

Abstract
Within the idiosyncratic enzyme active site environment, side chain and ligand pKa values can be profoundly perturbed relative to their values in aqueous solution. Whereas structural inspection of systems has often attributed perturbed pKa values to dominant contributions from placement near to charged groups or within hydrophobic pockets, Tyr57 of a P. putida ketosteroid isomerase (KSI) mutant, suggested to have a pKa perturbed by nearly 4 units to 6.3, is situated within a solvent-exposed active site devoid of cationic side chains, metal ions, or cofactors. Extensive comparisons among 45 variants with mutations in and around the KSI active site, along with protein semi-synthesis, 13C NMR spectroscopy, absorbance spectroscopy, and x-ray crystallography, was used to unravel the basis for this perturbed Tyr pKa. The results suggest that the origin of large energetic perturbations are more complex than suggested by visual inspection. For example, the introduction of positively charged residues near Tyr57 raises its pKa rather than lowers it; this effect, and part of the increase in the Tyr pKa from introduction of nearby anionic groups arise from accompanying active site structural rearrangements. Other mutations with large effects also cause structural perturbations or appear to displace a structured water molecule that is part of a stabilizing hydrogen bond network. Our results lead to a model in which three hydrogen bonds are donated to the stabilized ionized Tyr, with these hydrogen bond donors, two Tyr side chains and a water molecule, positioned by other side chains and by a water-mediated hydrogen bond network. These results support the notion that large energetic effects are often the consequence of multiple stabilizing interactions, rather than a single dominant interaction. Most generally, this work provides a case study for how extensive and comprehensive comparisons via site-directed mutagenesis in a tight feedback loop with structural analysis can greatly facilitate our understanding of enzyme active site energetics. The extensive dataset provided may also be a valuable resource for those wishing to extensively test computational approaches for determining enzymatic pKa values and energetic effects.
The numerous examples of ionizable groups with highly perturbed pKa values within enzyme active sites demonstrate that proteins can manipulate local environments, a property that is likely integral to the catalytic power of enzymes [e.g., refs. (1-18)]. Understanding the physical basis for perturbed pKa values in enzymes has been a longstanding goal in the study of enzyme action and in the computational prediction and design of new enzymes and ligand-binding interactions [e.g., refs. (1, 19-36)].
Structural analysis has shown residues with highly perturbed pKa values buried in hydrophobic pockets or positioned near charged residues, cofactors, or substrates, and a role for these surrounding groups is supported in several instances by effects of mutations on pKa values [e.g., refs. (1, 6, 7, 10, 11, 36-42)]. Nevertheless, energetics within protein interiors and active sites are complex (30, 43-52).
Surprisingly, a tyrosine residue with an unusually low pKa of 6.3 has been identified in the active site of a mutant of bacterial ketosteroid isomerase (KSI) from Pseudomonas putida, a solvent-exposed site devoid of metal ions, cofactors, and cationic amino acids (17, 18). Multiple computational pKa prediction engines predicted that the active site Tyr pKa’s are substantially increased by their environment, not decreased, relative to the solution pKa (17). Herein, we provide strong evidence that the ionizing residue is Tyr57, which has no nearby positive charges and is solvent exposed. Extensive site-directed mutagenesis in conjunction with structural studies provide evidence for stabilization of anionic Tyr57 via three positioned hydrogen bonds, two from neighboring positioned Tyr residues and one from a water molecule engaged in a hydrogen bond network with another water molecule and active site side chains.
EXPERIMENTAL PROCEDURES
Materials
All reagents were of the highest purity commercially available (≥97%). All buffers were prepared with reagent grade materials or better.
KSI mutagenesis, expression, and purification
Quik-Change site directed mutagenesis was used to introduce the mutations into the pKSI and tKSI genes encoded on pKK22-3 plasmids or pET21 plasmids. The mutations were confirmed by sequencing miniprep DNA from DH5α cells. Proteins were expressed and purified as previously described (44, 53).
KSI kinetics
Reactions to evaluate the effect of the R15K/D21N/D24C mutations on KSI activity were performed using 5(10)-estrene-3,17-dione and monitored continuously at 248 nm in a PerkinElmer Lambda 25 spectrophotometer. Reactions were conducted at 25 °C in 40 mM potassium phosphate, pH 7.2, 1 mM sodium EDTA, 2 mM DTT with 2% DMSO added as a cosolvent for substrate solubility. The values of kcat/KM were determined by plotting the values of the observed rate constant (kobs) measured under subsaturating substrate concentrations as a function of enzyme concentration.
Construction of the His-tagged SUMO-D24C-131 KSI plasmid
The sequence encoding residues 24-131 was PCR amplified out of the pKK223-3 plasmid containing KSI using a forward primer containing an AscI site followed by the KSI sequence starting at isoleucine 25 and a reverse primer containing the terminal KSI sequence, a stop codon, and a PacI site. Following digestion with the appropriate restriction enzymes, this PCR product was cloned between the AscI and PacI sites of a vector containing a His6-tag and ubiquitin-like (UBL) protein, SUMO (gift from Aaron Straight). QuikChange site-directed mutagenesis was used to mutate the residue at position 24 to a cysteine, generating a His-tagged SUMO-D24C-131 construct. The product was confirmed by sequencing miniprep DNA from DH5α cells.
Peptide synthesis and purification
A peptide comprising the N-terminal 23 amino acids of KSI with a C-terminal thioester for ligation was synthesized manually on β-mercaptopropionyl-Leu-PAM resin using BOC in situ neutralization protocols (54, 55). The peptide was deprotected using trifluoromethanesulfonic acid and thioanisole (54, 55). The peptide was purified by reverse phase HPLC using a gradient elution between A (water, 0.1% TFA) and B (9:1 acetonitrile/water, 0.09% TFA). Fractions containing the peptide product were pooled and lyophilized. The product mass was confirmed by electrospray mass spectrometry.
Expression and purification of the recombinant fragment containing an N-terminal cysteine 13Cζ-Tyr labeled at Y32, Y57, and Y119
The 13C-Tyr labeled fusion protein with Y32, Y57, and Y119 13Cζ-labeled was expressed in BL21(DE 3) cells grown in M9 minimal media supplemented with L-tyrosine (50 mg/L phenol-4-13C, 95-99%; Cambridge Isotope Laboratories, Inc.) and the remaining nineteen unlabeled amino acids. Cells were grown at 37 °C to an OD of ~0.6, followed by the addition of 0.4 mM IPTG and a further 10 h growth at 25 °C. Cells were harvested and resuspended in 20 mM sodium phosphate, pH 7.2, 150 mM NaCl (lysis buffer) and lysed by passage through a French pressure cell. Inclusion bodies containing the fusion protein were isolated by solubilization of membranes by addition of 1% Triton X-100, 20 mM sodium phosphate, pH 7.2, 150 mM NaCl, followed by centrifugation at 8000g. The inclusion bodies were then washed several times in 20 mM sodium phosphate, pH 7.2, 150 mM NaCl to remove detergent.
Inclusion bodies were resolubilized in 7 M urea, 20 mM sodium phosphate, pH 7.2, 150 mM NaCl. The samples were centrifuged to remove aggregated protein. The supernatant was loaded on a Ni-NTA column pre-equilibrated with 7 M urea, 20 mM sodium phosphate, pH 7.2, 150 mM NaCl. The column was washed with 7 M urea, 20 mM sodium phosphate, pH 7.2, 150 mM NaCl until the A280 dropped to ~0 (~10 column volumes). The product was eluted in one step using 250 mM imidazole, 7 M urea, 20 mM sodium phosphate, pH 7.2, 150 mM NaCl.
The eluted material was diluted at 4 °C by the dropwise addition in 20 mM sodium phosphate, pH 7.2, 150 mM NaCl to final urea concentration of 2 M to allow refolding of the SUMO protein. The material was concentrated using an Amicon centrifugal filter unit and then buffer exchanged by passing through a HiPrep 26/10 (GE Healthcare Life Sciences) desalting column pre-equilibrated with 2 M urea, 50 mM Tris·HCl, pH 8.0, 150 mM NaCl. The purity of the fusion protein was >95%, as determined by SDS-PAGE.
SUMO protease was added to cleave the fusion protein. The cleavage reaction was carried out in 2 M urea, 50 mM Tris·HCl, pH 8.0, 150 mM NaCl, 2 mM DTT at 30 °C for 2 hr. To minimize aggregation of the cleaved products, the concentration of the fusion protein in the cleavage reaction was below 100 μM. Cleavage efficiency was typically >95%, as determined by SDS-PAGE. Following reaction, solid urea was added directly to the mixture to a final concentration of 8 M in the reaction mixture. The mixture was centrifuged to remove aggregated material. Cleavage products were purified by loading the mixture on a Superose-12 gel filtration column pre-equilibrated with 7 M urea, 20 mM sodium phosphate, pH 7.2. Fractions containing the KSI fragment were identified by SDS-PAGE, pooled, concentrated to a final concentration ~2 mM using a 3 kDa cutoff centrifugal filter unit, and stored at 4 °C.
Native chemical ligation
The peptide containing a C-terminal thioester was ligated to the 13Cζ-Tyr labeled recombinant fragment containing an N-terminal cysteine using native chemical ligation (54). The lyophilized peptide was dissolved in 7 M urea, 20 mM sodium phosphate, pH 7.2, to a concentration of ~4 mM. The peptide and recombinant fragments were combined to give final concentrations of ~4 mM and ~2 mM, respectively, in 7 M urea, 20 mM sodium phosphate, pH 7.2. Sodium 4-mercaptophenylacetic acid was added to a final concentration of 1 M (56). The ligation was allowed to proceed for 2 hr at 25 °C. The ligation mixture was then refolded by a 20-fold dilution into 40 mM potassium phosphate, pH 7.2, 1 mM EDTA, 2 mM DTT followed by stirring for 1 hr at 4 °C. The refolded protein was purified by deoxycholate affinity chromatography, followed by buffer exchange into 40 mM potassium phosphate, pH 7.2,1 mM EDTA, 2 mM DTT in a 10 kDa cutoff concentrator. Final purity was >99% on a Coomassie-stained SDS-PAGE gel. Protein concentration was determined using a calculated molar extinction coefficient at 280 nm. The yield relative to the limiting recombinant fragment was ~40%, and 4.2 mg of pure KSI was recovered.
Absorbance spectra
Absorbance spectra of unliganded KSI were acquired in a 150 μL microcuvette with a Perkin-Elmer Lambda 25 absorbance spectrophotometer. The spectra of 10-50 μM enzyme were recorded at 25 °C in 10 mM buffer. The buffers used were the following: sodium acetate, pH 3.7-4.8; sodium phosphate, pH 4.7-9.1; sodium glycine, pH 8.7-11.0; sodium carbonate, pH 8.3-11.2. The ionic strength was held constant at 0.1 M with sodium chloride.
Determining difference absorbance spectra
Absorbance spectra were recorded from 320 to 220 nm at each pH. Difference absorbance spectra were obtained by subtraction of the absorbance spectra at the low pH. The change in extinction coefficient at 300 nm as a function of pH was fit to a titration curve to determine an apparent pKa. The change in extinction coefficient agreed with the expected change for ionization of a single tyrosine (Δε = 2300 M−1 cm−1) (57). Fits using the extinction coefficient change at 244, 290, 295, and 305 nm gave values in good agreement with the value determined using the extinction coefficient change at 300 nm. Reported pKa values are the average of two or more independent determinations. To evaluate if pH-dependent changes in the absorbance spectra independent of tyrosine ionization affect the determined pKa, individual spectra were normalized at 267 or 278 nm, the isosbestic points for tyrosine/tyrosinate, and the change in extinction coefficient determined at 290-305 nm was fit to a titration curve. The results were in good agreement with the values determined by normalizing at 320 nm.
Changes in Trp absorbance (maximum at 280 and 288 nm) from the two Trp residues in KSI could affect the observed absorbance change and limit accurate determination of tyrosyl pKa values and number of tyrosines ionizing (57-59). While differences in Trp absorbance could complicate interpretation of absorbance differences near 290 nm, absorbance differences at 244 nm have been suggested to be unaffected by changes in Trp absorbance (57-59). We therefore compared the change in extinction coefficient at 244 nm and at 295-305 nm to ensure minimal interference from Trp absorbance changes. Tyr pKa values and the number of ionized Tyr residues determined by the change in extinction coefficient at 244 nm were in good agreement with values determined at 295, 300, and 305 nm (±0.2 pKa units; extinction coefficient change consistent with one Tyr ionized), again suggesting that observed absorbance differences corresponded to ionization of a single Tyr and that changes in extinction Trp absorbance do not contribute significantly to the absorbance difference spectra at 295-305 nm.
NMR spectroscopy
13C NMR spectra of KSI were acquired at the Stanford Magnetic Resonance Laboratory on a 600-MHz Varian UNITYINOVA spectrometer running VNMR v6.1C. NMR samples consisted of 0.4-1.0 mM KSI in 40 mM potassium phosphate buffer, pH 7.2, 1 mM EDTA, 2 mM DTT, and 10% (v/v) D2O in a 10 mm Shigemi symmetrical microtube at 25 °C. Data were collected with 39000 points and a 1.0 s recycle delay for 4308-17232 scans and processed using a 10-Hz line broadening. Chemical shifts were referenced externally to a sample of sodium 3-trimethylsilylpropionate-2,2,3,3-d4 (0 ppm) in the same buffer conditions.
KSI x-ray crystallography
Single crystal diffraction data were collected at the SSRL beamline BL9-1 using a wavelength of 0.98 Å (60). The reflections were indexed and integrated and with the programs XDS (61); the intensities were scaled, merged and converted to amplitudes with SCALA and TRUNCATE (62). The phases were derived from the PDB entry 3CPO and refined with REFMAC5 (63, 64). Manual model building was carried out with COOT (65). Crystallographic refinement statistics are given in the Supporting Information.
RESULTS AND DISCUSSION
UNCOVERING A TYR RESIDUE WITH A PERTURBED pKa IN THE KSI ACTIVE SITE
Ketosteroid isomerase (KSI) catalyzes the double bond isomerization in steroid substrates via formation of a dienolate intermediate within an active site composed of an Asp general acid/base, an ‘oxyanion hole’ containing two hydrogen bond donors, a Tyr and Asp residue, a network of adjoining hydrogen-bonding residues, and neighboring hydrophobic residues that participate in substrate binding (Scheme 1) (66-68).
Scheme 1. Mechanism of KSI-Catalyzed Isomerization.
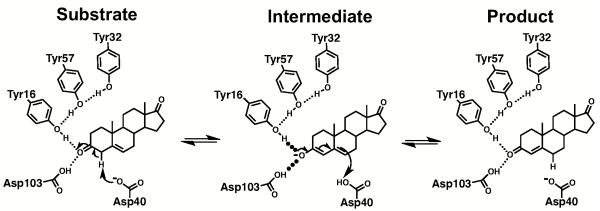
Studying a mutant of this enzyme, with the Asp general base mutated to Asn (Asp40Asn), Fafarman et al. used 13C NMR to identify a Tyr residue with a perturbed pKa and UV absorbance to measure its pKa value of 6.3 ± 0.1 (17). The 13C NMR spectrum of Asp40Asn KSI from Pseudomonas putida (pKSI) containing isotopically enriched Tyr residues showed four peaks at pH 7, with one peak shifted downfield near the chemical shift expected for ionized Tyr (17). In the simplest scenario, the four distinct peaks corresponded to the four tyrosine residues in pKSI: Tyr16, Tyr32, Tyr57, and Tyr119, and the one downfield peak corresponded to one tyrosine ionized at pH 7. 13C NMR experiments using a Tyr119Phe mutant indicated that Tyr119 is not the ionizing residue, as expected as this is a surface residue (17). Of the three remaining tyrosines, results with site-specific vibrational probes could be most simply accounted for if Tyr57 is the ionizing residue (17), but alternative assignments could not be ruled out. We used protein semi-synthesis to generate KSI bearing 13C-labeled Tyr at specific Tyr residues to probe the identity of the ionizing Tyr.
Protein semi-synthesis to probe the identity of the ionizing tyrosine
Three mutations were introduced in KSI to facilitate protein semi-synthesis: Arg15Lys, Asp21Asn, and Asp24Cys. The Arg15Lys mutation allowed the use of trifluoromethanesulfonic acid deprotection of all side chain protecting groups following solid phase peptide synthesis (SPPS); the Asp21Asn mutation eliminated potential aspartimide formation during SPPS; and the Asp24Cys mutation introduced a cysteine residue for native chemical ligation. The combined mutations had less than a two-fold effect on the activity of the WT enzyme (Figure S1 of the Supporting Information) and the 13C NMR spectrum of recombinant 13C Tyr labeled Arg15Lys/Asp21Asn/Asp24Asn/Asp40Asn was nearly identical to the spectrum of 13C Tyr labeled Asp40Asn (Figure S2 of the Supporting Information)(17).
Semi-synthetic KSI Asp40Asn bearing the mutations introduced for protein semi-synthesis was prepared by ligating a synthetic 23mer peptide containing natural abundance Tyr16 to a 108 residue recombinant peptide fragment containing 13Cζ-labeled Tyr57, Tyr32, and Tyr119, and refolding out of urea (Figure 1A). While semi-synthetic Asp40Asn containing 13C labels at all four Tyr residues gave a 13C NMR spectrum identical to that previously observed for fully recombinant Asp40Asn (Figure 1B & Figure S2 of the Supporting Information), semi-synthetic enzyme containing unlabeled Tyr16 results in a 13C NMR spectrum lacking a peak at 160.5 ppm but sill containing the most-downfield peak at 165.1 ppm (Figure 1B). With Tyr16 not 13C labeled and Tyr119 previously assigned as the 157.3 ppm peak, the only remaining options for the peaks at 159.5 ppm and 165.1 ppm are Tyr32 and Tyr57 (17). Tyr32 is in a deep enzyme pocket surrounded by hydrophobic groups, with Tyr57 as the only apparent hydrogen bond donor that could stabilize an anionic Tyr32. In contrast, Tyr57 is situated between two potential hydrogen bond donors, Tyr16 and Tyr32, and is solvent exposed (Figure S3 of Supporting Information). These features are expected to result in much greater stabilization of ionized Tyr57 than ionized Tyr32. Shortened distances between Tyr residues observed from x-ray crystallography are also consistent with deprotonation of the central Tyr57 above pH 7 (See Table S1 of the Supporting Information). We therefore attribute the tyrosine ionization to Tyr57 in unliganded Asp40Asn, consistent with the prediction of Fafarman et al. (17) and then set out to understand the origin of the large and unexpected pKa perturbation. The results obtained provide additional support for Tyr57 as the ionized Tyr residue and we assume this is the case for the remainder of this report.
Figure 1.

13C-NMR spectra of site-specifically labeled semi-synthetic R15K/D21N/D24C/D40N KSI to assign the identity of the ionizing Tyr residue. (A) A synthetic fragment (1-23) bearing unlabeled Tyr16 is ligated to a recombinant fragment (24-131) bearing 13Cζ-Tyr at residues 32, 57, and 119 (*Tyr). (B) The 13C-NMR spectrum for fully recombinant R15K/D21N/D24C/D40N KSI with 13Cζ-Tyr residues and semi-synthetic R15K/D21N/D24C/D40N KSI with unlabeled Tyr16. The spectrum of semi-synthetic KSI lacks the peak at 160.5 ppm allowing assignment of this peak to Tyr16. The peak at 157.3 ppm was previously assigned to Tyr119, and the peak at 165.1 was attributed to Tyr57 as described in the text.
DEVELOPING A MODEL FOR THE STABILIZED TYR57 ANION FROM STRUCTURAL ANALYSIS
To assess potential determinants underlying the low Tyr pKa in pKSI Asp40Asn we first evaluated published KSI x-ray structures. Unliganded wild type KSI (PDB ID 1OPY) shows Tyr57 positioned in a solvent accessible active site, not adjacent to any cationic side chains (Figure 2A). Previously identified cases of stabilized tyrosinates have nearby positive charges (1, 12, 69), but a structural feature or features other than a cationic group must be responsible for this unusually low Tyr pKa. Hydrogen bonds from neutral hydrogen bond donors have been suggested to perturb the pKa values of functional groups in proteins [e.g., refs. (1, 39)]. In particular, the unusually low pKa for aspartic acid in turkey ovomucoid third domain, RNase Sa, RNase T1, and α-lytic protease have been attributed to hydrogen bonds from neutral donors that stabilize the ionized carboxylate relative to its neutral form (pKa values of 2.4, <2.3, 0.6, and <1.5, respectively) (31, 39, 70-72).
Figure 2.

Structural features of the KSI active site. (A) Space filling representation of the KSI monomer shows that Tyr57 (green) is solvent accessible in the unbound wild type active site (PDB ID 8CHO). Red is oxygen, blue is nitrogen, and white is carbon. Tyr57 carbon atoms are shown in green. (B) 2F0-Fc lectron density map for the Met116Ala KSI structure shows density for two ordered water molecules in the active site (contoured at 1.0 σ). (C) Structural model of the active site residues in Met112Ala KSI. (D) Model for hydrogen bond stabilization of the Tyr57 anion in the Asp40Asn mutant that is tested herein. Ionized Tyr57 is red. Crystallographically observed water molecules are colored as in panel B. The figure was generated using Pymol (Schrödinger, LLC).
Consideration of KSI structures leads to a model involving three hydrogen bond donors to the Tyr57 anion (Figure 2). As noted above, Tyr57 has two Tyr residues adjacent to it, Tyr16 and Tyr32. The short O·O distances between Tyr57 and each of these Tyr residues suggests the presence of hydrogen bonds, and these hydrogen bonds appear to shorten upon ionization of Tyr57 (Supporting Table S1). A crystal structure of the Met116Ala mutant of KSI described below shows electron density consistent with two water molecules within the active site (Wat1 and Wat2, Figure 2B). Although Asp40 was present in this structure, it suggests a possible extended hydrogen-bonding network composed of Asp40Asn, Asp103, and a distinct crystallographically observed water molecule in the Asp40Asn mutant (Wat2; Figure 2B, C, D) (73, 74). Further support for the placement of Wat1 and Wat2 come from observation of Wat2 in the wild-type KSI structure (PDB ID 8CHO) and from observation of Wat1 in the Asp40Asn/Asp103Asn structure with bound ligand (PDB ID 3FZW). While the electron density map for the wild type structure is not available to evaluate if multiple water molecules are positioned in the unliganded wild-type KSI active site, the structural data support the model from the Met116Ala mutant of KSI.
Although it was reasonably straightforward to develop this structural model for stabilization of the Tyr57 anion, testing it and understanding its underlying energetics required multiple cycles of site-directed mutagenesis comparisons and structural data. The results described in the following sections with 45 KSI variants support the above model, with its multiple interactions contributing to stabilization of the Tyr57 anion, but also reveal complex energetic and structural effects.
ENERGETIC AND STRUCTURAL ANALYSIS OF MUTATIONAL EFFECTS
UV absorbance spectroscopy to determine Tyr pKa values
While NMR is powerful in identifying and assigning the ionized residue, we could not accurately determine pKa values by this method for two reasons. The NMR spectra exhibit peak broadening at low pH, presumably from exchange, and KSI is less soluble at low pH values, so that obtaining NMR data across an entire titration range is challenging, given the limited sensitivity of NMR. These traits prevented simple determination of chemical shift as a function of pH (17). We therefore turned to UV-Vis spectra as a function of pH to evaluate the effect of mutations on the Tyr pKa, as the tyrosinate ion has an absorbance peak at 295 nm that is absent for neutral tyrosine and not subject to complications from exchange (Figure 3) (17). This assay had the additional advantage of allowing rapid determination with modest amounts of sample, thereby facilitating the extensive analyses involving 45 KSI variants carried out herein.
Figure 3.

Evaluating tyrosine ionization by UV spectroscopy. (A) Absorbance spectra of tyrosine measured in 10 mM sodium acetate, pH 4.7 (red) and 1 M NaOH pH 14 (blue). (B) The difference absorbance spectra for tyrosine (pH 14 - pH 4.7; black) and Asp40Asn KSI (pH 10 - pH 4; green). The difference spectra show maxima for tyrosine at 244 nm (Δε = 1.3 × 104 M−1cm−1) and 295 nm (Δε = 2.8 × 103 M−1cm−1) and for Asp40Asn KSI at 244 nm (Δε = 1.4 × 104 M−1cm−1) and 293 nm (Δε = 2.6 × 103 M−1cm−1). The similar spectra shape and extinction coefficient differences provide support for the ionization of one tyrosine. The dashed line in Figures 4 and 10 represent the expected extinction coefficient change for the ionization of one tyrosine (green).
Figure 3A shows the tyrosine absorbance spectra at pH 4.7 (red line) and pH 14 (blue line), and Figure 3B shows the difference spectrum (black line). At low pH the absorbance maximum is centered near 280 nm, and at high pH ionized tyrosine gives maxima at 244 and 295 nm (57, 75). To determine KSI tyrosine pKa values, we recorded absorbance spectra across a pH series and calculated difference spectra relative to the pH 4 spectrum for that KSI variant. Figure 3B shows an overlay of the absorbance difference spectrum of Asp40Asn KSI (green line). An increase in absorbance with maximums centered near 244 nm and 295 nm is observed with extinction coefficient differences of 1.4 × 104 M−1cm−1 and 2.6 × 103 M−1cm−1, respectively, consistent with ionization of one tyrosine (see also Methods section).
Effect of mutations of the hydrogen bond donors directly adjacent to Tyr57 on the Tyr pKa
We first tested the effect from mutation of Tyr16 and Tyr32, the potential hydrogen bond donors directly adjacent to Tyr57 (Figure 2B, C, D). A simple expectation would be that groups closest to the ionizing hydroxyl group would have the largest pKa effects.
To eliminate potential complications from ionization of the active site Asp103 in the Tyr mutants (see below), we first mutated the Asp103 to Asn; we then determined the effects of the Tyr mutations in the Asp40Asn/Asp103Asn background. The Asp103Asn mutation has only a small effect on the Tyr57 pKa, as described below, and, remarkably, Tyr57 ionizes at a lower pH than Asp103, which resides within 6 Å of Tyr57 and is also exposed to solvent within the active site pocket (3, 73).1 Figure 4 shows the absorbance spectra for Asp40Asn/Asp103Asn as a function of pH (black line). Like the Asp40Asn mutant, a simple titration corresponding to formation of a single tyrosinate is observed with highly perturbed pKa of 7.2 ± 0.2. In contrast, mutation of Tyr57 or of either adjacent Tyr residues increased the pKa such that there was no stabilized tyrosine anion: its pKa is >10 (Figure 4 and Table 1) and thus at least as high as that of Tyr in an unstructured peptide [pKa = 10.2; (77)] (See also Figure S4 of the Supporting Information).
Figure 4.
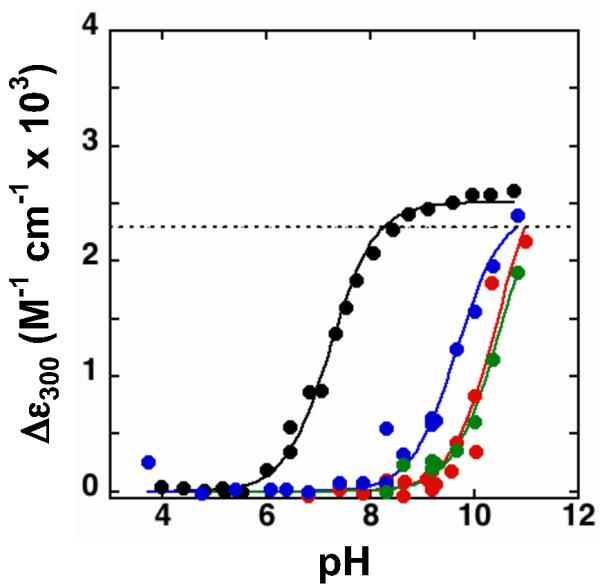
Changes in extinction coefficients at 300 nm relative to pH 4 for D40N/D103N (black), Y16F/D40N/D103N (red), Y32F/D40N/D103N (blue), and Y57F/D40N/D103N (green). Absorbance spectra were collected as described in the Experimental Section. The lines are a fit of the data to a titration curve, and give pKa values of 7.2 for D40N/D103N; >9 for Y32F/D40N/D103N; >10 for Y16F/D40N/D103N; and >10 for Y57F/D40N/D103N.
Table 1.
Effects of Tyr mutations on the Tyr pKa
| Mutant | pKa | ΔpKa |
|---|---|---|
| None | 7.2 | (0) |
| Tyr16Phe | >10 | >3 |
| Tyr32Phe | >9 | >2 |
| Tyr57Phe | >10 | >3 |
| Tyr16Ala | >10 | >3 |
| Tyr32Ala | >10 | >3 |
| Tyr57Ala | >10 | >3 |
None refers to Asp40Asn/Asp103Asn.
One model to account for the complete loss of a pKa perturbation upon mutation of either adjacent Tyr would be hindered solvation of Tyr57 due to the Phe residues replacing the neighboring Tyr residues. However, mutation these Tyr residues to Ala similarly eliminated the highly perturbed pKa value (Figure S5 of the Supporting Information & Table 1). Although it is possible that other properties of the site render it a highly non-aqueous-like environment, Tyr57 is solvent accessible and there is evidence from prior studies of Tyr16 to Ala mutants for an ability of solvent to access the newly created space (44, 45).
A more likely model for the effect of the Tyr16 mutations is conformational rearrangement of Tyr57 upon mutation of Tyr16 to disrupt the hydrogen bonds potentially remaining with Tyr32 and with Wat1. Indeed, a superposition of the previously determined Tyr16Ser/Asp40Asn and Asp40Asn structures shows that while the overall structures are superimposable, with a root-mean square deviation of 0.4 Å for the backbone atoms, the Tyr32 and Tyr57 side chains are displaced by 0.8 and 0.7 Å upon removal of Tyr16 (Figure 5). A rearrangement is also observed in the Tyr16Phe mutant (Figure S6A of the Supporting Information).
Figure 5.
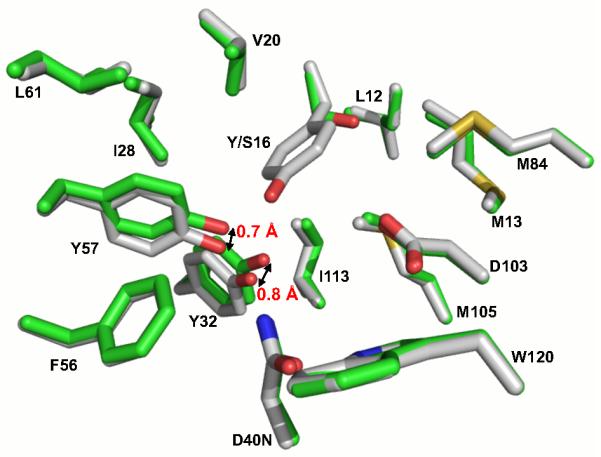
Crystal structure of Tyr16Ser/Asp40Asn shows that Ty32 and Tyr57 are displaced 0.8 and 0.7 Å, respectively, relative to Asp40Asn. Superposition of the previously determined 1.6 Å Tyr16Ser/Asp103Asn structure (PDB ID 3IPT, carbon atoms colored green) and the previously determined 2.0 Å Asp40Asn structure (PDB 1OGX, carbon atoms colored white). The bound equilenin is omitted from both structures for clarity. The overall root-mean-square deviation between the two structures for backbone atoms is 0.4 Å. The pKa comparison in the text uses the Ala mutant, whereas this structural comparison uses the Tyr16Ser version of KSI; prior studies show that the mutation of Tyr16 to Ala or Ser has the same functional effect within two-fold (44).
While a similar rearrangement is not observed in the Tyr32Phe mutant (Figure S6B of the Supporting Information), Tyr32 is buried deep in a hydrophobic pocket that might exclude water. A structure of the Tyr32Ala mutant is not available, where greater water access is expected, but there is evidence for rearrangements in this more severe mutant from its 30-fold rate decrease compared to the only three-fold effect from mutation to Phe [(78) and unpublished results].
Effect of mutations of the distal active site hydrogen bond donors on the Tyr pKa
The model of Figure 2 invokes a hydrogen bond network that includes the side chains at positions 40 and 103 and two water molecules. To learn about the nature and contributions of this presumed network we perturbed the hydrogen-bonding capability of the distal hydrogen-bonding groups Asp103 and Asp40Asn. We first determined the change in apparent Tyr pKa upon mutation of Asp103 to groups that either ablate (Ala, Leu) or perturb (Asn) the hydrogen-bonding ability of residue 103. Measurements were made in the Asp40Asn background, as this latter mutation is required to observe the perturbed Tyr57 pKa, as noted above and further assessed below.
Mutation of Asp103 to Ala, Leu, and Asn increased the apparent Tyr pKa by 0.6, 1.9, 0.9 pKa units, respectively (Figure 6A and Figure 7 of the Supporting Information & Table 2). These results suggest that the distal Asp103 hydrogen bond plays a role in lowering the pKa of Tyr57, although, as expected, its removal does not eliminate the preferential stabilization of the tyrosinate anion in the active site. The larger effect from mutation of Asp103 to Leu rather than Ala is consistent with a perturbation from introduction of the longer hydrophobic side chain (Figure 6B), and an x-ray structure of this mutant reveals that the localized active site water molecule (Wat2, Figure 2B) is displaced by >1 Å (Figure 6B) (73, 78). Wat2 in its normal position may help orient and/or polarize the groups directly donating hydrogen bonds to the Tyr57 anion (Figure 2B, C).
Figure 6.
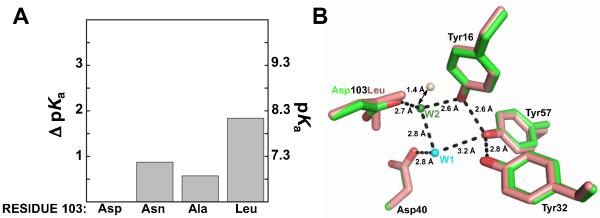
Effects of Asp103 mutations on the Tyr pKa. (A) The ΔpKa values are relative to the pKa value of Asp40Asn. Values are from Table 2. (B) Superposition of structural models of Met116Ala (green, PDB ID 3RGR) and Asp103Leu (salmon, PDB ID 1W00) suggests the position of an organized water molecule in the oxyanion hole is perturbed in Asp103Leu (salmon sphere) compared to wild type (green sphere). Wat1 is not observed in the Asp103Leu structure.
Figure 7.
Effects of Asp40 mutations of on the Tyr pKa The ΔpKa values are relative to the pKa value of Asp40Asn/Asp103Asn and are from Table 3.
Table 2.
Effects of Asp103 mutations on the Tyr pKa
| Residue 103 | pKa | ΔpKa |
|---|---|---|
| Asp | 6.3 | (0) |
| Ala | 6.9 | 0.6 |
| Leu | 8.2 | 1.9 |
| Asn | 7.2 | 0.9 |
Measurements were made in the Asp40Asn background.
The similar effect of mutation of Asp103 to Ala and Asn was surprising, as Asn can maintain hydrogen bonding (Figure 2D). However, the side chain truncation to Ala could allow access of additional water molecules and maintenance of the hydrogen bond from Wat2. Alternatively, mutation of protonated Asp to Asn could disrupt a hydrogen bond network involving Wat2, either by additional geometrical constraints of planarity of the amide hydrogen atoms or from steric effects or solvation requirements of an additional solvent molecule recruited to accept a hydrogen bond from the additional amide hydrogen atom.
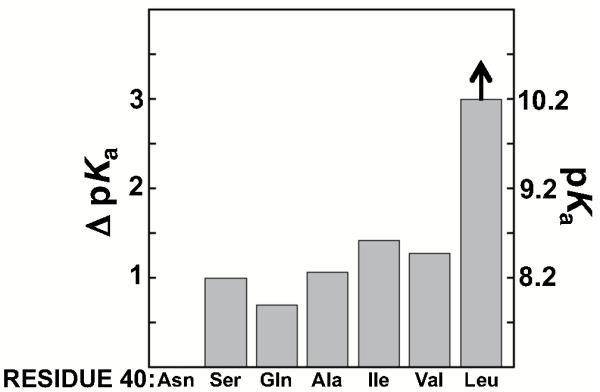
We next turned to the effects of residue 40 on the Tyr pKa. The low Tyr pKa is only observed upon mutating Asp40, the general base in the KSI reaction (Scheme 1); thus, we use Asn at position 40 as our reference point, and we address the Asp40 effects in a later section (“Effects of introducing charged residues at position 40 on the Tyr pKa”). We mutated Asn at position 40 to perturb (Ser, Gln) or ablate (Ala, Leu, Val, Ile) the hydrogen-bonding capability at residue 40, and measurements were made in the Asp103Asn background as above. All position 40 mutations increased the Tyr pKa relative to Asp40Asn. Mutation to Ala, Ser, and Gln increased the apparent Tyr pKa by about one unit, to 7.9-8.3 and mutation to Leu increased the Tyr pKa at least three units, to >10 (Figure 7 and Table 3). A downfield peak was observed in 13C NMR spectra at pH 10 for the Ala, Ser, and Gln mutants, consistent with an ionized Tyr, but was not observed for the Leu mutant, consistent with loss of the tyrosine anion in this mutant (Figure S8 of the Supporting Information). The increased pKa upon mutation to Ser, Gln, or Ala provides evidence for a specific involvement of Asn40 in stabilizing the tyrosinate at position 57 (Figure 2C), and the absence of a direct interaction with Tyr57 and the presence of an intervening positioned water molecule suggest the involvement of Asn40 in the hydrogen bond network depicted in Figure 2.
Table 3.
Effects of Asp40 mutations on the Tyr pKa
| Residue 40 | pKa | ΔpKa |
|---|---|---|
| Asp | 7.2 | (0) |
| Ala | 8.3 | 1.1 |
| Ser | 8.2 | 1.0 |
| Gln | 7.9 | 0.7 |
| Ile | 8.5 | 1.3 |
| Val | 8.5 | 1.3 |
| Leu | >10 | >3 |
Measurements were made in the Asp103Asn background.
The mutations to Leu, Val, and Ile each ablate hydrogen-bonding capability at position 40 and introduce branched, hydrophobic side chains, but the Leu mutant shows a significantly higher Tyr pKa than the others (Figure 7 & Table 3). Modeling Leu in the place of Asn40 shows that the Leu side chain would, in the absence of other rearrangements, clash with Phe56, which neighbors Tyr57, and may thus affect the position of Tyr57 and its ability to be stabilized as an anion. The larger Leu residue could also disrupt the position of one or both of the water molecules that help stabilize the Tyr57 anion (Wat1 & Wat2 in Figure 2C). In either case, removal of the Phe56 side chain might provide space for the Leu side chain and thereby prevent destabilization of the hydrogen bond network. Introduction of the Asp40Leu mutation with Phe56 mutated to Ala increased the Tyr pKa by only 0.6 pKa units, similar to the effect of the smaller branched amino acids at position 40 and much smaller than the >3 unit increase with Phe56 intact (Figure S9 of the Supporting Information & Tables 4, 5). [The Phe56Ala mutation alone had only a small effect on the Tyr pKa (Table S4 of Supporting Information & see below)].
Table 4.
Effects of mutating Asp40 to Leu in the Phe56 background on the Tyr pKa
| Residue mutated | pKa | ΔpKa |
|---|---|---|
| Asp40Asn | 6.3 | (0) |
| Asp40Leu | >10 | >3 |
Table 5.
Effects of mutating Asp40 to Leu in the Phe56Ala background on the Tyr pKa
| Residue mutated | pKa | ΔpKa |
|---|---|---|
| Asp40Asn/Phe56Ala | 6.9 | (0) |
| Asp40Leu/Phe56Ala | 7.5 | 0.6 |
Double mutant cycle to test the properties of the putative active site hydrogen bond network
Despite the absence of direct interaction of the side chains at positions 40 and 103 with Tyr57, the Tyr57 pKa is increased upon mutation of these residues, as described in the previous section. These results, the observed structural perturbation of a localized water molecule in an Asp103 mutant (Figure 6B), and additional results presented below support the model of a hydrogen bond network involving these side chains and the intervening water molecules shown in Figure 2. To learn more about this putative network we next determined the consequences of mutating residues 40 and 103 together.
Whereas the Asp103Ala mutation increased the Tyr pKa by 0.6 units when introduced in the Asp40Asn background, the same mutation in a background with residue 40 mutated to Ala had a 1.2 units larger effect (1.8 pKa unit total effect; Figure 8A and Table 6). These results indicate that there is a functional interaction between the distal-hydrogen bonding groups, and the simplest model from the energetic and structural data is that the Asp40Asn and Asp103 side chains and positioned water molecules participate in a robust hydrogen bond network that helps to stabilize ionized Tyr. The larger than additive effect supports the scenario depicted in Figure 8B in which the side chains are partially redundant such that the remaining side chain and water-mediated interactions are alone able to position the water molecules or Tyr hydrogen bond donors upon ablation of one distal hydrogen-bonding group; however, ablation of both distal hydrogen-bonding groups destabilizes the network sufficiently so that it and its stabilizing effect is abolished. The loss of ~3 kcal/mol stabilization of the tyrosinate upon ablation of the hydrogen-bonding capability at position 40 and 103 provides additional support for a substantial role of the distal hydrogen bonding groups in stabilizing the Tyr57 anion.
Figure 8.

Probing the side chain and water hydrogen bond network that helps stabilize the ionized Tyr. (A) Effects on the Tyr pKa for the single Asp40Ala and Asp103Ala mutations and double Asp40Ala/Asp103Ala mutation. The dashed line represents the expected Tyr pKa if the Ala mutations are additive. Values are from Table 6. (B) Model for the larger than additive effect for mutation of both Asp40 and Asp103 compared to the single mutations. In this model the water molecules substantially rearrange or become fully mobile only after both mutations are introduced.
Table 6.
Effects of mutating Asp40 and Asp103 to Ala on the Tyr pKa
| Residue mutated | pKa | ΔpKa |
|---|---|---|
| None | 6.3 | (0) |
| Asp40 | 7.1 | 0.8 |
| Asp103 | 6.9 | 0.6 |
| Asp40/Asp103 | 8.7 | 2.4 |
None refers to Asp40Asn.
Effect of neighboring group mutations on the Tyr pKa
To further delineate the interactions important for the lowered Tyr pKa we determined the consequence of mutations to residues that surround the hydrogen bond network (Figure 9A). As described above, Phe56 packs against Tyr57 and mutation to Ala had a small effect on the apparent Tyr pKa (Table 7). We also mutated Phe86, Met105, and Met116. Phe86 packs against Asp103, and Met105 and Met116 pack against Tyr16 and Asp103 (Figure 9A). Despite these close interactions, the Phe86Ala and Met105Ala mutations had no measurable effect on the apparent Tyr pKa, and the Met116Ala mutation unexpectedly decreased the apparent the Tyr pKa, though by less than 0.5 units (Figure 9B; Table 7). An overlay of the crystal structures of the Met116Ala mutant (see Table S2 of the Supporting Information for statistics: PDB ID 3RGR) with wild-type KSI shows that the residues engaged in the hydrogen-bonding network are essentially superimposable (Figure 9C). A structure of the homologous KSI from Comamonas testosteroni, tKSI, similarly shows no rearrangement of active Tyr residues upon mutation of the neighbor Phe56 residue (Phe56Gly tKSI, Figure 9D)(79). These results indicate that subtractive mutations of individual surrounding residues are not sufficient to disrupt the positioning of neighboring Tyr hydrogen bond donors, despite the clear need for positioning of these residues to stabilize the Tyr57 anion. In general, important effects that are encoded in redundant structural networks are not revealed by single mutations.
Figure 9.

Effects of neighboring group mutations on the Tyr pKa. (A) The structural model showing the positions of residues neighboring the active site hydrogen bond network that have been mutated. Same model as in Figure 2C with peripheral residues added (orange). (B) The changes in pKa upon mutation of the peripheral residues to Ala relative to Asp40Asn KSI. “-” refers to the Asp40Asn variant. Values are from Table 7. (C) Structural overlays of the Met116Ala mutant (carbon atoms colored green) and wild-type pKSI (carbon atoms colored white). (D) Structural overlay of tKSI Phe56Gly mutant (carbon atoms colored green) with wild-type tKSI (carbon atoms colored white), show that the residues engaged in the hydrogen-bonding network are essentially superimposable. pKSI numbering used for tKSI for simplicity.
Table 7.
Effects of mutating neighboring residue to Ala on the Tyr pKa
| Residue mutated | pKa | ΔpKa |
|---|---|---|
| None | 6.3 | (0) |
| Phe56 | 6.9 | 0.6 |
| Phe86 | 6.3 | 0 |
| Met105 | 6.3 | 0 |
| Met116 | 5.9 | −0.4 |
None refers to Asp40Asn.
Recreating the hydrogen bond network in tKSI
The homologous KSI enzymes, tKSI and pKSI, share 34% overall sequence identity (80). The active site hydrogen bonding residues in tKSI are identical to those in pKSI and their positioning is the same, except that the residue corresponding to Tyr32 is replaced with Phe (Figure 10A; for simplicity pKSI numbering is used throughout)(81). If positioning of the active site residues and water molecules, as depicted in Figure 2, is sufficient for stabilization of the Tyr57 anion in pKSI, then introduction of an analogous Tyr in tKSI should reproduce this positioned network and thus the stabilized Tyr57 anion equivalent.
Figure 10.

Recreating the potential hydrogen bond network in tKSI. (A) Superposition of structural models of the wild-type pKSI (green, PDB ID 1OPY) and wild-type tKSI (grey, PDB ID 8CHO); while the active site residues are the same, the surrounding residues are different. (B) Relative extinction coefficients at 300 nm for pKSI D40N/D103N (black), tKSI D40N/D103N (red), and tKSI F32Y/D40N/D103N (blue) (pKSI numbering used throughout). Absorbance spectra were collected as described in the Experimental Section. The lines are fits of the data to the ionization of one or two Tyr residues, and give Tyr pKa values of 7.2 for pKSI D40N/D103N; >10 for tKSI D40N/D103N; and 7.3 for tKSI F32Y/D40N/D103N. Values are from Table 8.
As expected due to the absence of the second Tyr hydrogen bond donor, no perturbed pKa was observed in Asp40Asn tKSI with the wild-type Phe32 present (Figure 10B). We recreated the potential active site hydrogen-bond arrangement by mutating Phe32 to Tyr, and, to eliminate potential complications from Asp103 ionization, measured the pKa value in the Asp103Asn background as for pKSI above. Remarkably, introduction of Tyr at this position in tKSI gave a pKa indistinguishable from that for pKSI (7.3 ± 0.2 and 7.2 ± 0.2, respectively; Figure 10B & Table 8). These results provide further strong evidence for a role of this Tyr in reducing the pKa of its neighbor. Further, despite differences in the identity of the second shell residues, no significant difference is observed in the Tyr57 pKa, consistent with the observed absence of pKa perturbations from mutation of the surrounding residues in pKSI (Figure 9).
Table 8.
Effects of Tyr mutations on the Tyr pKa in tKSI
| tKSI mutant | pKa | ΔpKa |
|---|---|---|
| D40N D103N | >10 | (0) |
| F32Y D40N D103N | 7.3 | >−3 |
For simplicity, pKSI numbering is used throughout.
USING THE PERTURBED TYR IONIZATION TO EXPLORE THE EFFECTS OF NEARBY CHARGED RESIDUES
Effects of introducing charged residues at position 40 on the Tyr pKa
In the simplest scenario cationic residues at position 40 would decrease the Tyr pKa. Indeed, other Tyr residues with substantially decreased pKa values are found near cationic groups (Table S3 of the Supporting Information) (1, 12, 69, 82, 83).
As noted above, the presence of an anionic Asp residue at position 40 greatly increases the Tyr57 pKa, relative to Asp40Asn (and other neutral residues; Figure 7 and Table 3). However, the converse was not observed. Mutation of residue 40 to positively charged side chains (His, Lys, and Arg in the Asp103Asn background, as described above, to eliminate possible complications from its ionization) increased rather than decreased the Tyr pKa relative to Asp40Asn, with effects of 1.0 - 2.0, pKa units (Figure 11 & Table 9). These results indicate that the presence of nearby positive charge alone is not enough to lower the Tyr57 pKa value in the KSI active site and suggest that distinct factors can be involved, and that the features responsible, which are present in Asp40Asn KSI, are disrupted upon introduction of positively charged residues at position 40.
Figure 11.
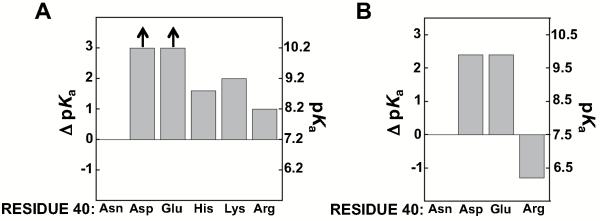
Effect of charged residues at position 40 on the Tyr pKa. (A) Effects of charged residues in the Asn103 background. The ΔpKa values are relative to the pKa value for Asp40Asn/Asp103Asn KSI. Values are from Table 9. (B) Effects of charged mutations in the Phe56Ala/Asp103Asn background. The number on the right at pKa zero represents the pKa for the parent form of KSI used in each comparison. Values are from Table 10.
Table 9.
Effects of charged Asp40 residues on the Tyr pKa in the Asp103Asn background
| Residue 40 | pKa | ΔpKa |
|---|---|---|
| Asn | 7.2 | (0) |
| Asp | >10 | >3 |
| Glu | >10 | >3 |
| Arg | 8.2 | 1.0 |
| His | 8.8 | 1.6 |
| Lys | 9.2 | 2.0 |
The simplest model to account for these unexpected observations is that the positively charged mutations introduce active site structural rearrangements, relative to Asp40Asn, that disrupt the position of the groups stabilizing the Tyr57 anion. A crystal structure of the Asp40His/Asp103Asn in the related tKSI shows that, while the Tyr positions are unaffected, the Phe56 side chain is displaced, relative to tKSI Asp40Asn/Asp103Asn (Figure 12A and Table S2: PDB ID 3MKI). As described above, Phe56 is directly adjacent to Tyr57 and to the crystallographically observed water molecule, Wat2 (Figure 9). We also compared the previously determined wild-type KSI and Asp40Asn structures to evaluate if a negatively charged residue at position 40 might also affect the arrangement of active site residues relative to uncharged Asp40Asn. The structural overlay shows that, while the active site Tyr residues are superimposable, the Phe56 side chain is displaced by 1.9 Å (Figure 12B). Mutations that affect the position of Phe56 may thus affect the positioned water molecule (or the position of Tyr57 is a manner too subtle to ascertain in the x-ray structures) and thereby destabilize the tyrosine anion.
Figure 12.
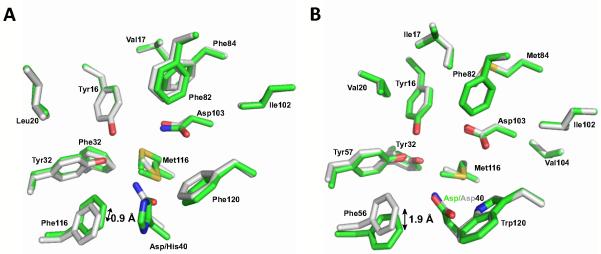
Introduction of charged residues at position 40 cause structural rearrangements of Phe56 relative to Asp40Asn. (A) Superposition of the 2.0 Å Asp40His/Asp103Asn structure determined herein (PDB ID 3MYT, carbon atoms colored green) and the previously determined 2.3 Å Asp40Asn structure (PDB 1QJG, carbon atoms colored white) shows that Phe56 is displaced 0.9 Å upon introduction of the His residue. (B) Superposition of the previously determined structures of the 1.1 Å wild-type KSI structure (PDB ID 1OH0, carbon atoms colored green) and the 2.0 Å Asp40Asn structure (PDB 1OGX, carbon atoms colored white) shows that the Phe56 side chain is displaced 1.9 Å upon changing residue 40 from Asp to Asn. pKSI numbering is used for simplicity.
Effects of charged mutations at residue 40 with the Phe56 side chain removed
Reversion of residue 40 from Asn to Asp in the Phe56 background increased the Tyr pKa by ≥3 units (Figure 11 and Table 9), and a pKa increase of ≥3 units was observed upon introducing anionic Glu in the Phe56 background (Table 9). In the Phe56Ala/Asp103Asn background, the Asp40Arg mutation decreased the Tyr pKa by 1.5 units relative to Asn40Asn (Figure 11 and Table 10), in contrast to the increase observed with Phe56 intact (Figure 11A). Together with the above structural data, these results suggest that rearrangement of Phe56 upon mutation of residue 40 to positively charged residues destabilizes ionized Tyr, obscuring any favorable effect from introducing a nearby positive charge. This residue also appears to rearrange in the presence of Asp40 (Figure 12B). Indeed, reversion of Asn40 to Asp40 in the Phe56Ala/Asp103Asn background gave a smaller destabilizing effect of 2.4 pKa units, suggesting that factors in addition to introduction of a negative charge at residue 40 contribute to the >3 pKa unit increase for reverting Asn40 to Asp40 with Phe56 intact (Figure 11 and Table 9). The additional effect presumably arises from structural rearrangement of Phe56, consistent with effects noted above (“Effects of introducing charged residues at position 40 on the Tyr pKa”).
Table 10.
Effects of charged Asp40 residues on the Tyr pKa in the Phe56Ala/Asp103Asn background
| Residue 40 | pKa | ΔpKa |
|---|---|---|
| Asn | 7.4 | (0) |
| Asp | 9.8 | 2.4 |
| Glu | 9.8 | 2.4 |
| Arg | 5.9 | −1.5 |
Given that addition of the positively charged side chains at position 40 reduced the pKa by ~2 units in the absence of the structurally rearranging Phe56 side chain, we tested whether the remainder of the groups interacting with Tyr57 are used to stabilize the Tyr57 anion when an active site positive charge is present. In other words, would a nearby positive charge alone be sufficient to stabilize ionized Tyr57 in an active site that likely does not suffer from structural rearrangements upon introducing charged groups. To eliminate potential complications from rearrangement of Phe56 and ionization of Asp103, Tyr16 and Tyr32 were individually mutated to Phe or Ala in the Asp40Arg/Phe56Ala/Asp103Ala background. These Tyr mutations eliminated the ionized Tyr (pKa > 10; Table S4 and Figure S10 of the Supporting Information), suggesting that even with this nearby positive charge that can help stabilize the Tyr anion, the adjacent Tyr residues are needed to stabilize the Tyr anion sufficiently to observe it below pH 10. This simplest model to account for these results is that the hydrogen bonds from the neighboring Tyr residues and possibly the positioned active site water molecule (W2) contribute to Tyr anion stabilization such that the positively charged active site residue at position 40 is not fully responsible for the pKa perturbation.
IMPLICATIONS
Structural analysis has allowed us to develop a model for the substantial stabilization of the anionic form of an active site Tyr residue, and extensive mutagenesis coupled with structural analysis has provided strong support for this model (Figure 2). Two neighboring Tyr residues are positioned to donate hydrogen bonds to stabilize anionic Tyr57 and a third hydrogen bond is donated from a water molecule positioned within a hydrogen bond network that involves a second positioned water molecule, two distal side chains, and one of the Tyr hydrogen bond donors. This model accounts for the ability of the KSI active site to lower the pKa of Tyr57 from an expected solution value of 10.2 to 6.3, a stabilization of 5 kcal/mol that occurs without a nearby charged residue.
Whereas mutation of Tyr57 has only a small effect on KSI catalysis (84), and is not present in its anionic form in the normal reaction cycle, our multifaceted dissection of contributions to the 5 kcal/mol stabilization may have revealed properties that are also hallmarks of enzymatic catalysis. These include positioning of residues, networks of interactions, and stabilization (here of an anion and in catalysis of a transition state) via multiple features that contribute in a fundamentally non-additive manner and can thus be difficult to uncover from individual mutations and without complimentary structural information (43, 85-92).
The high tractability of this system also allowed us to explore the effects from the presence nearby charged residues. Whereas introduction of a nearby anion (the wild type general base Asp40 for Asn at this position) gave the expected result of a large increase in the Tyr pKa, the introduction of positively charged side chains at the same position also increased, rather than decreased, the pKa. Structural analysis suggested a possible complication from a nearby Phe residue, Phe56, which alters its position in response to introduction of either negatively or positively charged residues at position 40, leading to overestimation of the destabilizing effect from introduction of an anionic residue and obscuring the stabilizing effect from introduction of a cationic residue (Figure 12). Removal of the Phe56 side chain -and thus its disruptive reorientation-led to the expected simpler behavior: i. Introduction of the anionic Asp40 increased the Tyr57 pKa (less than with Phe56 present) with the Tyr anion still experiencing net stabilization relative to solution due to the stabilizing features that remain surrounding it (Table 10; Figure 11); ii. Introduction of positively charged side chains at position 40 stabilizes the Tyr anion, as expected (Table 10; Figure 11).
Our ability to dissect these energetic contributions highlights the power of extensive mutagenesis carried out in a tight feedback loop with structure. For example, had we just mutated one or the other of the Tyr and saw the pKa revert to >10 we might have concluded, incorrectly, that that a single Tyr was the origin of all of the 5 kcal/mol of stabilization and that this hydrogen bond was especially strong. Had we not had extensive structural information we might never have come upon the model of Figure 2 involving water molecules positioned within a hydrogen bond network. And had we not identified the conformational rearrangement of Phe56 from x-ray structures we might have incorrectly concluded that there was no significant electrostatic stabilization of the Tyr57 anion from introduction of a nearby positive charge (at position 40) in this system (Table 10; Figure 11).
Despite the strong structural and functional evidence for the model of Figure 2, we have not achieved a quantitative understanding of this system. For example, how much of the stabilization arises from the pre-positioning of the neighboring Tyr residues and Wat1, rendering formation of the anion less costly than in solution where conformational rearrangement of the hydrogen bonding waters will be required? How much solvent rearrangement occurs beyond the first shell upon ionization in solution, and how much does the hydrogen-bonded network with Wat1 and Wat2 eliminate or reduce the need for such rearrangement? Are there three or only two hydrogen bonds, on average, from solvent to a tyrosinate anion in solution? Does the stronger hydrogen bond donation ability of the Tyr hydroxyl groups, relative to water (solution pKa values of 10 vs 16) contribute substantially to the stabilization, or are such effects small or negligible in the protein environment?
These quantitative questions pose a grand challenge to computational chemistry. We have presented 45 equilibrium values within a single environment, with substantial support by x-ray crystallographic structures. It is not surprising that the common, off-the-shelf pKa prediction algorithms fail for this system, given the involvement of discrete water molecules (17, 34, 93-95). But even the contributions from the neighboring Tyr residues, in mutants with the water network disrupted, are not accounted for.
We suggest that there is considerably more potential to unravel the energetic underpinnings of such complex systems such as KSI and similarly highly tractable systems, in comparison to systems where only a limited number of measurements can be compared to computation or where multiple measurements are distributed over the entire protein rather than focused in a particular region. We believe that such studies must, as has occurred in the protein folding community (96-99), make predictions -in this case of new pKa values and, ultimately, also of accompanying conformational and/or dynamic changes. We would be pleased to carry out experimental tests after such predictions are made, in order to deepen the connection between experiment and computation and allow true blind tests of emerging methods, and we urge interested computational chemists to contact us.
Supplementary Material
Figure 13.
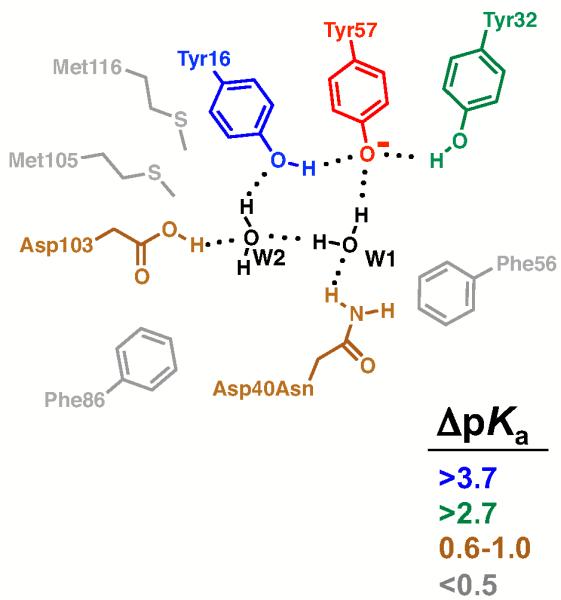
Summary of the effects of Ala mutations on the Tyr57 pKa. Ionized Tyr57 is red, and residues are colored according to the change in the Tyr pKa value upon mutation of the residue to Ala (ΔpKa, relative to Asp40Asn). The effects at residue 40 are for mutation from Asn to other neutral residues; mutation to charged residue can have larger effects (Tables 9-10). Crystallographically observed water molecules are colored black. Mutation of Tyr57 eliminates the observed pKa, as expected (Table 1).
ACKNOWLEDGMENT
We thank Paul Sigala and Aaron Fafarman for helpful discussions; Aaron Straight for the SUMO-fusion protein plasmid; Corey Liu for assistance with NMR experiments; and members of the Herschlag for comments on the manuscript.
Abbreviations
- KSI
ketosteroid isomerase
- pKSI
KSI from Pseudomonas putida
- tKSI
KSI from Comamonas testosteroni
- PDB
Protein Data Bank
- SPPS
solid phase peptide synthesis
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SUMO
small ubiquitin-like modifier
Footnotes
This work was funded by a NSF grant to D.H. (MCB-1121778). J.P.S. was supported in part by a NIH Postdoctoral Fellowship. Portions of this research were carried out at the Stanford Magnetic Resonance Laboratory, which is supported in part by the Stanford University Medical School, and at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences. The project described was partially supported by Grant Number 5 P41 RR001209 from the National Center for Research Resources (NCRR), a component of the NIH.
The solution pKa of the Asp side chain is ~4, whereas that for a Tyr side chain is 10 (76). Despite this difference of over five orders of magnitude in the stability of their respective anions in aqueous solution, Tyr is observed to deprotonate instead of Asp103 (17).
SUPPORTING INFORMATION AVAILABLE. The Supporting Information contains the following figures and tables: Effect of the R15K/D21N/D24C mutations on KSI activity (Figure S1); 13C NMR spectra of recombinant KSI bearing mutations for semi-synthesis (Figure S2); space filling representation of KSI showing the location of Tyr57 and Tyr32 in the active site (Figure S3); 13C NMR spectra of the Tyr mutants (Figure S4); change in extinction coefficient at 300 nm for the Tyr16Ala and Tyr32Ala mutations (Figure S5); superposition of the x-ray structures of the Tyr16Phe and Tyr32Phe mutants with wild-type KSI (Figure S6); change in extinction coefficient at 300 nm for the Asp103 mutations (Figure S7); 13C NMR spectra of the Asp40 mutants (Figure S8); effects of the Asp40Leu mutation on the Tyr pKa in the Phe56 and Phe56Ala backgrounds (Figure S9); change in extinction coefficient at 300 nm for mutation of Tyr16 and Tyr32 to Phe and Ala in the D40R/F56A/D103A background (Figure S10); Tyr O·O distances in crystal structures bearing an expected neutral and anionic Tyr at position 57 (Table S1); crystallographic data and refinement statistics for the Met116Ala and tKSI Asp40His/Asp103Asn structures (Table S2); summary of previously reported Tyr residues with low pKa values (Table S3); summary of effects of mutations on the Tyr pKa reported herein (Table S4). This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Harris TK, Turner GJ. Structural basis of perturbed pKa values of catalytic groups in enzyme active sites. IUBMB Life. 2002;53:85–98. doi: 10.1080/15216540211468. [DOI] [PubMed] [Google Scholar]
- 2.Zscherp C, Schlesinger R, Tittor J, Oesterhelt D, Heberle J. In situ determination of transient pKa changes of internal amino acids of bacteriorhodopsin by using time-resolved attenuated total reflection Fourier-transform infrared spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 1999;96:5498–5503. doi: 10.1073/pnas.96.10.5498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thornburg LD, Henot F, Bash DP, Hawkinson DC, Bartel SD, Pollack RM. Electrophilic assistance by Asp-99 of 3-oxo-Δ5-steroid isomerase. Biochemistry. 1998;37:10499–10506. doi: 10.1021/bi980099a. [DOI] [PubMed] [Google Scholar]
- 4.Stivers JT, Abeygunawardana C, Mildvan AS, Hajipour G, Whitman CP. 4-Oxalocrotonate tautomerase: pH dependence of catalysis and pKa values of active site residues. Biochemistry. 1996;35:814–823. doi: 10.1021/bi9510789. [DOI] [PubMed] [Google Scholar]
- 5.Karp DA, Stahley MR, Garcia-Moreno B. Conformational consequences of ionization of Lys, Asp, and Glu buried at position 66 in staphylococcal nuclease. Biochemistry. 2010;49:4138–4146. doi: 10.1021/bi902114m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang PF, McLeish MJ, Kneen MM, Lee G, Kenyon GL. An unusually low pKa for Cys282 in the active site of human muscle creatine kinase. Biochemistry. 2001;40:11698–11705. doi: 10.1021/bi011208f. [DOI] [PubMed] [Google Scholar]
- 7.Dyson HJ, Jeng MF, Tennant LL, Slaby I, Lindell M, Cui DS, Kuprin S, Holmgren A. Effects of buried charged groups on cysteine thiol ionization and reactivity in Escherichia coli thioredoxin: structural and functional characterization of mutants of Asp 26 and Lys 57. Biochemistry. 1997;36:2622–2636. doi: 10.1021/bi961801a. [DOI] [PubMed] [Google Scholar]
- 8.Oda Y, Yamazaki T, Nagayama K, Kanaya S, Kuroda Y, Nakamura H. Individual ionization constants of all the carboxyl groups in ribonuclease HI from Escherichia coli determined by NMR. Biochemistry. 1994;33:5275–5284. doi: 10.1021/bi00183a034. [DOI] [PubMed] [Google Scholar]
- 9.McIntosh LP, Hand G, Johnson PE, Joshi MD, Korner M, Plesniak LA, Ziser L, Wakarchuk WW, Withers SG. The pKa of the general acid/base carboxyl group of a glycosidase cycles during catalysis: a 13C-NMR study of bacillus circulans xylanase. Biochemistry. 1996;35:9958–9966. doi: 10.1021/bi9613234. [DOI] [PubMed] [Google Scholar]
- 10.Gladysheva T, Liu J, Rosen BP. His-8 lowers the pKa of the essential Cys-12 residue of the ArsC arsenate reductase of plasmid R773. J. Biol. Chem. 1996;271:33256–33260. doi: 10.1074/jbc.271.52.33256. [DOI] [PubMed] [Google Scholar]
- 11.Czerwinski RM, Harris TK, Massiah MA, Mildvan AS, Whitman CP. The structural basis for the perturbed pKa of the catalytic base in 4-oxalocrotonate tautomerase: kinetic and structural effects of mutations of Phe-50. Biochemistry. 2001;40:1984–1995. doi: 10.1021/bi0024714. [DOI] [PubMed] [Google Scholar]
- 12.Sun S, Toney MD. Evidence for a two-base mechanism involving tyrosine-265 from arginine-219 mutants of alanine racemase. Biochemistry. 1999;38:4058–4065. doi: 10.1021/bi982924t. [DOI] [PubMed] [Google Scholar]
- 13.Brown LS, Lanyi JK. Determination of the transiently lowered pKa of the retinal Schiff base during the photocycle of bacteriorhodopsin. Proc. Natl. Acad. Sci. U.S.A. 1996;93:1731–1734. doi: 10.1073/pnas.93.4.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Highbarger LA, Gerlt JA, Kenyon GL. Mechanism of the reaction catalyzed by acetoacetate decarboxylase. Importance of lysine 116 in determining the pKa of active-site lysine 115. Biochemistry. 1996;35:41–46. doi: 10.1021/bi9518306. [DOI] [PubMed] [Google Scholar]
- 15.Kokesh FC, Westheimer FH. A reporter group at the active site of acetoacetate decarboxylase. II. Ionization constant of the amino group. J. Am. Chem. Soc. 1971;93:7270–7274. doi: 10.1021/ja00755a025. [DOI] [PubMed] [Google Scholar]
- 16.Lodi PJ, s JR. Neutral imidazole is the electrophile in the reaction catalyzed by triosephosphate isomerase: structural origins and catalytic implications. Biochemistry. 1991;30:6948–6956. doi: 10.1021/bi00242a020. [DOI] [PubMed] [Google Scholar]
- 17.Fafarman AT, Sigala PA, Schwans JP, Fenn TD, Herschlag D, Boxer SG. Quantitative, directional measurement of electric field heterogeneity in the active site of ketosteroid isomerase. Proc. Natl. Acad. Sci. U.S.A. 2012;109:E299–308. doi: 10.1073/pnas.1111566109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sigala PA, Fafarman AT, Schwans JP, Fried SD, Fenn TD, Caaveiro JM, Pybus B, Ringe D, Petsko GA, Boxer SG, Herschlag D. Quantitative dissection of hydrogen bond-mediated proton transfer in the ketosteroid isomerase active site. Proc. Natl. Acad. Sci. U.S.A. 2013;110:E2552–2561. doi: 10.1073/pnas.1302191110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elcock AH. Prediction of functionally important residues based solely on the computed energetics of protein structure. J. Mol. Biol. 2001;312:885–896. doi: 10.1006/jmbi.2001.5009. [DOI] [PubMed] [Google Scholar]
- 20.Kamerlin SC, Haranczyk M, Warshel A. Progress in ab initio QM/MM free-energy simulations of electrostatic energies in proteins: accelerated QM/MM studies of pKa, redox reactions and solvation free energies. J. Phys. Chem. B. 2009;113:1253–1272. doi: 10.1021/jp8071712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Russell ST, Warshel A. Calculations of electrostatic energies in proteins. The energetics of ionized groups in bovine pancreatic trypsin inhibitor. J. Mol. Biol. 1985;185:389–404. doi: 10.1016/0022-2836(85)90411-5. [DOI] [PubMed] [Google Scholar]
- 22.Warshel A. Calculations of enzymatic reactions: calculations of pKa, proton transfer reactions, and general acid catalysis reactions in enzymes. Biochemistry. 1981;20:3167–3177. doi: 10.1021/bi00514a028. [DOI] [PubMed] [Google Scholar]
- 23.Sandberg L, Edholm O. A fast and simple method to calculate protonation states in proteins. Proteins. 1999;36:474–483. doi: 10.1002/(sici)1097-0134(19990901)36:4<474::aid-prot12>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 24.Nielsen JE, McCammon JA. Calculating pKa values in enzyme active sites. Protein Sci. 2003;12:1894–1901. doi: 10.1110/ps.03114903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nielsen JE, McCammon JA. On the evaluation and optimization of protein X-ray structures for pKa calculations. Protein Sci. 2003;12:313–326. doi: 10.1110/ps.0229903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nielsen JE, Vriend G. Optimizing the hydrogen-bond network in Poisson-Boltzmann equation-based pKa calculations. Proteins. 2001;43:403–412. doi: 10.1002/prot.1053. [DOI] [PubMed] [Google Scholar]
- 27.Mehler EL, Guarnieri F. A self-consistent, microenvironment modulated screened coulomb potential approximation to calculate pH-dependent electrostatic effects in proteins. Biophys. J. 1999;77:3–22. doi: 10.1016/S0006-3495(99)76868-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vizcarra CL, Mayo SL. Electrostatics in computational protein design. Curr. Opin. Chem. Biol. 2005;9:622–626. doi: 10.1016/j.cbpa.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 29.Juffer AH. Theoretical calculations of acid-dissociation constants of proteins. Biochem. Cell Biol. 1998;76:198–209. doi: 10.1139/bcb-76-2-3-198. [DOI] [PubMed] [Google Scholar]
- 30.Antosiewicz J, McCammon JA, Gilson MK. The determinants of pKas in proteins. Biochemistry. 1996;35:7819–7833. doi: 10.1021/bi9601565. [DOI] [PubMed] [Google Scholar]
- 31.Forsyth WR, Antosiewicz JM, Robertson AD. Empirical relationships between protein structure and carboxyl pKa values in proteins. Proteins. 2002;48:388–403. doi: 10.1002/prot.10174. [DOI] [PubMed] [Google Scholar]
- 32.Nakamura H. Roles of electrostatic interaction in proteins. Q. Rev. Biophys. 1996;29:1–90. doi: 10.1017/s0033583500005746. [DOI] [PubMed] [Google Scholar]
- 33.Isom DG, Castaneda CA, Cannon BR, Garcia-Moreno EB. Large shifts in pKa values of lysine residues buried inside a protein. Proc. Natl. Acad. Sci. U.S.A. 2011;108:5260–5265. doi: 10.1073/pnas.1010750108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bas DC, Rogers DM, Jensen JH. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins. 2008;73:765–783. doi: 10.1002/prot.22102. [DOI] [PubMed] [Google Scholar]
- 35.Li H, Robertson AD, Jensen JH. Very fast empirical prediction and rationalization of protein pKa values. Proteins. 2005;61:704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 36.Mehler EL, Fuxreiter M, Simon I, Garcia-Moreno EB. The role of hydrophobic microenvironments in modulating pKa shifts in proteins. Proteins. 2002;48:283–292. doi: 10.1002/prot.10153. [DOI] [PubMed] [Google Scholar]
- 37.Inoue M, Yamada H, Yasukochi T, Kuroki R, Miki T, Horiuchi T, Imoto T. Multiple role of hydrophobicity of tryptophan-108 in chicken lysozyme: structural stability, saccharide binding ability, and abnormal pKa of glutamic acid-35. Biochemistry. 1992;31:5545–5553. doi: 10.1021/bi00139a017. [DOI] [PubMed] [Google Scholar]
- 38.Joshi MD, Sidhu G, Nielsen JE, Brayer GD, Withers SG, McIntosh LP. Dissecting the electrostatic interactions and pH-dependent activity of a family 11 glycosidase. Biochemistry. 2001;40:10115–10139. [PubMed] [Google Scholar]
- 39.Thurlkill RL, Grimsley GR, Scholtz JM, Pace CN. Hydrogen bonding markedly reduces the pK of buried carboxyl groups in proteins. J. Mol. Biol. 2006;362:594–604. doi: 10.1016/j.jmb.2006.07.056. [DOI] [PubMed] [Google Scholar]
- 40.Laurents DV, Huyghues-Despointes BM, Bruix M, Thurlkill RL, Schell D, Newsom S, Grimsley GR, Shaw KL, Trevino S, Rico M, Briggs JM, Antosiewicz JM, Scholtz JM, Pace CN. Charge-charge interactions are key determinants of the pK values of ionizable groups in ribonuclease Sa (pI=3.5) and a basic variant (pI=10.2) J. Mol. Biol. 2003;325:1077–1092. doi: 10.1016/s0022-2836(02)01273-1. [DOI] [PubMed] [Google Scholar]
- 41.Adams J, Johnson K, Matthews R, Benkovic SJ. Effects of distal point-site mutations on the binding and catalysis of dihydrofolate reductase from Escherichia coli. Biochemistry. 1989;28:6611–6618. doi: 10.1021/bi00442a012. [DOI] [PubMed] [Google Scholar]
- 42.Pey AL, Rodriguez-Larrea D, Gavira JA, Garcia-Moreno B, Sanchez-Ruiz JM. Modulation of buried ionizable groups in proteins with engineered surface charge. J. Am. Chem. Soc. 2010;132:1218–1219. doi: 10.1021/ja909298v. [DOI] [PubMed] [Google Scholar]
- 43.Kraut DA, Carroll KS, Herschlag D. Challenges in enzyme mechanism and energetics. Annu. Rev. Biochem. 2003;72:517–571. doi: 10.1146/annurev.biochem.72.121801.161617. [DOI] [PubMed] [Google Scholar]
- 44.Kraut DA, Sigala PA, Fenn TD, Herschlag D. Dissecting the paradoxical effects of hydrogen bond mutations in the ketosteroid isomerase oxyanion hole. Proc. Natl. Acad. Sci. U.S.A. 2010;107:1960–1965. doi: 10.1073/pnas.0911168107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schwans JP, Sunden F, Gonzalez A, Tsai Y, Herschlag D. Evaluating the catalytic contribution from the oxyanion hole in ketosteroid isomerase. J. Am. Chem. Soc. 2011;133:20052–20055. doi: 10.1021/ja208050t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Knowles JR. Tinkering with enzymes: What are we learning? Science. 1987;236:1252–1258. doi: 10.1126/science.3296192. [DOI] [PubMed] [Google Scholar]
- 47.Lim WA, Farruggio DC, Sauer RT. Structural and energetic consequences of disruptive mutations in a protein core. Biochemistry. 1992;31:4324–4333. doi: 10.1021/bi00132a025. [DOI] [PubMed] [Google Scholar]
- 48.Lim WA, Sauer RT. Alternative packing arrangements in the hydrophobic core of lambda repressor. Nature. 1989;339:31–36. doi: 10.1038/339031a0. [DOI] [PubMed] [Google Scholar]
- 49.Baase WA, Liu L, Tronrud DE, Matthews BW. Lessons from the lysozyme of phage T4. Protein Sci. 2010;19:631–641. doi: 10.1002/pro.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dao-pin S, Anderson DE, Baase WA, Dahlquist FW, Matthews BW. Structural and thermodynamic consequences of burying a charged residue within the hydrophobic core of T4 lysozyme. Biochemistry. 1991;30:11521–11529. doi: 10.1021/bi00113a006. [DOI] [PubMed] [Google Scholar]
- 51.Mooers BH, Baase WA, Wray JW, Matthews BW. Contributions of all 20 amino acids at site 96 to the stability and structure of T4 lysozyme. Protein Sci. 2009;18:871–880. doi: 10.1002/pro.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang XJ, Wozniak JA, Matthews BW. Protein flexibility and adaptability seen in 25 crystal forms of T4 lysozyme. J. Mol. Biol. 1995;250:527–552. doi: 10.1006/jmbi.1995.0396. [DOI] [PubMed] [Google Scholar]
- 53.Kraut DA, Sigala PA, Pybus B, Liu CW, Ringe D, Petsko GA, Herschlag D. Testing electrostatic complementarity in enzyme catalysis: hydrogen bonding in the ketosteroid isomerase oxyanion hole. PLoS Biol. 2006;4:501–519. doi: 10.1371/journal.pbio.0040099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kent SB. Chemical synthesis of peptides and proteins. Annu. Rev. Biochem. 1988;57:957–989. doi: 10.1146/annurev.bi.57.070188.004521. [DOI] [PubMed] [Google Scholar]
- 55.Hackeng TM, Griffin JH, Dawson PE. Protein synthesis by native chemical ligation: Expanded scope by using straightforward methodology. Proc. Natl. Acad. Sci. U.S.A. 1999;96:10068–10073. doi: 10.1073/pnas.96.18.10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Johnson EC, Kent SB. Insights into the mechanism and catalysis of the native chemical ligation reaction. J. Am. Chem. Soc. 2006;128:6640–6646. doi: 10.1021/ja058344i. [DOI] [PubMed] [Google Scholar]
- 57.Edelhoch H. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry. 1967;6:1948–1954. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
- 58.Nagel RL, Ranney HM, Kucinskis LL. Tyrosine ionization in human carbon monoxide and deoxyhemoglobins. Biochemistry. 1966;5:1934–1942. doi: 10.1021/bi00870a021. [DOI] [PubMed] [Google Scholar]
- 59.Donovan JW, Laskowski MJ, Scheraga HA. The effects of charged groups on the chromophores of lysozyme and of amino acids. J. Am. Chem. Soc. 1961;83:2686–2694. [Google Scholar]
- 60.Soltis SM, Cohen AE, Deacon A, Eriksson T, Gonzalez A, McPhillips S, Chui H, Dunten P, Hollenbeck M, Mathews I, Miller M, Moorhead P, Phizackerley RP, Smith C, Song J, van dem Bedem H, Ellis P, Kuhn P, McPhillips T, Sauter N, Sharp K, Tsyba I, Wolf G. New paradigm for macromolecular crystallography experiments at SSRL: automated crystal screening and remote data collection. Acta Crystallogr. D Biol. Crystallogr. 2008;64:1210–1221. doi: 10.1107/S0907444908030564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Collaborative Computational Project, N The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 63.Krissinel EB, Winn MD, Ballard CC, Ashton AW, Patel P, Potterton EA, McNicholas SJ, Cowtan KD, Emsley P. The new CCP4 Coordinate Library as a toolkit for the design of coordinate-related applications in protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2250–2255. doi: 10.1107/S0907444904027167. [DOI] [PubMed] [Google Scholar]
- 64.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 65.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 66.Pollack RM. Enzymatic mechanisms for catalysis of enolization: ketosteroid isomerase. Bioorg. Chem. 2004;32:341–353. doi: 10.1016/j.bioorg.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 67.Pollack RM, Thornburg LD, Wu ZR, Summers MF. Mechanistic insights from the three-dimensional structure of 3-oxo-Δ5-steroid isomerase. Arch. Biochem. Biophys. 1999;370:9–15. doi: 10.1006/abbi.1999.1384. [DOI] [PubMed] [Google Scholar]
- 68.Ha NC, Choi G, Choi KY, Oh BH. Structure and enzymology of Δ5-3-ketosteroid isomerase. Curr. Opin. Struct. Biol. 2001;11:674–678. doi: 10.1016/s0959-440x(01)00268-8. [DOI] [PubMed] [Google Scholar]
- 69.Liu Y, Thoden JB, Kim J, Berger E, Gulick AM, Ruzicka FJ, Holden HM, Frey PA. Mechanistic roles of tyrosine 149 and serine 124 in UDP-galactose 4-epimerase from Escherichia coli. Biochemistry. 1997;36:10675–10684. doi: 10.1021/bi970430a. [DOI] [PubMed] [Google Scholar]
- 70.Forsyth WR, Robertson AD. Insensitivity of perturbed carboxyl pKa values in the ovomucoid third domain to charge replacement at a neighboring residue. Biochemistry. 2000;39:8067–8072. doi: 10.1021/bi992967p. [DOI] [PubMed] [Google Scholar]
- 71.Click TH, Kaminski GA. Reproducing basic pKa values for turkey ovomucoid third domain using a polarizable force field. J. Phys. Chem. B. 2009;113:7844–7850. doi: 10.1021/jp809412e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Everill P, Sudmeier JL, Bachovchin WW. Direct NMR Observation and pKa Determination of the Asp(102) Side Chain in a Serine Protease. J. Am. Chem. Soc. 2012;134:2348–2354. doi: 10.1021/ja210091q. [DOI] [PubMed] [Google Scholar]
- 73.Kim SW, Cha SS, Cho HS, Kim JS, Ha NC, Cho MJ, Joo S, Kim KK, Choi KY, Oh BH. High-resolution crystal structures of Δ5-3-ketosteroid isomerase with and without a reaction intermediate analogue. Biochemistry. 1997;36:14030–14036. doi: 10.1021/bi971546+. [DOI] [PubMed] [Google Scholar]
- 74.Sigala PA, Caaveiro JM, Ringe D, Petsko GA, Herschlag D. Hydrogen bond coupling in the ketosteroid isomerase active site. Biochemistry. 2009;48:6932–6939. doi: 10.1021/bi900713j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Crammer JL, Neuberger A. The state of tyrosine in egg albumin and in insulin as determined by spectrophotometric titration. Biochem. J. 1943;37:302–310. doi: 10.1042/bj0370302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grimsley GR, Scholtz JM, Pace CN. A summary of the measured pK values of the ionizable groups in folded proteins. Protein Sci. 2009;18:247–251. doi: 10.1002/pro.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Richarz R, Wuthrich K. C-13 NMR Chemical shifts of the common amino acid residues measured in aqueous solutions of the linear tetrapeptides H-Gly-Gly-X-L-Ala-OH. Biopolymers. 1978;17:2133–2141. [Google Scholar]
- 78.Jang DS, Cha HJ, Cha SS, Hong BH, Ha NC, Lee JY, Oh BH, Lee HS, Choi KY. Structural double-mutant cycle analysis of a hydrogen bond network in ketosteroid isomerase from Pseudomonas putida biotype B. Biochem. J. 2004;382:967–973. doi: 10.1042/BJ20031871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schwans JP, Sunden F, Lassila JK, Gonzalez A, Tsai Y, Herschlag D. Use of anion-aromatic interactions to position the general base in the ketosteroid isomerase active site. Proc. Natl. Acad. Sci. U.S.A. 2013;110:11308–11313. doi: 10.1073/pnas.1206710110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim SW, Choi KY. Identification of active site residues by site-directed mutagenesis of Δ5-3-ketosteroid isomerase from Pseudomonas putida biotype B. J. Bacteriol. 1995;177:2602–2605. doi: 10.1128/jb.177.9.2602-2605.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cho H-S, Choi G, Choi KY, Oh B-H. Crystal structure of Δ5-3-ketosteroid isomerase from Pseudomonas testosteroni. Biochemistry. 1998;37:8325–8330. doi: 10.1021/bi9801614. [DOI] [PubMed] [Google Scholar]
- 82.Gerratana B, Cleland WW, Frey PA. Mechanistic roles of Thr134, Tyr160, and Lys 164 in the reaction catalyzed by dTDP-glucose 4,6-dehydratase. Biochemistry. 2001;40:9187–9195. doi: 10.1021/bi0108249. [DOI] [PubMed] [Google Scholar]
- 83.Pundak S, Roche RS. Tyrosine and tyrosinate fluorescence of bovine testes calmodulin: calcium and pH dependence. Biochemistry. 1984;23:1549–1555. doi: 10.1021/bi00302a032. [DOI] [PubMed] [Google Scholar]
- 84.Kim DH, Jang DS, Nam GH, Choi G, Kim JS, Ha NC, Kim MS, Oh BH, Choi KY. Contribution of the hydrogen-bond network involving a tyrosine triad in the active site to the structure and function of a highly proficient ketosteroid isomerase from Pseudomonas putida biotype B. Biochemistry. 2000;39:4581–4589. doi: 10.1021/bi992119u. [DOI] [PubMed] [Google Scholar]
- 85.Jencks WP. Catalysis in Chemistry and Enzymology. 2 ed. Dover, New York: 1987. [Google Scholar]
- 86.Blow D. So do we understand how enzymes work? Structure Fold. Des. 2000;8:R77–81. doi: 10.1016/s0969-2126(00)00125-8. [DOI] [PubMed] [Google Scholar]
- 87.Narlikar GJ, Herschlag D. Direct demonstration of the catalytic role of binding interactions in an enzymatic reaction. Biochemistry. 1998;37:9902–9911. doi: 10.1021/bi980495t. [DOI] [PubMed] [Google Scholar]
- 88.Wang S, Karbstein K, Peracchi A, Beigelman L, Herschlag D. Identification of the hammerhead ribozyme metal ion binding site responsible for rescue of the deleterious effect of a cleavage site phosphorothioate. Biochemistry. 1999;38:14363–14378. doi: 10.1021/bi9913202. [DOI] [PubMed] [Google Scholar]
- 89.Huang Z, Wagner CR, Benkovic SJ. Nonadditivity of mutational effects at the folate binding site of Escherichia coli dihydrofolate reductase. Biochemistry. 1994;33:11576–11585. doi: 10.1021/bi00204a020. [DOI] [PubMed] [Google Scholar]
- 90.Fersht AR. Structure and Mechanism in Protein Science. 2 ed. W.H. Freeman and Company; New York: 1999. [Google Scholar]
- 91.Horovitz A. Non-additivity in protein-protein interactions. J. Mol. Biol. 1987;196:733–735. doi: 10.1016/0022-2836(87)90045-3. [DOI] [PubMed] [Google Scholar]
- 92.Horovitz A. Double-mutant cycles: a powerful tool for analyzing protein structure and function. Fold. Des. 1996;1:R121–126. doi: 10.1016/S1359-0278(96)00056-9. [DOI] [PubMed] [Google Scholar]
- 93.Gordon JC, Myers JB, Folta T, Shoja V, Heath LS, Onufriev A. H++: a server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005;33:W368–371. doi: 10.1093/nar/gki464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kieseritzky G, Knapp EW. Optimizing pKa computation in proteins with pH adapted conformations. Proteins. 2008;71:1335–1348. doi: 10.1002/prot.21820. [DOI] [PubMed] [Google Scholar]
- 95.Nielsen JE, Gunner MR, Garcia-Moreno BE. The pKa cooperative: a collaborative effort to advance structure-based calculations of pKa values and electrostatic effects in proteins. Proteins. 2011;79:3249–3259. doi: 10.1002/prot.23194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Moretti R, Fleishman SJ, Agius R, Torchala M, Bates PA, Kastritis PL, Rodrigues JP, Trellet M, Bonvin AM, Cui M, Rooman M, Gillis D, Dehouck Y, Moal I, Romero-Durana M, Perez-Cano L, Pallara C, Jimenez B, Fernandez-Recio J, Flores S, Pacella M, Kilambi KP, Gray JJ, Popov P, Grudinin S, Esquivel-Rodriguez J, Kihara D, Zhao N, Korkin D, Zhu X, Demerdash ON, Mitchell JC, Kanamori E, Tsuchiya Y, Nakamura H, Lee H, Park H, Seok C, Sarmiento J, Liang S, Teraguchi S, Standley DM, Shimoyama H, Terashi G, Takeda-Shitaka M, Iwadate M, Umeyama H, Beglov D, Hall DR, Kozakov D, Vajda S, Pierce BG, Hwang H, Vreven T, Weng Z, Huang Y, Li H, Yang X, Ji X, Liu S, Xiao Y, Zacharias M, Qin S, Zhou HX, Huang SY, Zou X, Velankar S, Janin J, Wodak SJ, Baker D. Community-wide evaluation of methods for predicting the effect of mutations on protein-protein interactions. Proteins. 2013 doi: 10.1002/prot.24356. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fleishman SJ, Whitehead TA, Strauch EM, Corn JE, Qin S, Zhou HX, Mitchell JC, Demerdash ON, Takeda-Shitaka M, Terashi G, Moal IH, Li X, Bates PA, Zacharias M, Park H, Ko JS, Lee H, Seok C, Bourquard T, Bernauer J, Poupon A, Aze J, Soner S, Ovali SK, Ozbek P, Tal NB, Haliloglu T, Hwang H, Vreven T, Pierce BG, Weng Z, Perez-Cano L, Pons C, Fernandez-Recio J, Jiang F, Yang F, Gong X, Cao L, Xu X, Liu B, Wang P, Li C, Wang C, Robert CH, Guharoy M, Liu S, Huang Y, Li L, Guo D, Chen Y, Xiao Y, London N, Itzhaki Z, Schueler-Furman O, Inbar Y, Potapov V, Cohen M, Schreiber G, Tsuchiya Y, Kanamori E, Standley DM, Nakamura H, Kinoshita K, Driggers CM, Hall RG, Morgan JL, Hsu VL, Zhan J, Yang Y, Zhou Y, Kastritis PL, Bonvin AM, Zhang W, Camacho CJ, Kilambi KP, Sircar A, Gray JJ, Ohue M, Uchikoga N, Matsuzaki Y, Ishida T, Akiyama Y, Khashan R, Bush S, Fouches D, Tropsha A, Esquivel-Rodriguez J, Kihara D, Stranges PB, Jacak R, Kuhlman B, Huang SY, Zou X, Wodak SJ, Janin J, Baker D. Community-wide assessment of protein-interface modeling suggests improvements to design methodology. J. Mol. Biol. 2011;414:289–302. doi: 10.1016/j.jmb.2011.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Runthala A. Protein structure prediction: challenging targets for CASP10. J. Bio. Struct. & Dynamics. 2012;30:607–615. doi: 10.1080/07391102.2012.687526. [DOI] [PubMed] [Google Scholar]
- 99.Nugent T, Cozzetto D, Jones DT. Evaluation of predictions in the CASP10 model refinement category. Proteins. 2013 doi: 10.1002/prot.24377. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.