

Abstract
Bacterial live vector vaccines represent a vaccine development strategy that offers exceptional flexibility. In this approach, genes encoding protective antigens of unrelated bacterial, viral or parasitic pathogens are expressed in an attenuated bacterial vaccine strain that delivers these foreign antigens to the immune system, thereby eliciting relevant immune responses. Rather than expressing these antigens using low copy expression plasmids, here we pursue expression of foreign proteins from the live vector chromosome. Our strategy is designed to compensate for the inherent disadvantage of loss of gene dosage (vs. plasmid-based expression) by integrating antigen-encoding gene cassettes into multiple chromosomal sites already inactivated in an attenuated Salmonella enterica serovar Typhi vaccine candidate. We tested expression of a cassette encoding the green fluorescent protein (GFPuv) integrated separately into native guaBA, htrA or clyA chromosomal loci. Using single integrations, we show that expression levels of GFPuv are significantly affected by the site of integration, regardless of the inclusion of additional strong promoters within the incoming cassette. Using cassettes integrated into both guaBA and htrA, we observe cumulative synthesis levels from two integration sites superior to single integrations. Most importantly, we observe that GFPuv expression increases in a growth phase-dependent manner, suggesting that foreign antigen synthesis may be “tuned” to the physiology of the live vaccine. We expect this novel platform expression technology to prove invaluable in the development of a wide variety of multivalent live vector vaccines, capable of expressing multiple antigens from both chromosomal and plasmid-based expression systems within a single strain.
Keywords: Salmonella, chromosomal expression, foreign gene, live vector, vaccine
Introduction
Excellent progress has been made over the past 20 y in the adaptation of attenuated bacterial vaccine strains for expression of foreign antigens to create multivalent live vector vaccines. Significant effort has been devoted to the creation of expression technologies which either directly or indirectly address the important problem of metabolic stress often associated with expression of foreign immunogens.1,2 It is now recognized that inappropriate synthesis of high levels of foreign protein to induce an antigen-specific protective immune response can adversely affect the fitness and growth rate of an already attenuated vaccine strain, resulting in over-attenuation and loss of immunity directed at both the live vector and foreign antigen. If these target immunogens are encoded by multicopy expression plasmids, these undesirable metabolic fluxes can result in plasmid loss in the absence of selective pressure, which ultimately defeats the strategy of live vector-mediated delivery of vaccine antigens. We and others have developed effective genetic stabilization systems for enhancing the retention of multicopy plasmids encoding regulated synthesis of foreign antigens, without the further requirement to select with antibiotics.3-5 Antigen export systems have also been developed by our group and others to reduce proteolytic degradation of foreign antigens within the cytoplasm and more effectively deliver these antigens to the immune system to enhance immunogenicity.6-9 It is clear that a wide variety of genetic techniques and technologies are now readily available for efficient delivery of one or more antigens using live vector vaccines. However, inclusion of all of the genes encoding these foreign antigens within a single multicopy plasmid can lead to large plasmids which ultimately prove to be genetically unstable, reducing both antigen synthesis and the ensuing immune responses.10 One approach to stable delivery of multiple antigens has been integration of foreign genes into the live vector chromosome, but the decrease in gene dosage again leads to reduced antigen synthesis and lower antigen-specific immunity.11
We hypothesize that delivery of sufficiently immunogenic levels of chromosomally encoded antigens can be accomplished through strategic integration of identical gene cassettes into multiple locations within the live vector chromosome, thereby compensating for loss of copy number afforded by stable low copy plasmids while avoiding further attenuation of the vaccine strain. Integration of multiple identical cassettes avoids the use of strong constitutive promoters to enhance antigen synthesis from a single gene copy, an approach which does not necessarily lead to adequate antigen synthesis or immune responses.12,13 In this report, we employ a synthetic codon-optimized gene cassette encoding the green fluorescent protein GFPuv as an easily quantifiable model antigen to explore expression levels after single integrations into chromosomal sites encoding synthesis of guanine nucleotide (guaBA), a serine protease expressed under environmental stress (htrA) or a cryptic hemolysin naturally found in S. Typhi (clyA). Using flow cytometry to measure GFPuv-mediated fluorescence within individual bacteria of a growing population, we compare fluorescence levels from single integrations, compared with multiple chromosomal integrations within a single vaccine strain and further examine how fluorescence varies with growth rate.
Results
Construction of CVD 910
We have previously constructed the attenuated vaccine candidate, CVD 908-htrA, derived from Ty2 and carrying deletions in aroC, aroD and htrA, which proved to be safe and highly immunogenic in Phase 2 clinical trials.14 Here, we have constructed a new vaccine strain, CVD 910, carrying deletions in guaBA and htrA. We have chosen to replace ΔaroC ΔaroD with the single deletion ΔguaBA for two important reasons: (1) previous work by our group has shown that ΔguaBA alone sufficiently attenuates Ty2, resulting in a live vector strain capable of eliciting impressive humoral immunity to a plasmid-encoded foreign antigen using the murine intranasal model of immunogenicity;15 (2) transcriptional control of the guaBA locus is controlled by growth rate, independent of guanine-mediated repression,16 theoretically allowing expression of properly integrated foreign gene cassettes to be increased as live vectors grow in the host. We constructed deletion cassettes, targeting guaBA and htrA, for use with the λ Red-mediated site-directed mutagenesis method,17 and have used each cassette to successfully delete either guaBA or htrA from wildtype S. Typhi Ty2. Introduction of both deletion mutations into a single strain resulted in the creation of CVD 910. We then performed a preliminary assessment of attenuation of CVD 910 by comparing the minimum lethal dose causing death in 50% of a group of BALB/c mice (LD50) for CVD 910 vs. CVD 908-htrA, using the hog gastric mucin intraperitoneal murine challenge model. For this model, we broadly follow the guidelines recommended in the Code of Federal Regulations for Food and Drugs, Title 21, Part 620.13 (c-d), 1986 for intraperitoneal challenge of mice with S. Typhi. Using this method, we have determined the LD50 for both CVD 910 and CVD 908-htrA to be approximately 5 × 105 CFU (data not shown), vs. an LD50 of ~10 CFU for wildtype Ty2.18
Chromosomal integration of gfpuv cassettes into CVD 910
We took advantage of both the deleted guaBA and htrA chromosomal genes of CVD 910 to express GFPuv from independently controlled cassettes in which the osmotically regulated PompC promoter was genetically fused to a promoterless gfpuv gene. The resulting PompC-gfpuv cassette was integrated into either the guaBA or htrA loci such that only the open reading frame was replaced, but the original promoters for both chromosomal loci were preserved, as depicted schematically in Figure 1. For example, integration of PompC-gfpuv into the guaBA locus to create CVD 910-GG would result in transcription of gfpuv controlled both by osmolarity (via PompC) and growth rate (via PguaBA). Similarly, integration of the same cassette into htrA to create CVD 910-HG would result in synthesis of GFPuv controlled both by osmolarity (PompC) and heat shock/environmental stress (PhtrA).19 In addition, we tested a third chromosomal integration, CVD 910-CG, in which PompC-gfpuv replaced clyA, encoding a cryptic hemolysin from Ty2 whose transcription is normally controlled by low pH.20 Interestingly, when the resulting strains were grown overnight at 37°C in liquid cultures and analyzed for fluorescence by flow cytometry, observed fluorescence intensity was found to be strongly influenced by the site of integration, regardless of osmotic induction of PompC. As shown in the fluorescence histograms of Figure 2, under inducing conditions of 200 mM NaCl, strains with PompC-gfpuv integrated into either guaBA or htrA displayed remarkably uniform bacterial populations with mean fluorescence intensities of 28.65 and 21.59 respectively, while integration into clyA resulted in a very low mean fluorescence intensity of 7.53, barely above the background autofluorescence of 5.94 detected for CVD 910 alone. Having established substantial expression of GFPuv from two independent chromosomal loci, we then tested the hypothesis that integration of PompC-gfpuv into both guaBA and htrA together would result in additive expression of fluorescence. Analysis of fluorescence from the resulting strain, CVD 910–2G, revealed an uninduced (50 mM NaCl) mean fluorescence intensity of 36.01, which increased to 48.21 after induction with 200 mM NaCl. In this experiment, uninduced fluorescence intensities for CVD 910-GG and CVD 910-HG were 25.35 and 15.85 respectively, while induced fluorescence levels were 32.46 and 24.03 respectively. It is immediately evident that for overnight liquid cultures, cumulative fluorescence observed with 2 copies of gfpuv integrated into CVD 910–2G is approximately equivalent to the combined fluorescence levels for individual copies of integrated gfpuv observed in CVD 910-GG and CVD 910-HG, under both uninduced and induced osmotic conditions.

Figure 1. Schematic depiction of the strategy for chromosomal integration of the foreign antigen cassette PompC-gfpuv, encoding the model fluorescent antigen GFPuv. Details of this approach are thoroughly presented in the Materials and Methods. Briefly, an osmotically controlled GFPuv-encoding cassette (tandem white circle and hatched thick arrow) was constructed and linked to an aph marker encoding resistance to kanamycin (shaded thick arrow), flanked by FRT recombination sites (black triangles). The incoming PompC-gfpuv-aph cassette was integrated into the live vector chromosome using the λ Red recombination system, followed by removal of the aph marker using FLP recombinase, to yield the final live vector strain bearing no genes encoding resistance to antibiotics. The bacterial chromosome is represented by 5′-proximal and 3′-terminal darkened rectangles, and the black circle labeled with a “P” represents the wildtype chromosomally encoded promoter of the deleted target open reading frame (i.e either guaBA or htrA).
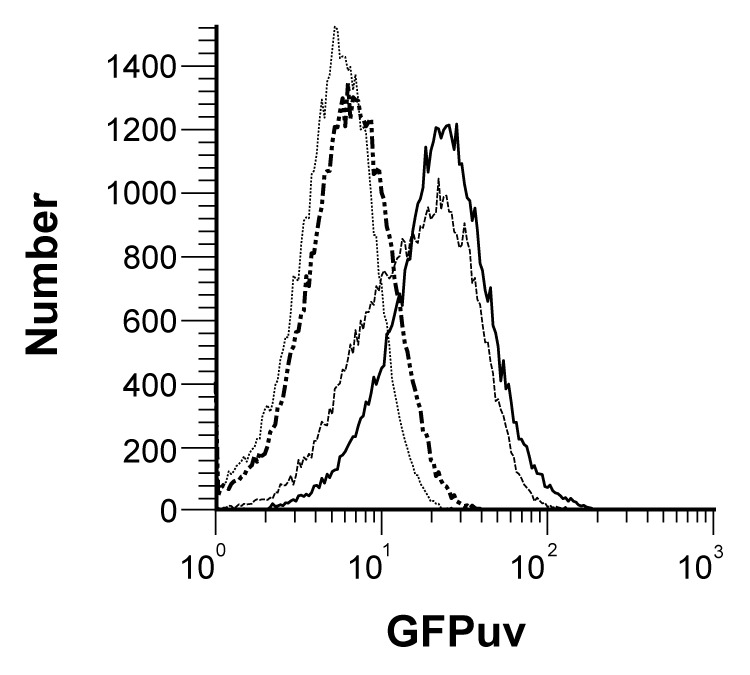
Figure 2. Flow cytometry histograms of GFPuv-mediated fluorescence encoded by PompC-gfpuv gene cassettes integrated into either the guaBA (thick solid line), htrA (thin hatched line) or clyA (thick broken line) sites of the attenuated S. Typhi live vector vaccine candidate CVD 910, compared with the vaccine strain alone (thin dotted line). Fluorescence intensities are measured for individual bacterial cells grown under inducing conditions of 200 mM NaCl in rich medium at 37°C/250 rpm for 16 h.
Growth-phase regulated expression of GFPuv in CVD 910–2G
We have previously hypothesized that regulated but sustained expression of foreign antigens delivered by live vectors will reduce any metabolic burden associated with antigen synthesis, thereby allowing live vectors to persist longer in immunized hosts and prolong delivery of candidate vaccine antigens to the immune system.21 However, we have suspected that despite recent improvements, tightly regulated and appropriately timed antigen expression using plasmid-based expression technologies still remains elusive in many cases, with leaky expression potentially contributing to over-attenuation of live vector vaccine strains. Therefore, one of the goals of the current work was to investigate the feasibility of linking foreign antigen expression to the growth phase of the live vector, such that expression would be reduced when bacteria are adapting to a significant change in environmental conditions (i.e., lag phase), but would be strongly induced after bacteria have successfully adapted their metabolism to new energy sources and environmental conditions (i.e., exponential growth transitioning into stationary phase). We first compared chromosomally encoded GFPuv expression in CVD 910–2G to a previously described live vector CVD 908-htrAssb(pGEN206),3 in which GFPuv is expressed independently of growth phase from a low copy (~5 copies per chromosomal equivalent) stabilized expression plasmid. Overnight starter cultures of CVD 910–2G and CVD 908-htrAssb(pGEN206) were grown at 37°C for approximately 16 h and then diluted 1:100 into 100 ml of fresh medium in 250 ml baffle flasks. To reduce the influence of osmolarity on growth phase and more clearly establish any link between observed fluorescence and induction of PguaBA and PhtrA during growth, all strains were grown under non-inducing conditions of 50 mM NaCl. Fresh cultures were incubated at 37°C/ 250 rpm, and 5 ml aliquots were removed every hour for 7 h to measure both OD600 and fluorescence intensities by flow cytometry. As expected, plasmid-based expression in CVD 908-htrAssb(pGEN206) significantly slowed the growth kinetics of the live vector when compared with either CVD 910 or CVD 910–2G, even under non-inducing conditions of 50mM NaCl (Table 1). Initial fluorescence intensities in lag phase started out quite high at 1262.66, dipped during exponential phase to 686.27, and then rose again to 1131.59 in stationary phase. In sharp contrast, the kinetics of GFPuv expression in CVD 910–2G was closely linked to the growth phase of the culture, with a low mean fluorescence intensity of 81.19 measured in the lag phase, which gradually increased with cell density to a maximum fluorescence intensity of 200.06 as the culture reached stationary phase. We stress here that the observed variation of fluorescence with growth phase, as quantitated by flow cytometry, is not an aggregate effect of increasing cell numbers, but instead reflects the level of GFPuv synthesis within individual bacteria in a growth-rate dependent manner. These data support the feasibility of chromosomal expression of a foreign antigen from multiple integration sites, and the possibility of antigen expression synchronized with growth-rate, a possibility not supported by plasmid-based expression in these experiments.
Table 1. Chromosomal vs. plasmid-based expression of GFPuv in attenuated Salmonella Typhi live vectors.
| Time (hr) | CVD 910 | CVD 910–2G (guaBA::gfpuv htrA::gfpuv) |
CVD 908htrAssb (pGEN206S2) |
|||
|---|---|---|---|---|---|---|
| OD600a | MFI b | OD600 | MFI | OD600 | MFI | |
| 0 | 0.04 | ND c | 0.04 | ND | 0.04 | ND |
| 1 | 0.07 | ND | 0.08 | 81.19 | 0.06 | 1262.66 |
| 2 | 0.27 | ND | 0.3 | 96.77 | 0.14 | 1196.59 |
| 3 | 0.71 | ND | 0.71 | 105.59 | 0.38 | 721.34 |
| 4 | 1.36 | ND | 1.36 | 182.77 | 0.72 | 686.27 |
| 5 | 1.88 | ND | 1.86 | ND | 1.25 | ND |
| 6 | 2.18 | 6.34 | 2.18 | 169.87 | 1.67 | 891.53 |
| 7 | 2.29 | ND | 2.29 | 200.06 | 1.95 | 1131.59 |
We then repeated this experiment to compare GFPuv expression from double integrations in CVD 910–2G to single integration expression levels in CVD 910-GG and CVD 910-HG. As summarized in Table 2, we again observed growth phase-dependent expression of fluorescence intensity, increasing from an initial lag phase level of 38.31 to a high of 161.65 in stationary phase. Interestingly, fluorescence levels during the 3 h lag phase for the double integration did not reflect the sum of fluorescence observed with single integrations during this period, but became additive as the cultures progressed into exponential and stationary phases. Fluorescence intensities from single integrations did not seem to reflect the same dependence on growth phase as observed for the double integration; intensities for the guaBA integration in CVD 910-GG progressed from 74.94 to 96.31 during growth while htrA-controlled fluorescence in CVD 910-HG progressed from 32.90 to 68.94. Despite this anomaly, the data reported here suggest that integration of foreign antigen cassettes into multiple loci within a live vector chromosome can be accomplished without further attenuation of the vaccine strain, and that this multiple integration strategy results in superior expression levels of foreign antigens vs. conventional integration into a single locus.
Table 2. Growth-phase regulated chromosomal expression of GFPuv in CVD 910 attenuated Salmonella Typhi live vectors.
| Time (hr) | CVD 910 | CVD 910-GG (guaBA::gfpuv) |
CVD 910-HG (htrA::gfpuv) |
CVD 910–2G (guaBA::gfpuv htrA::gfpuv) |
||||
|---|---|---|---|---|---|---|---|---|
| OD600a | MFI b | OD600 | MFI | OD600 | MFI | OD600 | MFI | |
| 0 | 0.04 | ND c | 0.04 | ND | 0.03 | ND | 0.02 | ND |
| 1 | 0.09 | ND | 0.09 | 74.94 | 0.06 | 32.9 | 0.06 | 38.31 |
| 2 | 0.33 | ND | 0.3 | 71.03 | 0.24 | 49.08 | 0.24 | 72.53 |
| 3 | 0.81 | ND | 0.72 | 70.58 | 0.68 | 56.12 | 0.6 | 95.41 |
| 4 | 1.45 | ND | 1.31 | 75.26 | 1.29 | 60 | 1.36 | 121.95 |
| 5 | 1.96 | ND | 1.86 | 84.81 | 1.84 | 66.55 | 1.86 | 138.01 |
| 6 | 2.24 | 5.87 | 2.17 | 96.31 | 2.19 | 68.94 | 2.16 | 161.65 |
aCultures grown under non-inducing conditions in 50 mM NaCl; bMean Fluorescence Intensity; cNot Determined
Discussion
Live vectors engineered for delivery of foreign antigens to the host immune system have performed exceedingly well in experimental animal models, but have been only modestly successful in clinical trials.21 Given the advances in the development of powerful plasmid-based expression technologies designed to deliver ample levels of foreign protein, it is unlikely that the lack of antigen-specific immunity observed in clinical trials is due to insufficient antigen synthesis following immunization. To the contrary, we hypothesize that inappropriate antigen synthesis occurring in vivo results in sufficient shock to the metabolism of the live vector to over-attenuate the strain and destroy immunogenicity. Although various attempts have been made to control the timing of foreign protein synthesis, using tightly regulated promoters to control transcription of genes in response to host environmental signals, improved immunogenicity in animals has not translated into improvements in clinical trials.13,22,23 Here we present an alternative to this dilemma, in which we attempt to circumvent over-attenuation by linking antigen synthesis with the growth rate of the live vector vaccine, such that synthesis is initially reduced after immunization, but steadily increases as the vaccine strain adjusts to prevailing environmental conditions and undergoes limited replication within the host.
We are pursuing an expression strategy designed to facilitate efficient expression of multiple foreign antigens within a single live vector vaccine strain, which complements plasmid-based methods by distributing the location of foreign gene cassettes between the chromosome and an expression plasmid. This approach offers the flexibility of independently adjusting the copy number of potentially toxic foreign genes by integrating a designated number of copies into the chromosome. By appropriate integration of foreign genes into chromosomal loci whose induction of expression is intimately associated with the physiology and growth rate of the vaccine strain, it becomes theoretically possible to “tune” foreign antigen synthesis to the metabolic state of the live vector.
To investigate this approach, we examined expression of the model fluorescent antigen GFPuv, and compared plasmid-based expression from a low copy genetically stabilized expression plasmid to stable expression from both the guaBA and htrA sites in the chromosome of an attenuated S. Typhi live vector vaccine strain. High levels of plasmid-encoded fluorescence were observed in live vectors, which adversely impacted the growth rate even under non-inducing conditions. In contrast, gradually increasing levels of fluorescence were observed for chromosomally encoded GFPuv, which correlated well with growth rate and exerted no apparent effect on fitness when compared with the growth of empty live vector CVD 910. As expected, chromosomal expression of GFPuv from two sites was not equivalent to expression from a plasmid with a copy number of five per chromosomal equivalent, but our results clearly suggest that with appropriate integration of additional copies of gfpuv, we may be able to achieve high level expression of a foreign antigen which remains linked to growth rate without further attenuating the live vector. Toward this goal, we have integrated a third copy of gfpuv into the rpoS site (which is naturally inactivated in the parent Ty2 S. Typhi strain used to construct CVD 910); initial results show an additional increase in GFPuv fluorescence which continues to correlate well with growth rate without further attenuation of the vaccine (data not shown). In addition, we have applied these observations to development of a multivalent live vector vaccine against plague, in which one key protective antigen LcrV (an essential type III secretion virulence protein) is integrated into both guaBA and htrA while the remaining protective capsular antigen F1 is expressed from a low copy plasmid. Mouse immunogenicity and challenge experiments are underway to determine if a multivalent live vector vaccine can be successfully engineered using both chromosomal and plasmid-based expression of foreign antigens to confer protection against potentially devastating infections with Yersinia pestis.
Materials and Methods
Bacterial strains and culture conditions
The attenuated S. enterica serovar Typhi live vector vaccine strain CVD 910 used in these studies is an auxotrophic derivative of wild-type strain Ty2, with deletions in guaBA and htrA. To improve the clinical acceptability of our live vector vaccine strains, all genetic and bacteriologic manipulations of the live vectors were performed using an animal product-free medium equivalent to Luria-Bertani medium, comprised of 10 g/liter of Soytone (Teknova; S9052), 5 g/liter Hy-Yest 412 (Sigma; Y1001) and 3 g/liter NaCl (American Bioanalytical; AB01915), supplemented with 0.002% guanine (Sigma; G6779).
Construction of chromosomal integrations
We constructed deletion cassettes for use with the λ Red-mediated site-directed mutagenesis method17 to delete either guaBA, htrA or clyA from wildtype S. Typhi Ty2. Cassettes encoding upstream and downstream flanking chromosomal sequences were constructed using primer pairs listed in Table 3 and purified chromosomal DNA from Ty2 as the template DNA. These cassettes were used to exchange chromosomal targets with a Tn5 neomycin phosphotransferase cassette (aph), encoding resistance to kanamycin and recombined into the chromosome using the λ Red recombination system encoded by pKD46. Final removal of the kanamycin resistance cassette was accomplished using FLP recombinase encoded by pCP20. We confirmed the integrity of the intended chromosomal deletion mutations by DNA sequence analysis of the chromosomal locus from each strain using PCR primers listed in Table 3. For chromosomal expression of GFPuv, we chose to integrate a gene cassette in which an osmotically regulated ompC promoter (PompC ref. 24) was linked to gfpuv and inserted 5′-proximal to the aph resistance marker of chromosomal deletion cassettes. As shown in Figure 1, care was taken to preserve the natural chromosomal promoters controlling transcription of chromosomally encoded targets, with the intent that synthesis of GFPuv would ultimately be controlled both by osmolarity (via PompC) as well as growth rate in the case of the guaBA locus,16 heat shock/environmental stress in the case of the htrA locus,19 or possibly low pH for clyA.20
Table 3. Primers used in the construction and testing of live vector strains expressing chromosomally encoded GFPuv.
| Primer | Sequencea |
|---|---|
| 5guaBA-for | 5′- GAA TTC TAG CTG CTC ATA CTT CTG CTG CA -3′ |
| 5guaBA-rev | 5′- GCT AGC CAA TTG GGG CAA TAT CTC ACC TGG -3′ |
| 3guaBA-for | 5′- GGA TCC ACT AGT GTC GAT AAC CCT TCC TGT GT -3′ |
| 3guaBA-rev | 5′- CTC GAG ACA GCA CCT ACA AGT CTG GCA TG -3′ |
| guaBA PCR-for | 5′- GCG CTG ACC ACC GGA ATA CGG CTG -3′ |
| guaBA PCR-rev | 5′- CAT GGC ATG GAT GAG GCA ACC GCG AAG C -3′ |
| 5htrA-for | 5′- GAA TTC GTA CCT TCA ATC AGG CGT TAC TGG AAG ATG -3′ |
| 5htrA-rev | 5′- GCT AGC CAA TTG CGA TTA ACA GGT AAC GCA AAA TTG CTG TGT ACG TCA G -3′ |
| 3htrA-for | 5′- GGA TCC ACT AGT CTG CGT AAG ATT CTC GAC AGC AAG CCG TCG GT -3′ |
| 3htrA-rev | 5′- CTC GAG CCA GCA TCA TTT CGG CAG TCA TAC ACA CCA GTT CGC -3′ |
| htrA PCR-for | 5′-GTG TCG CCG ATC TTG AAG ACG CGG TAG AG -3′ |
| htrA PCR-rev | 5′- CTA TCG ACG CCA AGC TGG CCG CTG TCG AC -3 |
| 5clyA-for | 5′- TAG TAA TGA GAA TTC GCT GGT ATT GAT CGG CTC TCC GGT AGA GAT TAG CGA -3′ |
| 5clyA-rev | 5′- GCT AGC CAA TTG TGC CTC TTT AAA TAT ATA AAT TGC AAT TAA GTA CCT G -3′ |
| 3clyA-for | 5′- GGA TCC ACT AGT GAT ACA TTT TCA TTC GAT CTG TGT ACT TTT AAC GCC CGA TAG CG -3′ |
| 3clyA-rev | 5′- TGA TAG TAA CTC GAG ACA ATC CAT AAG AAA GGT CAG GCA CAC TGG GAA GGC GAC ATC -3′ |
| clyA PCR-for | 5′- CAT GAT GGT ATC CAG TAT GGC ACA AGC -3′ |
| clyA PCR-rev | 5′- GTA ATC GAC AAC ATG CTA CAT CCA TCG -3′ |
| 5FRT-aph-for | 5′- GAA TTC GCT AGC GCT GGA GCT GCT TCG AAG TTC -3′ |
| 3FRT-aph-rev | 5′- CTC GAG TTC CGG GGA TCC GTC GAC CTG CAG TTC -3′ |
| 5gfpuv | 5′- CAA TTG TGT GGT AGC ACA GAA TAA TGA AAA GT -3′ |
| 3gfpuv | 5′- GCT AGC TCA TTA TTT GTA GAG CTC ATC CAT -3′ |
a Relevant restriction sites are underlined.
Flow cytometry
GFPuv-expressing strains were grown overnight at 37°C on rich solid medium supplemented with guanine. 2–3 fluorescing colonies were then inoculated into 20 ml of supplemented liquid medium and incubated with shaking at 250 rpm overnight at 37°C. Overnight starter cultures were then diluted 1:100 into fresh supplemented liquid medium and incubated at 37°C, 250 rpm. For growth curve studies, 5 ml volumes were periodically removed from incubating cultures, from which bacteria from 4 ml were pelleted, while the remaining 1 ml volume was used to measure the optical density at 600 nm (OD600). Pelleted bacteria were resuspended in 1 ml of PBS, and cells then diluted 1:1,000 in PBS prior flow analysis. Quantitation of GFPuv fluorescence was analyzed using a MoFlo Legacy flow cytometer/cell sorter system (Beckman Coulter) with the argon laser exciting bacteria at 488 nm and emissions detected at 525 nm. Forward vs. side light scatter, measured with logarithmic amplifiers, was used to gate on bacteria. A minimum of 50,000 events were acquired from each sample at a collection rate of approximately 3,500 events per second. The mean fluorescence intensity was determined using Summit software (Beckman Coulter). Background autofluorescence was determined using the negative control S. Typhi vaccine strain CVD 910.
Acknowledgments
A portion of the data from this manuscript was presented at the 6th Annual Vaccine Renaissance Conference of the Institute for Immunology and Informatics in Providence, RI, October 15–17, 2012. This research was supported by grant U01 AI077911 (J.E. Galen, PI).
Glossary
Abbreviations:
- CVD 910-GG
CVD 910guaBA::gfpuv
- CVD 910-HG
CVD 910htrA::gfpuv, CVD 910-2G, CVD 910guaBA::gfpuv htrA::gfpuv
- GFPuv
codon optimized green fluorescent protein
- LD50
50% lethal dose
Submitted
12/04/12
Accepted
12/12/12
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/vaccines/article/23248
References
- 1.Wang S, Li Y, Shi H, Sun W, Roland KL, Curtiss R., 3rd Comparison of a regulated delayed antigen synthesis system with in vivo-inducible promoters for antigen delivery by live attenuated Salmonella vaccines. Infect Immun. 2011;79:937–49. doi: 10.1128/IAI.00445-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galen JE, Levine MM. Can a ‘flawless’ live vector vaccine strain be engineered? Trends Microbiol. 2001;9:372–6. doi: 10.1016/S0966-842X(01)02096-0. [DOI] [PubMed] [Google Scholar]
- 3.Galen JE, Wang JY, Chinchilla M, Vindurampulle C, Vogel JE, Levy H, et al. A new generation of stable, nonantibiotic, low-copy-number plasmids improves immune responses to foreign antigens in Salmonella enterica serovar Typhi live vectors. Infect Immun. 2010;78:337–47. doi: 10.1128/IAI.00916-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cranenburgh RM, Lewis KS, Hanak JA. Effect of plasmid copy number and lac operator sequence on antibiotic-free plasmid selection by operator-repressor titration in Escherichia coli. J Mol Microbiol Biotechnol. 2004;7:197–203. doi: 10.1159/000079828. [DOI] [PubMed] [Google Scholar]
- 5.Nakayama K, Kelley SM, Curtiss R., III Construction of an Asd+ expression-cloning vector: stable maintenance and high level expression of cloned genes in a Salmonella vaccine strain. Bio/Technology. 1988;6:693–7. doi: 10.1038/nbt0688-693. [DOI] [Google Scholar]
- 6.Galen JE, Zhao L, Chinchilla M, Wang JY, Pasetti MF, Green J, et al. Adaptation of the endogenous Salmonella enterica serovar Typhi clyA-encoded hemolysin for antigen export enhances the immunogenicity of anthrax protective antigen domain 4 expressed by the attenuated live-vector vaccine strain CVD 908-htrA. Infect Immun. 2004;72:7096–106. doi: 10.1128/IAI.72.12.7096-7106.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galen JE, Chinchilla M, Pasetti MF, Wang JY, Zhao L, Arciniega-Martinez I, et al. Mucosal immunization with attenuated Salmonella enterica serovar Typhi expressing protective antigen of anthrax toxin (PA83) primes monkeys for accelerated serum antibody responses to parenteral PA83 vaccine. J Infect Dis. 2009;199:326–35. doi: 10.1086/596066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gentschev I, Dietrich G, Goebel W. The E. coli alpha-hemolysin secretion system and its use in vaccine development. Trends Microbiol. 2002;10:39–45. doi: 10.1016/S0966-842X(01)02259-4. [DOI] [PubMed] [Google Scholar]
- 9.Kang HY, Curtiss R., 3rd Immune responses dependent on antigen location in recombinant attenuated Salmonella typhimurium vaccines following oral immunization. FEMS Immunol Med Microbiol. 2003;37:99–104. doi: 10.1016/S0928-8244(03)00063-4. [DOI] [PubMed] [Google Scholar]
- 10.Smith MA, Bidochka MJ. Bacterial fitness and plasmid loss: the importance of culture conditions and plasmid size. Can J Microbiol. 1998;44:351–5. doi: 10.1139/w98-020. [DOI] [PubMed] [Google Scholar]
- 11.Husseiny MI, Hensel M. Evaluation of Salmonella live vaccines with chromosomal expression cassettes for translocated fusion proteins. Vaccine. 2009;27:3780–7. doi: 10.1016/j.vaccine.2009.03.053. [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez C, Hone DM, Noriega FR, Tacket CO, Davis JR, Losonsky G, et al. Salmonella typhi vaccine strain CVD 908 expressing the circumsporozoite protein of Plasmodium falciparum: strain construction and safety and immunogenicity in humans. J Infect Dis. 1994;169:927–31. doi: 10.1093/infdis/169.4.927. [DOI] [PubMed] [Google Scholar]
- 13.Hohmann EL, Oletta CA, Loomis WP, Miller SI. Macrophage-inducible expression of a model antigen in Salmonella typhimurium enhances immunogenicity. Proc Natl Acad Sci USA. 1995;92:2904–8. doi: 10.1073/pnas.92.7.2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tacket CO, Sztein MB, Wasserman SS, Losonsky G, Kotloff KL, Wyant TL, et al. Phase 2 clinical trial of attenuated Salmonella enterica serovar typhi oral live vector vaccine CVD 908-htrA in U.S. volunteers. Infect Immun. 2000;68:1196–201. doi: 10.1128/IAI.68.3.1196-1201.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang JY, Pasetti MF, Noriega FR, Anderson RJ, Wasserman SS, Galen JE, et al. Construction, genotypic and phenotypic characterization, and immunogenicity of attenuated DeltaguaBA Salmonella enterica serovar Typhi strain CVD 915. Infect Immun. 2001;69:4734–41. doi: 10.1128/IAI.69.8.4734-4741.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Husnain SI, Thomas MS. The UP element is necessary but not sufficient for growth rate-dependent control of the Escherichia coli guaB promoter. J Bacteriol. 2008;190:2450–7. doi: 10.1128/JB.01732-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–5. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tacket CO, Hone DM, Curtiss R, 3rd, Kelly SM, Losonsky G, Guers L, et al. Comparison of the safety and immunogenicity of ΔaroC Δ aroD and Δ cya Δ crp Salmonella typhi strains in adult volunteers. Infect Immun. 1992;60:536–41. doi: 10.1128/iai.60.2.536-541.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewis C, Skovierova H, Rowley G, Rezuchova B, Homerova D, Stevenson A, et al. Salmonella enterica Serovar Typhimurium HtrA: regulation of expression and role of the chaperone and protease activities during infection. Microbiology. 2009;155:873–81. doi: 10.1099/mic.0.023754-0. [DOI] [PubMed] [Google Scholar]
- 20.Fuentes JA, Jofré MR, Villagra NA, Mora GC. RpoS- and Crp-dependent transcriptional control of Salmonella Typhi taiA and hlyE genes: role of environmental conditions. Res Microbiol. 2009;160:800–8. doi: 10.1016/j.resmic.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 21.Galen JE, Pasetti MF, Tennant SM, Ruiz-Olvera P, Sztein MB, Levine MM. Salmonella enterica serovar Typhi live vector vaccines finally come of age. Immunol Cell Biol. 2009;87:400–12. doi: 10.1038/icb.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu X, Husseiny MI, Goldwich A, Hensel M. Efficacy of intracellular activated promoters for generation of Salmonella-based vaccines. Infect Immun. 2010;78:4828–38. doi: 10.1128/IAI.00298-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Everest P, Frankel G, Li J, Lund P, Chatfield S, Dougan G. Expression of LacZ from the htrA, nirB and groE promoters in a Salmonella vaccine strain: influence of growth in mammalian cells. FEMS Microbiol Lett. 1995;126:97–101. doi: 10.1111/j.1574-6968.1995.tb07398.x. [DOI] [PubMed] [Google Scholar]
- 24.Galen JE, Nair J, Wang JY, Wasserman SS, Tanner MK, Sztein MB, et al. Optimization of plasmid maintenance in the attenuated live vector vaccine strain Salmonella typhi CVD 908-htrA. Infect Immun. 1999;67:6424–33. doi: 10.1128/iai.67.12.6424-6433.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]