated to allow straightforward detailed assessment of any model within PMP and making a model’s utility more transparent. Model quality estimation is an essential component of modeling to indicate if a model is expected to be sufficiently accurate for a given application. However, so far, no consensus has emerged in the community as to which confidence measures should be reported in publications describing theoretical models. Additionally, the coordinates of these models are often not available to the reader. Hence, PMP aims to address these issues in the following way: as next category in CAMEO, we will implement the evaluation of model quality estimation methods. These measures will then serve as the basis for the Model Validation Task Force (11) to establish a community-wide standard for validation and archiving of theoretical models in publications of models in peer-reviewed journals. In parallel, PMP will establish a public archive of macromolecular models, which can currently not be deposited to PDB.
Acknowledgements
We would like to thank Helen Berman, John Westbrook and the SBKB team for stimulating discussions and inspiration. We are grateful to Morena Spreafico, Aleksandra Kos and the whole team for constructive discussions and past and continuous testing of new PMP releases. We are indebted to Marco Biasini, Stefan Bienert, Andrew Waterhouse and Tobias Schmidt for their invaluable contributions concerning the structure annotation functionality within CAMEO.
Funding
NIH and National Institute of General Medical Sciences (U01 GM093324-01 to Protein Model Portal and CAMEO); SIB Swiss Institute of Bioinformatics toward the development of CAMEO, SWISS-MODEL, OpenStructure and the operation of the [BC]2 Basel Computational Biology Center. Funding for open access charge: SIB Swiss Institute of Bioinformatics.
Conflict of interest. None declared.
References
- 1.Stevens RC, Yokoyama S, Wilson IA. Global efforts in structural genomics. Science. 2001;294:89–92. doi: 10.1126/science.1066011. [DOI] [PubMed] [Google Scholar]
- 2.Terwilliger TC, Stuart D, Yokoyama S. Lessons from structural genomics. Annu. Rev. Biophys. 2009;38:371–383. doi: 10.1146/annurev.biophys.050708.133740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gabanyi MJ, Adams PD, Arnold K, et al. The structural biology knowledgebase: a portal to protein structures, sequences, functions, and methods. J. Struct. Funct. Genomics. 2011;12:45–54. doi: 10.1007/s10969-011-9106-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker D, Sali A. Protein structure prediction and structural genomics. Science. 2001;294:93–96. doi: 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- 5.Schwede T, Sali A, Eswar N, et al. 2008. Protein structure modeling. In: T.S.–M.P. (eds). Computational Structural Biology. World Scientific Publishing, Singapore, pp. 3–34. [Google Scholar]
- 6.Mariani V, Kiefer F, Schmidt T, et al. Assessment of template based protein structure predictions in CASP9. Proteins. 2011;79(Suppl. 10):37–58. doi: 10.1002/prot.23177. [DOI] [PubMed] [Google Scholar]
- 7.Guex N, Peitsch MC, Schwede T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis. 2009;30(Suppl. 1):S162–S173. doi: 10.1002/elps.200900140. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y, Thiele I, Weekes D, et al. Three-dimensional structural view of the central metabolic network of Thermotoga maritima. Science. 2009;325:1544–1549. doi: 10.1126/science.1174671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rose PW, Bi C, Bluhm WF, et al. The RCSB protein data bank: new resources for research and education. Nucleic Acids Res. 2013;41:D475–D482. doi: 10.1093/nar/gks1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berman HM, Kleywegt GJ, Nakamura H, et al. The future of the protein data bank. Biopolymers. 2013;99:218–222. doi: 10.1002/bip.22132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwede T, Sali A, Honig B, et al. Outcome of a workshop on applications of protein models in biomedical research. Structure. 2009;17:151–159. doi: 10.1016/j.str.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stroud RM, Choe S, Holton J, et al. 2007 Annual progress report synopsis of the center for structures of membrane proteins. J. Struct. Funct. Genomics. 2009;10:193–208. doi: 10.1007/s10969-008-9058-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiao R, Anderson S, Aramini J, et al. The high-throughput protein sample production platform of the Northeast Structural Genomics Consortium. J. Struct. Biol. 2010;172:21–33. doi: 10.1016/j.jsb.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sauder MJ, Rutter ME, Bain K, et al. High throughput protein production and crystallization at NYSGXRC. Methods Mol. Biol. 2008;426:561–575. doi: 10.1007/978-1-60327-058-8_37. [DOI] [PubMed] [Google Scholar]
- 15.Pieper U, Webb BM, Barkan DT, et al. ModBase, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 2011;39:D465–D474. doi: 10.1093/nar/gkq1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kiefer F, Arnold K, Kunzli M, et al. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009;37:D387–D392. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vroling B, Sanders M, Baakman C, et al. GPCRDB: information system for G protein-coupled receptors. Nucleic Acids Res. 2011;39:D309–D319. doi: 10.1093/nar/gkq1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eswar N, Webb B, Marti-Renom MA, et al. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 2007 doi: 10.1002/0471140864.ps0209s50. Chapter 2, Unit 2.9. [DOI] [PubMed] [Google Scholar]
- 21.Rykunov D, Steinberger E, Madrid-Aliste CJ, et al. Improved scoring function for comparative modeling using the M4T method. J. Struct. Funct. Genomics. 2009;10:95–99. doi: 10.1007/s10969-008-9044-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwede T, Bordoli L, Kiefer F, et al. Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 2009;4:1–13. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- 23.Arnold K, Bordoli L, Kopp J, et al. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 24.Eramian D, Eswar N, Shen MY, et al. How well can the accuracy of comparative protein structure models be predicted? Protein Sci. 2008;17:1881–1893. doi: 10.1110/ps.036061.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen MY, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15:2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Melo F, Sanchez R, Sali A. Statistical potentials for fold assessment. Protein Sci. 2002;11:430–448. doi: 10.1002/pro.110430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McGuffin LJ. The ModFOLD server for the quality assessment of protein structural models. Bioinformatics. 2008;24:586–587. doi: 10.1093/bioinformatics/btn014. [DOI] [PubMed] [Google Scholar]
- 28.McGuffin LJ, Roche DB. Rapid model quality assessment for protein structure predictions using the comparison of multiple models without structural alignments. Bioinformatics. 2010;26:182–188. doi: 10.1093/bioinformatics/btp629. [DOI] [PubMed] [Google Scholar]
- 29.Benkert P, Kunzli M, Schwede T. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009;37:W510–W514. doi: 10.1093/nar/gkp322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phillips GN, Jr, Fox BG, Markley JL, et al. Structures of proteins of biomedical interest from the Center for eukaryotic structural genomics. J. Struct. Funct. Genomics. 2007;8:73–84. doi: 10.1007/s10969-007-9023-6. [DOI] [PubMed] [Google Scholar]
- 31.The UniProt Consortium. Reorganizing the protein space at the universal protein resource (UniProt) Nucleic Acids Res. 2012;40:D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Velankar S, McNeil P, Mittard-Runte V, et al. E-MSD: an integrated data resource for bioinformatics. Nucleic Acids Res. 2005;33:D262–D265. doi: 10.1093/nar/gki058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu C, Nebert DW. Update on genome completion and annotations: protein information resource. Hum. Genomics. 2004;1:229–233. doi: 10.1186/1479-7364-1-3-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prlic A, Down TA, Kulesha E, et al. Integrating sequence and structural biology with DAS. BMC Bioinformatics. 2007;8:333. doi: 10.1186/1471-2105-8-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwede T, Benkert P, Kunzli M. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009;37:W510–W514. doi: 10.1093/nar/gkp322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 37.Chothia C, Lesk AM. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986;5:823–826. doi: 10.1002/j.1460-2075.1986.tb04288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.The UniProt Consortium. Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Res. 2011;39:D214–D219. doi: 10.1093/nar/gkq1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hunter S, Apweiler R, Attwood TK, et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37:D211–D215. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jmol: an open-source Java viewer for chemical structures in 3D. http://www.jmol.org/
- 41.Brown NP, Leroy C, Sander C. MView: a web-compatible database search or multiple alignment viewer. Bioinformatics. 1998;14:380–381. doi: 10.1093/bioinformatics/14.4.380. [DOI] [PubMed] [Google Scholar]
- 42.Chivian D, Baker D. Homology modeling using parametric alignment ensemble generation with consensus and energy-based model selection. Nucleic Acids Res. 2006;34:e112. doi: 10.1093/nar/gkl480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Biasini M, Mariani V, Haas J, et al. OpenStructure: a flexible software framework for computational structural biology. Bioinformatics. 2010;26:2626–2628. doi: 10.1093/bioinformatics/btq481. [DOI] [PMC free article] [PubMed] [Google Scholar]