

Abstract
Smith-Lemli-Opitz syndrome (SLOS) is an autosomal recessive genetic condition with a broad phenotype that results from deficiency of the final enzyme of the cholesterol synthesis pathway. This defect causes low or low-normal plasma cholesterol levels and increased 7- and 8-dehydrocholesterol (DHC) levels. Many therapies for SLOS and other disorders of sterol metabolism have been proposed, and a few of them have been undertaken in selected patients, but robust prospective clinical trials with validated outcome measures are lacking. We review the current literature and expert opinion on treatments for SLOS and other selected sterol disorders, including dietary cholesterol therapy, statin treatment, bile acid supplementation, medical therapies and surgical interventions, as well as directions for future therapies and treatment research.
Keywords: Smith-Lemli-Opitz Syndrome, sterol disorders, cholesterol supplementation, simvastatin, behavior
Introduction
Smith-Lemli-Opitz syndrome (SLOS) is an autosomal recessive sterol biosynthesis disorder characterized by multiple, variable major and minor malformations and intellectual disability [Smith et al., 1964; Cunniff et al., 1997; Battaile and Steiner, 2000; Porter, 2008]. Children and adults with SLOS typically exhibit low or low-normal plasma cholesterol concentrations along with elevated concentrations of 7-dehydrocholesterol (7DHC) and 8DHC, the immediate precursor of cholesterol and its isomer, respectively [Tint et al., 1994; Batta et al., 1995]. The unique sterol profile in SLOS is the result of deficient activity of 7DHC Δ7-reductase (DHCR7) [Shefer et al., 1995], which converts 7DHC to cholesterol in the last step of the cholesterol biosynthesis pathway (see Fig 1). The most common therapies being studied or applied clinically include dietary cholesterol supplementation and 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (HMG CoA reductase inhibitors, also known as statins). Other supportive therapies are also employed. This article reviews the current evidence and expert opinions on treatment for SLOS and other sterol disorders, with an emphasis on clinical and biochemical perspectives.
Figure 1.
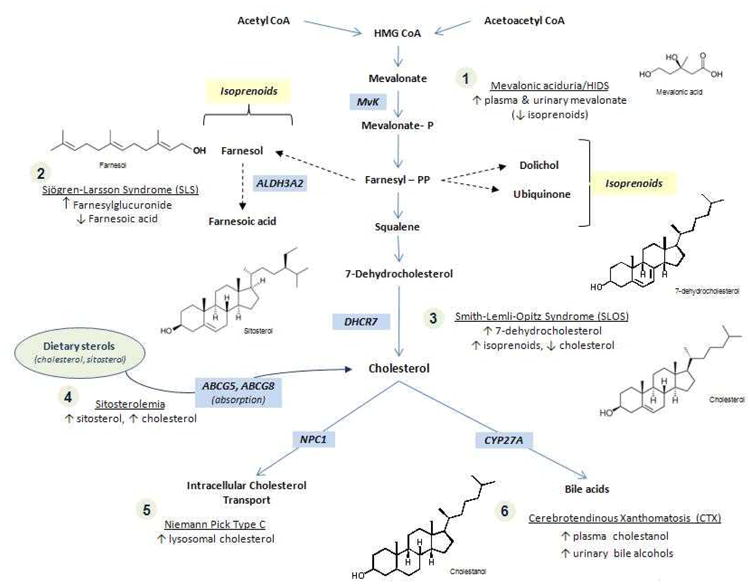
Cholesterol biosynthesis and relevant disease corresponding to enzyme deficiencies.
Biochemical Treatments in SLOS
Both cholesterol deficiency and excess 7DHC and 8DHC likely contribute to the pathogenesis of SLOS. Therefore, the therapeutic goal for the treatment of SLOS has been to enhance cholesterol production and/or accretion, and to decrease the accumulation of potentially toxic cholesterol precursors such as 7DHC and 8DHC. Clinical goals of treatment largely focus on improved development and behaviors such as irritability, hyperactivity, self-injurious behavior and sleep difficulties, but also improved linear growth and weight gain.
Cholesterol supplementation
Individuals with SLOS synthesize significantly less cholesterol than controls (9 vs 20 mg/kg/day, respectively) [Steiner et al., 2000]. The primary therapeutic approach to SLOS treatment has therefore focused on cholesterol supplementation as the most logical treatment choice. Individuals with SLOS also produce appreciable amounts of 7DHC and 8DHC, while control subjects have undetectable levels of these metabolites [Steiner et al., 2000]. Cholesterol supplementation not only increases plasma cholesterol in most individuals with SLOS [Chan et al., 2009; Elias et al., 1997; Linck et al., 2000], but it often decreases 7DHC and 8DHC levels via feedback inhibition of HMG CoA reductase as well [Pappu et al., 2002; Linck et al., 2000].
There are multiple reports of dietary cholesterol treatment for SLOS [Elias et al., 1997; Irons et al., 1997; Linck et al., 2000; Chan et al., 2009]. The most common forms of dietary cholesterol supplementation are egg yolks and/or cholesterol suspension, which is a crystalline or powder form of cholesterol that is either dissolved in oil or aqueous solution or sprinkled on food. There is also a relatively new formulation of cholesterol, SLOesterol™, which is a powdered crystalline form developed specifically as a potential treatment for SLOS. For an individual with SLOS who dislikes the taste or is allergic to egg, this may be a reasonable alternative. Less commonly, increased delivery of dietary cholesterol is attempted by use of a high cholesterol diet, including butter, cream, and meat.
One trial of cholesterol supplementation found a significant increase (164%) in plasma cholesterol levels in patients with SLOS after dietary therapy for 6-15 months [Irons et al., 1997]. This same investigation found no change in 7DHC and 8DHC concentrations over a relatively short time period. Most of these subjects were taking a crystalline form of cholesterol.
Another small trial evaluated the use of egg yolk supplementation for up to 23 months and found a significant decrease in both 7DHC and 8DHC concentrations at 9-23 months [Linck et al., 2000]. Cholesterol concentration increased early on in the study, and these increases persisted at subsequent measurements. In the only study that compared the response to egg yolk cholesterol versus cholesterol suspensions there was a trend toward significant differences in absorption of egg yolk (27%) compared to crystalline cholesterol (21%) [Lin et al., 2005]. Dietary cholesterol usually increases circulating cholesterol concentration, but also decreases sterol synthesis via feedback inhibition [Chan et al., 2009]. This may be one reason why dietary cholesterol supplementation does not universally lead to the desired increased plasma cholesterol concentrations in SLOS.
In addition to variation in the use of natural sources such as egg yolk and preparations such as cholesterol suspension, previous trials have also employed different doses of cholesterol. These have ranged from 20-300 mg/kg/day [DeBarber et al., 2011]. Typical doses are 30 mg/kg/day of egg yolk versus 150-300 mg/kg/day of suspension [DeBarber et al., 2011]. Higher doses of the crystalline form of cholesterol may be required because of decreased absorption [Lin et al., 2005]. Providing cholesterol through either egg yolk or cholesterol suspension raises cholesterol levels and lowers 7DHC and 8DHC levels in many affected individuals [Chan et al., 2009; Tierney et al., 2010]. In one investigation comparing high and low doses of cholesterol in various forms (some crystalline, some food-based), there was a significant decrease in the fractional synthesis rate (FSR) of cholesterol in subjects with moderately high cholesterol intake (27-47 mg/kg/day) compared to those with low intake (0.5-5 mg/kg/day) [Chan et al., 2009]. This suggests that higher cholesterol doses may result in greater feedback inhibition, which may be beneficial in reducing potentially toxic levels of 7DHC and 8DHC. Cholesterol homeostasis involves complex pathways, of which synthesis is only one small part; degradation and metabolism also play an important role in maintenance of the plasma level and overall accretion. Studies are currently underway to assess the balance between cholesterol synthesis, utilization, metabolism, and accretion. The current practice of measuring the plasma cholesterol level may not necessarily reflect tissue cholesterol content.
Although the biochemical goals of increasing cholesterol and decreasing 7DHC and 8DHC are logical, the beneficial effects of cholesterol supplementation on many clinical outcomes are far from established. Improvements in central nervous system (CNS) functioning, particularly regarding behavior and development, are difficult to measure in small, short-term studies. To date, no randomized controlled trials have shown improvement in these features in SLOS. One of the reasons for this lack of demonstrable effect may be that dietary cholesterol, according to currently accepted evidence, does not cross the blood brain barrier (BBB) [Jira et al., 2000; Bjorkhem and Meaney, 2004]. Approximately 25% of total body cholesterol in adults is present in the central nervous system in the form of myelin [Bjorkhem and Meaney, 2004]. Cholesterol in the brain is produced locally and recycled efficiently, with minimal losses to the circulation. The BBB keeps plasma cholesterol levels from affecting the levels in the brain and vice versa [Bjorkhem and Meaney, 2004]. Dietary cholesterol supplementation does not appear to increase cholesterol levels in the cerebrospinal fluid (CSF) in patients with SLOS, and the CSF sterol profile of low cholesterol and high 7DHC and 8DHC is similar to that in the blood [van Rooij et al., 1997]. In another investigation, two patients had no improvement in the CSF sterol [(7DHC + 8DHC)/cholesterol] ratio from dietary cholesterol supplementation alone [Jira et al., 2000]. Some investigators have questioned whether the BBB functions normally in SLOS, and one in vitro investigation found that there may be less adhesion in the BBB in SLOS from the downstream effects of the aberrant sonic hedgehog pathway [Alvarez et al., 2011]. In addition, the CSF sterol profile may not reflect the sterol environment in and around CNS cells. Even if dietary cholesterol is able to make its way to the brain to exert a therapeutic effect, there may be developmental brain abnormalities in some individuals that are fixed and not amenable to such therapy.
Many investigations of the possible benefits of cholesterol supplementation have also focused on improvement in growth and physical function. Early observational studies showed apparent benefits in many non-CNS domains including linear growth, weight gain, pubertal development, gastrointestinal symptoms, and decreased number of infections [Elias et al., 1997; Irons et al., 1994; Nwokoro and Mulvihill, 1997]. However, these studies lacked objective, standardized clinical outcome measures and relied on parent and practitioner impressions. There is some objective evidence however, that cholesterol supplementation may be helpful in ameliorating skin photosensitivity [Azurdia et al., 2001]. Additional observational investigations found improved tone and achievement of ambulation, as well as developmental, cognitive and behavioral changes [Irons et al., 1997; Tierney et al., 2000; Tierney et al., 2001], but these were also hampered by the lack of well-defined clinical outcome measures. To analyze the possible benefit of cholesterol supplementation on symptoms of autism spectrum disorder (ASD), analysis of the prevalence of ASD was examined when cholesterol supplementation was begun at various ages in 2 cohorts. Using the Autism Diagnostic Interview – Revised, cut-offs for ASD were met in 22% of subjects who began supplementation at age 5 years or younger, versus 88% of those who began supplementation after 5 years of age [Tierney et al., 2001]. However, there was an age discrepancy between the 2 groups and the older children may simply have had more time to develop more obvious signs of autism. In spite of its limitations, the differences in outcome in this study may represent a positive effect of early treatment. The only published double blind, placebo-controlled trial of cholesterol supplementation found no measurable reduction of behavioral abnormalities over a period of about 2.5 months [Tierney et al., 2010]. An unblinded longitudinal study also found that six years of egg yolk supplementation did not improve developmental progress in 14 children with SLOS [Sikora et al., 2004]. Randomized controlled trials with standardized cholesterol dosing, laboratory monitoring of sterol levels and well-defined outcome measures are needed to assess more objectively the potential role of cholesterol on improvements in health, development and behavior of patients with SLOS.
Statins
Statins have also been suggested for treatment of SLOS. Simvastatin is a 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor that crosses the BBB. It inhibits the cholesterol pathway proximal to the enzymatic defect in SLOS. In unaffected individuals taking statins, there is reduction in endogenous cholesterol synthesis. Although it might seem counterintuitive to use a cholesterol lowering medication such as simvastatin in a syndrome of cholesterol deficiency, investigators hope that by blocking the cholesterol biosynthesis pathway proximal to the location of the defect in SLOS, the abnormally high levels of potentially toxic 7DHC and 8DHC can be reduced [Jira et al., 2000]. Statins upregulate DHCR7 in animal models and in vitro in human cell lines, which could be therapeutic if a similar upregulation can be achieved in vivo [Correa-Cerro et al., 2006; Wassif et al., 2005]. SLOS fibroblast cell lines with residual DHCR7 enzymatic activity treated with simvastatin increased fractional cholesterol synthesis and decreased 7DHC levels [Wassif et al., 2005]. The seemingly incongruous increase in cholesterol from simvastatin in SLOS might be due in part to sterol regulatory element-binding proteins (SREBPs). SREBP-2 regulates the metabolism of cholesterol, and in an individual with SLOS prescribed simvastatin, SREBP-2 increased the expression of DHCR7 [Porter and Herman, 2011]. Statins also have antioxidant effects that may provide additional benefits [Shishehbor et al., 2003].
One study showed a rise in plasma cholesterol level and a decrease in the 7DHC/cholesterol ratio with use of simvastatin without cholesterol supplementation [Jira et al., 2000]. Improvement in anthropometric measurements, including length, weight, and head circumference were also observed. However, only two patients were included in analysis, and these observations were not reproduced in other studies [Haas et al., 2007; Chan et al., 2009].
Significantly reduced levels of the cholesterol precursors 7DHC and 8DHC were observed in a group of patients treated with cholesterol supplementation and simvastatin compared to a group who received only cholesterol supplementation [Haas et al., 2007]. However, no anthropometric improvements were observed in the simvastatin-treated group. Several subjects showed increased aggression and self-mutilation while on simvastatin that improved once treatment was stopped.
Side effects during statin therapy occur rarely and include rhabdomyolysis and elevated in transaminase levels. Individuals with SLOS receiving simvastatin should be monitored for elevation of plasma creatine kinase (CK), and transaminase levels 6 weeks after initiation of treatment or dosage adjustment, and then every 3-4 months while on therapy. Reversible elevation of CK during statin therapy was noted in one individual, increased transaminase levels and photosensitivity in a second, and sleep disturbance in a third [Haas et al., 2007]. Treatment of an individual with SLOS with statins had to be discontinued due to a marked elevation in transaminase levels in a study reported by Starck et al., [2002]. In summary, statin therapy theoretically provides more benefit for the CNS manifestations of SLOS when compared with cholesterol supplementation alone, but it is not generally recommended for use in SLOS outside of research studies. Other potential areas of investigation to deliver cholesterol to the brain include direct delivery of cholesterol to the brain, partial disruption of the BBB, neural stem cell transplantation into the brain, and development of synthetic sterols that can cross the BBB.
Bile acid and downstream hormone supplementation
Individuals with SLOS can exhibit reduced production of bile acids [Natowicz and Evans, 1994; Honda et al., 1999]; however, mildly affected individuals have normal bile acid synthesis [Steiner et al., 2000]. There is evidence that tauroursodeoxycholic acid (TUDCA), a taurine conjugated bile acid with antioxidant, antiapoptotic and neuroprotective properties crosses the BBB, and is therefore potentially therapeutic [Rodrigues et al., 2002]. However, supplementation with bile acids is not a widely used or generally recommended treatment for SLOS [Nwokoro and Mulvihill, 1997; Irons et al., 1997]. In spite of near total deficiency of normal bile acids in some patients, fat malabsorption and fat-soluble vitamin deficiencies are rare [Kelley and Hennekam, 2000]. Deficiency of adrenal steroid hormones is also rare in SLOS, and in mild to moderately severe cases of SLOS, the hypothalamic-pituitary-adrenal (HPA) axis functions normally; stress steroid supplementation is not required in these cases [Bianconi et al., 2011]. ACTH stimulation testing and stress steroid dosing for illnesses and other stresses should be considered in severe phenotypes only.
Surgical Intervention, Anesthesia, and Supportive Care in SLOS
Craniofacial, cardiovascular, skeletal, urogenital, pulmonary, ophthalmologic, gastrointestinal and other anomalies observed in SLOS frequently require surgical correction under anesthesia. Individuals with SLOS appear to be at increased risk for complications during anesthesia and perioperatively. Micrognathia, palatal anomalies, and prominent incisors can cause difficulties in airway management. In a series of 20 anesthetic exposures in 14 patients, mask airway was reported to be adequate, and fiberoptic intubation of trachea was found to be reliable in airway management [Quezado et al., 2002]. Suctioning of gastric contents and use of strict fasting guidelines can minimize postoperative risks of aspiration from gastroesophageal reflux. Anatomical cardiac and pulmonary defects are additional anesthetic considerations related to the severity of the individual's anomalies. Many children with SLOS have behavioral abnormalities requiring sedation to control aggressive behaviors in the perioperative period. Although children with SLOS may not respond well to sedatives and anesthetic agents [Ryan et al., 1998], most have an uncomplicated anesthetic course [Choi and Nowaczyk, 2000; Quezado et al., 2002].
In spite of reports of hyperthermia and muscle rigidity in some patients with the use of volatile anesthetics, there is insufficient evidence to link SLOS with malignant hyperthermia [Choi and Nowaczyk, 2000]. Thorough preoperative evaluation by an anesthesiologist and meticulous perioperative monitoring are critical to maximize safety.
Structural and functional gastrointestinal problems such as cleft palate, micrognathia, microgastria, pyloric stenosis, malrotation, gastroesophageal reflux and oral motor dysfunction contribute to poor growth in SLOS children. Many affected children require nasogastric tube feedings early in life and eventually gastrostomy tube placement for optimum nutritional support. In less severely affected patients the need for gastrostomy feeding may be temporary. Fundoplication for gastroesophageal reflux is not recommended routinely, but it can be considered if failure to thrive or vomiting are refractory to medical treatment or if reflux complications such as esophagitis, gastrointestinal bleeding, or aspiration are present. Milk protein sensitivity can present with symptoms similar to gastroesophageal reflux, and therefore should be investigated in refractory cases requiring surgery [Kelley and Hennekam, 2000].
Behavioral Modification and Medication
Behavioral modification
Behavioral challenges observed in SLOS include pervasive irritability [Kelley, 1997], hyperactivity, aggressive behavior [Ryan et al., 1998], self-injury [Tint et al., 1994; Opitz, 1999], and behavioral symptoms of autism [Tierney et al., 2001; Sikora et al., 2006]. To date, only the effects of dietary interventions on aberrant behavior have been reported. Case reports based on parental report and clinical observations suggest improved disposition and aberrant behavior in SLOS after cholesterol supplementation [Elias et al., 1997; Martin et al., 2001]. However, emerging research using more rigorous methods suggests cholesterol supplementation does not alter significantly the aberrant behavior [Tierney et al., 2010]; thus, the benefits of this intervention on behavioral abnormalities in SLOS remain unclear. While additional research is needed to understand better the effects of cholesterol supplementation and other medical interventions on aberrant behavior in SLOS, the evaluation of recognized behavioral interventions used in other neurodevelopmental disorders is also warranted. Though not yet evaluated in SLOS specifically, there are many effective interventions for behavioral disturbances associated with such disorders.
Medications for Behavioral Challenges in SLOS
Psychotropic medications, especially atypical antipsychotics, are commonly prescribed pharmacological treatments for many neurodevelopmental disorders, including autism and intellectual disability [Zarcone et al., 2001; McPheeters et al., 2011]. These medications have been used with some effectiveness in SLOS. Certain antipsychotic drugs (specifically, clozapine, chlorpromazine, haloperidol and the antidepressant, imipramine) activate the sterol regulatory element-binding proteins (SREBPs) and the transcription of SREBP-controlled genes such as DHCR7 in mice [Lauth et al., 2010] possible directly effecting endogenous DHCR7 activity. Aberrant behavior remains one of the most pervasive challenges that families of patients with SLOS face, and further studies to understand the effectiveness of medications as well as their alternative mechanisms of action are of utmost importance.
Future Studies in SLOS
While cholesterol supplementation and statin therapy are the most commonly studied treatments for SLOS, additional therapeutic maneuvers are being explored. These include antioxidants, prenatal cholesterol supplementation, and gene therapy.
7DHC can undergo lipid peroxidation and maintain an oxidative free radical chain reaction [Korade et al., 2010]. UV sensitivity, a common phenomenon in SLOS, may be the result of oxidative stress generated by 7DHC or 7DHC derived metabolites [Valencia et al., 2006; Xu et al., 2012]. In the AY9944-induced rat SLOS model, generation of cytotoxic oxysterols from 7DHC oxidation was proposed to contribute to retinal cell death exacerbated by intense light, with antioxidant therapy demonstrating possible ameliorative effects [Vaughan et al., 2006]. Because antioxidants have a reasonably good safety record, they may be promising agents for treatment of oxidative injury in SLOS.
Some of the consequences of the genetic defect in SLOS are established during prenatal development, which has led investigators to explore the feasibility of prenatal treatment. Manipulation of maternal cholesterol mass and maternal to fetal cholesterol transport have the potential to improve outcomes in fetuses affected with SLOS. Fetuses with SLOS are similar to unaffected fetuses in their capacity to extract cholesterol from trophoblasts [Jenkins et al., 2008], and cholesterol supplementation in early pregnancy has potential therapeutic efficacy [Woollett, 2011]. Modulation of maternal to fetal cholesterol transport also has the potential for in utero treatment of SLOS. Abca1, a cholesterol transporter in the placenta, contributes to transport of maternal cholesterol to the developing fetus. In a SLOS mouse model, in utero treatment with TO901317, an LXR-agonist that induces Abca1, increases cholesterol content in Dhcr7−/− embryos [Lindegaard et al., 2008]. In humans, antenatal cholesterol supplementation through fetal intravenous and intraperitoneal transfusions of fresh frozen plasma has been reported [Irons et al., 1999]. The in utero transfusions resulted in increased levels of fetal cholesterol, as measured in blood samples obtained by cordocentesis.
Restoration of normal enzyme activity by gene therapy or modification of gene expression in SLOS has the potential to arrest or potentially reverse many of its symptoms. Watson and colleagues recently described restoration of DHCR7 activity in T93M/Δ mouse (a null mutation with partial deletion of DHCR7 on one allele and a point mutation on the other) liver by treatment with adenovirus-associated viral vector containing human DHCR7 [Matabosch et al., 2010]. Preliminary biochemical analysis showed improvement in the 7DHC to cholesterol ratio. If effective in humans, this approach may offer an alternative to exogenous cholesterol treatment. However, even if this strategy is effective for newborns and older individuals, all the adverse effects of SLOS are unlikely to be eliminated because some are established during prenatal development.
Overview of treatment in other sterol disorders
Cerebrotendinous Xanthomatosis
Cerebrotendinous xanthomatosis (CTX) is a sterol and bile acid synthesis disorder caused by deficiency of mitochondrial sterol 27-hydroxylase. The enzymatic defect results in reduced synthesis of chenodeoxycholic acid (CDCA) from cholesterol and excessive formation and accumulation of cholestanol in plasma and tissues, especially brain and tendon (Fig 1). The bile of an individual with CTX is devoid of CDCA. Typical clinical presentation includes early cataracts and diarrhea, followed by cerebellar and pyramidal signs, intellectual disability, and tendon, CNS white matter and cerebellar xanthomas [Federico and Dotti, 2003].
Based on initial reports of clinical and metabolic improvement after CDCA supplementation [Berginer et al., 1984] clinical trials with CDCA were performed. Currently, oral bile acid supplementation with CDCA is the standard treatment for CTX [Federico and Dotti, 2003]. It provides a source of primary bile acid and restores normal feedback inhibition of endogenous cholesterol and bile acid synthesis leading to reduced production of cholestanol [Salen et al., 1975]. Long-term CDCA therapy normalizes bile acid synthesis, and improves plasma and cerebrospinal fluid levels of cholestanol as well as neurophysiologic parameters [Mondelli et al., 2001; Tokimura et al., 1992]. CNS magnetic resonance imaging abnormalities were found to stabilize after 2-5 years of CDCA therapy [Barkhof et al., 2000], and metabolic and structural improvements in the central nervous system have been demonstrated [Seidel et al., 2011; Selva-O'Callaghan et al., 2011].
Statin therapy was also evaluated in the treatment of CTX. Statins reduce cholesterol synthesis, thereby decreasing the amount of precursors in bile acid synthesis. Statins had little effect in CTX when used alone [Salen et al., 1994], but appear to be effective in reducing cholestanol load when used in combination with CDCA [Nakamura et al., 1991; Kuriyama et al., 1994; Verrips et al., 1999]. Lovastatin did not affect abnormal bile acid synthesis, whereas cholestyramine led to increased cholestanol levels [Batta et al., 2004]. Thus, it does not appear that statins provide added benefit to CDCA supplementation in treatment for CTX.
Low-density lipoprotein (LDL) is a carrier of serum cholestanol. LDL-apheresis can selectively decrease LDL and cholestanol in serum, and therefore has been used to treat a small numbers of individuals with CTX. However, despite reduction after each apheresis, serum cholestanol returns to pre-apheresis levels within 2 weeks. LDL-apheresis in combination with CDCA and statins has been more effective compared to LDL-apheresis alone or pharmacological agents alone [Dotti et al., 2004; Ito et al., 2003]. Demonstration of oxidative stress in the brain of a patient with CTX suggests a therapeutic potential of combination therapy with CDCA and antioxidants [Gonzalez-Cuyar et al., 2007]. Hydrophilic bile acids therapy with ursodeoxycholic acid or ursocholic acid failed to inhibit abnormal bile acid synthesis in patients with CTX. In young children, common side effects of CDCA treatment include diarrhea and liver dysfunction [Yasuhara et al., 1985]. Cholic acid supplementation may be an alternative to CDCA for infants and young children with CTX [Setchell and Heubi, 2006].
In addition to systemic treatments targeting the metabolic defect, patients with CTX may require specific treatments for other clinical consequences of the disease such as large tendon tumors. Caution is advised when contemplating surgical debridement of these tumors as regrowth can occur on a larger scale than originally present. Large tendon tumor excisions may require reconstruction with allografts [Huang et al., 2011]. Current consensus is to avoid surgical manipulation of xanthomas unless absolutely necessary [Salen G, personal communication]. Improvement in oromandibular jaw-opening dystonia has been achieved by botulinum toxin in one patient with CTX [Posada and Ramos, 2011]. Patients with CTX may be more prone to anesthesia and airway difficulty due to tendon xanthomata of the neck and require specific attention to this aspect of anaesthetic care [Habaragamuwa and Bajekal, 2010].
Early diagnosis and treatment of CTX is essential to prevent or slow the progression of neurological symptoms. The development of a newborn screening program for CTX would increase early detection rate and control of this treatable disease [DeBarber et al., 2010].
Mevalonate Kinase Deficiency Syndromes (MKD)
Mevalonate kinase deficiency (MKD) is a term used to describe syndromes due to MVK mutations including the most severe form, mevalonic aciduria (MA), and hyperimmunoglobulinemia D with periodic fever syndrome (HIDS) on the mildest end. MA is autosomal recessive in inheritance and characterized by early onset failure to thrive and neurological symptoms with recurrent febrile episodes. Death in childhood is typical. Hyper-IgD syndrome (HIDS) consists of the same recurrent febrile episodes and immunological concerns, but typically without neurologic involvement and a much more benign course. There is no current treatment approved by the FDA for either of these conditions. Bone marrow transplants have been shown to eliminate the febrile episodes [Neven et al., 2007]. Liver transplantation was performed in a child with mevalonic aciduria who developed severe end-stage liver disease [Chaudhury et al., 2012]. This resulted in improved liver and neurological function. The child required an allogenic hematopoietic stem cell transplant after the liver transplant due to persistent autoinflammatory episodes. Quality of life for this individual was reported to have substantially improved after both transplants. Etanercept, a TNF- α antagonist, and anakinra, a IL-1 receptor antagonist, have shown benefit with regards to decreasing the duration and severity of the fever attacks [Bodar et al., 2011; Takada et al., 2003] Statins have not been helpful for MA and can even exacerbate the condition (Gibson, KM personal communication).
SC4MOL gene mutation (SMO Deficiency)
The most recently discovered sterol disorder is an autosomal recessive syndrome resulting from mutations in the sterol-C4-methyl oxidase–like gene (SC4MOL). This gene encodes a sterol-C4-methyl oxidase (SMO). Clinical features in the affected individuals described to date include psoriaform dermatitis, arthralgias, congenital cataracts, microcephaly, and intellectual disability [He et al., 2011]. There is no disease-specific treatment for SMO deficiency, and treatment is primarily supportive. Topical statins and cholesterol may improve skin findings (Vockley G, personal communication).
Desmosterolosis
Desmosterolosis is an inborn error of cholesterol synthesis due to mutations in the gene that encodes 24-dehydrocholesterol reductase (DHCR24) [FitzPatrick et al., 1998; Andersson et al., 2002; Zolotushko et al., 2011; Schaaf et al., 2011]. No disease-specific treatment is available, and the focus is on symptomatic care, similar to that in SLOS. A study that evaluated the effects of vitamins and cofactors did not find any significant decrease in desmosterol levels using riboflavin, nicotinamide, and thiamine [Schaaf et al., 2011].
No disease specific therapies have been evaluated nor are available for lathosterolosis, X-linked dominant chondrodysplasia puncata, CHILD syndrome or CK-syndrome nor for the disorders of dolichol synthesis or metabolism.
Summary
In spite of significant progress in understanding the effects of altered sterol metabolism, there remains no clearly effective treatment for the most debilitating symptoms of SLOS and other sterol disorders. There is clear evidence that deficient synthesis of cholesterol is the major determinant of clinical severity in SLOS, but there also appear to be deleterious effects from excess 7DHC and 8DHC. Restoration of enzyme activity, during prenatal life if possible, is the optimum treatment strategy, but significant challenges remain in our ability to effect such changes at any time during the life cycle. For the time being therefore, it seems most reasonable to pursue treatments that alter the biochemical abnormalities that are associated with SLOS. Cholesterol supplementation, statin treatment, and similar pharmacologic approaches have each shown some promise for improved outcomes. There is a real need for larger scale intervention trials of SLOS treatment, with rigorous inclusion and exclusion criteria, careful monitoring of medication dose and timing, and highly objective outcome measures. These well-established supportive therapies, combined with developing technologies for treatment of the fundamental genetic defect of SLOS, have promise for improved health and development in affected individuals.
Acknowledgments
The authors thank Mary Lou Oster-Granite for her review of the manuscript. RDS is supported by the NIH Rare Diseases Clinical Research Network (RDCRN) via the Sterol and Isoprenoid Research (STAIR) consortium, HHS grant U54 HD061939, and by HHS grant R01 HL 073980.
Footnotes
The authors have no conflicts of interest to declare.
References
- Alvarez JI, Dodelet-Devillers A, Kebir H, Ifergan I, Fabre PJ, Terouz S, Sabbagh M, Wosik K, Bourbonniere L, Bernard M, van Horssen J, de Vries HE, Charron F, Prat A. The hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science. 2011;334:1727–1731. doi: 10.1126/science.1206936. [DOI] [PubMed] [Google Scholar]
- Andersson HC, Kratz L, Kelley R. Desmosterolosis presenting with multiple congenital anomalies and profound developmental delay. Am J Med Genet. 2002;113:315–319. doi: 10.1002/ajmg.b.10873. [DOI] [PubMed] [Google Scholar]
- Asmus JM, Wacker DP, Harding J, Berg WK, Derby KM, Kocis E. Evaluation of antecedent stimulus parameters for the treatment of escape-maintained aberrant behavior. J Appl Behav Anal. 1999;32:495–513. doi: 10.1901/jaba.1999.32-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Authors. National standards report. Randolf, MA: National Autism Center; 2009. [Google Scholar]
- Azurdia RM, Anstey AV, Rhodes LE. Cholesterol supplementation objectively reduces photosensitivity in the Smith-Lemli-Opitz syndrome. Br J Dermatol. 2001;144:143–145. doi: 10.1046/j.1365-2133.2001.03964.x. [DOI] [PubMed] [Google Scholar]
- Barkhof F, Verrips A, Wesseling P, van Der Knaap MS, van Engelen BG, Gabreels FJ, Keyser A, Wevers RA, Valk J. Cerebrotendinous xanthomatosis: The spectrum of imaging findings and the correlation with neuropathologic findings. Radiology. 2000;217:869–876. doi: 10.1148/radiology.217.3.r00dc03869. [DOI] [PubMed] [Google Scholar]
- Batta AK, Salen G, Tint GS. Hydrophilic 7 beta-hydroxy bile acids, lovastatin, and cholestyramine are ineffective in the treatment of cerebrotendinous xanthomatosis. Metabolism. 2004;53:556–562. doi: 10.1016/j.metabol.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Batta AK, Tint GS, Shefer S, Abuelo D, Salen G. Identification of 8-dehydrocholesterol (cholesta-5,8-dien-3 beta-ol) in patients with Smith-Lemli-Opitz syndrome. J Lipid Res. 1995;36:705–713. [PubMed] [Google Scholar]
- Battaile KP, Steiner RD. Smith-Lemli-Opitz syndrome: The first malformation syndrome associated with defective cholesterol synthesis. Mol Genet Metab. 2000;71:154–162. doi: 10.1006/mgme.2000.3020. [DOI] [PubMed] [Google Scholar]
- Berginer VM, Salen G, Shefer S. Long-term treatment of cerebrotendinous xanthomatosis with chenodeoxycholic acid. N Engl J Med. 1984;311:1649–1652. doi: 10.1056/NEJM198412273112601. [DOI] [PubMed] [Google Scholar]
- Bianconi SE, Conley SK, Keil MF, Sinaii N, Rother KI, Porter FD, Stratakis CA. Adrenal function in Smith-Lemli-Opitz syndrome. Am J Med Genet A. 2011;155A:2732–2738. doi: 10.1002/ajmg.a.34271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkhem I, Meaney S. Brain cholesterol: Long secret life behind a barrier. Arterioscler Thromb Vasc Biol. 2004;24:806–815. doi: 10.1161/01.ATV.0000120374.59826.1b. [DOI] [PubMed] [Google Scholar]
- Bodar EJ, Kuijk LM, Drenth JP, van der Meer JW, Simon A, Frenkel J. On-demand anakinra treatment is effective in mevalonate kinase deficiency. Ann Rheum Dis. 2011;70:2155–2158. doi: 10.1136/ard.2011.149922. [DOI] [PubMed] [Google Scholar]
- Chan YM, Merkens LS, Connor WE, Roullet JB, Penfield JA, Jordan JM, Steiner RD, Jones PJ. Effects of dietary cholesterol and simvastatin on cholesterol synthesis in Smith-Lemli-Opitz syndrome. Pediatr Res. 2009;65:681–685. doi: 10.1203/PDR.0b013e31819ea4eb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury S, Hormaza L, Mohammad S, Lokar J, Ekong U, Alonso EM, Wainwright MS, Kletzel M, Whitington PF. Liver transplantation followed by allogeneic hematopoietic stem cell transplantation for atypical mevalonic aciduria. American Journal of Transplantation. 2012;12:1627–1631. doi: 10.1111/j.1600-6143.2011.03989.x. [DOI] [PubMed] [Google Scholar]
- Choi PT, Nowaczyk MJ. Anesthetic considerations in Smith-Lemli-Opitz syndrome. Can J Anaesth. 2000;47:556–561. doi: 10.1007/BF03018947. [DOI] [PubMed] [Google Scholar]
- Correa-Cerro LS, Wassif CA, Kratz L, Miller GF, Munasinghe JP, Grinberg A, Fliesler SJ, Porter FD. Development and characterization of a hypomorphic Smith-Lemli-Opitz syndrome mouse model and efficacy of simvastatin therapy. Hum Mol Genet. 2006;15:839–851. doi: 10.1093/hmg/ddl003. [DOI] [PubMed] [Google Scholar]
- Cunniff C, Kratz LE, Moser A, Natowicz MR, Kelley RI. Clinical and biochemical spectrum of patients with RSH/Smith-Lemli-Opitz syndrome and abnormal cholesterol metabolism. Am J Med Genet. 1997;68:263–269. [PubMed] [Google Scholar]
- DeBarber AE, Connor WE, Pappu AS, Merkens LS, Steiner RD. ESI-MS/MS quantification of 7alpha-hydroxy-4-cholesten-3-one facilitates rapid, convenient diagnostic testing for cerebrotendinous xanthomatosis. Clin Chim Acta. 2010;411:43–48. doi: 10.1016/j.cca.2009.09.036. [DOI] [PubMed] [Google Scholar]
- DeBarber AE, Eroglu Y, Merkens LS, Pappu AS, Steiner RD. Smith-Lemli-Opitz syndrome. Expert Rev Mol Med. 2011;13:e24. doi: 10.1017/S146239941100189X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotti MT, Lutjohann D, von Bergmann K, Federico A. Normalisation of serum cholestanol concentration in a patient with cerebrotendinous xanthomatosis by combined treatment with chenodeoxycholic acid, simvastatin and LDL apheresis. Neurol Sci. 2004;25:185–191. doi: 10.1007/s10072-004-0320-6. [DOI] [PubMed] [Google Scholar]
- Elias ER, Irons MB, Hurley AD, Tint GS, Salen G. Clinical effects of cholesterol supplementation in six patients with the Smith-Lemli-Opitz syndrome (SLOS) Am J Med Genet. 1997;68:305–310. doi: 10.1002/(sici)1096-8628(19970131)68:3<305::aid-ajmg11>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Federico A, Dotti MT. Cerebrotendinous xanthomatosis: Clinical manifestations, diagnostic criteria, pathogenesis, and therapy. J Child Neurol. 2003;18:633–638. doi: 10.1177/08830738030180091001. [DOI] [PubMed] [Google Scholar]
- FitzPatrick DR, Keeling JW, Evans MJ, Kan AE, Bell JE, Porteous MEM, Mills K, Winter RM, Clayton PT. Clinical phenotype of desmosterolosis. Am J Med Genet. 1998;75:145–152. [PubMed] [Google Scholar]
- Gonzalez-Cuyar LF, Hunter B, Harris PL, Perry G, Smith MA, Castellani RJ. Cerebrotendinous xanthomatosis: Case report with evidence of oxidative stress. Redox Rep. 2007;12:119–124. doi: 10.1179/135100007X200173. [DOI] [PubMed] [Google Scholar]
- Haas D, Garbade SF, Vohwinkel C, Muschol N, Trefz FK, Penzien JM, Zschocke J, Hoffmann GF, Burgard P. Effects of cholesterol and simvastatin treatment in patients with Smith-Lemli-Opitz syndrome (SLOS) J Inherit Metab Dis. 2007;30:375–387. doi: 10.1007/s10545-007-0537-7. [DOI] [PubMed] [Google Scholar]
- Habaragamuwa BW, Bajekal R. Cerebrotendinous xanthomatosis and anaesthesia. Br J Anaesth. 2010;105:237–238. doi: 10.1093/bja/aeq188. [DOI] [PubMed] [Google Scholar]
- He M, Kratz LE, Michel JJ, Vallejo AN, Ferris L, Kelley RI, Hoover JJ, Jukic D, Gibson KM, Wolfe LA, Ramachandran D, Zwick ME, Vockley J. Mutations in the human SC4MOL gene encoding a methyl sterol oxidase cause psoriasiform dermatitis, microcephaly, and developmental delay. J Clin Invest. 2011;121:976–984. doi: 10.1172/JCI42650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda A, Salen G, Shefer S, Batta AK, Honda M, Xu G, Tint GS, Matsuzaki Y, Shoda J, Tanaka N. Bile acid synthesis in the Smith-Lemli-Opitz syndrome: Effects of dehydrocholesterols on cholesterol 7alpha-hydroxylase and 27-hydroxylase activities in rat liver. J Lipid Res. 1999;40:1520–1528. [PubMed] [Google Scholar]
- Huang L, Miao XD, Yang DS, Tao HM. Bilateral achilles tendon enlargement. Orthopedics. 2011;34:e960–4. doi: 10.3928/01477447-20111021-28. [DOI] [PubMed] [Google Scholar]
- Irons M, Elias ER, Abuelo D, Bull MJ, Greene CL, Johnson VP, Keppen L, Schanen C, Tint GS, Salen G. Treatment of Smith-Lemli-Opitz syndrome: Results of a multicenter trial. Am J Med Genet. 1997;68:311–314. [PubMed] [Google Scholar]
- Irons M, Elias ER, Tint GS, Salen G, Frieden R, Buie TM, Ampola M. Abnormal cholesterol metabolism in the Smith-Lemli-Opitz syndrome: Report of clinical and biochemical findings in four patients and treatment in one patient. Am J Med Genet. 1994;50:347–352. doi: 10.1002/ajmg.1320500409. [DOI] [PubMed] [Google Scholar]
- Irons MB, Nores J, Stewart TL, Craigo SD, Bianchi DW, D'Alton ME, Tint GS, Salen G, Bradley LA. Antenatal therapy of Smith-Lemli-Opitz syndrome. Fetal Diagn Ther. 1999;14:133–137. doi: 10.1159/000020906. [DOI] [PubMed] [Google Scholar]
- Ito S, Kuwabara S, Sakakibara R, Oki T, Arai H, Oda S, Hattori T. Combined treatment with LDL-apheresis, chenodeoxycholic acid and HMG-CoA reductase inhibitor for cerebrotendinous xanthomatosis. J Neurol Sci. 2003;216:179–182. doi: 10.1016/j.jns.2003.07.005. [DOI] [PubMed] [Google Scholar]
- Jenkins KT, Merkens LS, Tubb MR, Myatt L, Davidson WS, Steiner RD, Woollett LA. Enhanced placental cholesterol efflux by fetal HDL in Smith-Lemli-Opitz syndrome. Mol Genet Metab. 2008;94:240–247. doi: 10.1016/j.ymgme.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jira PE, Wevers RA, de Jong J, Rubio-Gozalbo E, Janssen-Zijlstra FS, van Heyst AF, Sengers RC, Smeitink JA. Simvastatin. A new therapeutic approach for Smith-Lemli-Opitz syndrome. J Lipid Res. 2000;41:1339–1346. [PubMed] [Google Scholar]
- Kelley RI. A new face for an old syndrome. Am J Med Genet. 1997;68:251–256. doi: 10.1002/(sici)1096-8628(19970131)68:3<251::aid-ajmg1>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Kelley RI, Hennekam RC. The Smith-Lemli-Opitz syndrome. J Med Genet. 2000;37:321–335. doi: 10.1136/jmg.37.5.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korade Z, Xu L, Shelton R, Porter NA. Biological activities of 7-dehydrocholesterol-derived oxysterols: Implications for Smith-Lemli-Opitz syndrome. J Lipid Res. 2010;51:3259–3269. doi: 10.1194/jlr.M009365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakowiak PA, Wassif CA, Kratz L, Cozma D, Kovářová M, Harris G, Grinberg A, Yang Y, Hunter AGW, Tsokos M, Kelley RI, Porter FD. Lathosterolosis: An inborn error of human and murine cholesterol synthesis due to lathosterol 5-desaturase deficiency. Human Molecular Genetics. 2003;12:1631–1641. doi: 10.1093/hmg/ddg172. [DOI] [PubMed] [Google Scholar]
- Kuriyama M, Tokimura Y, Fujiyama J, Utatsu Y, Osame M. Treatment of cerebrotendinous xanthomatosis: Effects of chenodeoxycholic acid, pravastatin, and combined use. J Neurol Sci. 1994;125:22–28. doi: 10.1016/0022-510x(94)90237-2. [DOI] [PubMed] [Google Scholar]
- Lin DS, Steiner RD, Flavell DP, Connor WE. Intestinal absorption of cholesterol by patients with Smith-Lemli-Opitz syndrome. Pediatr Res. 2005;57:765–770. doi: 10.1203/01.PDR.0000157723.98422.B5. [DOI] [PubMed] [Google Scholar]
- Linck LM, Lin DS, Flavell D, Connor WE, Steiner RD. Cholesterol supplementation with egg yolk increases plasma cholesterol and decreases plasma 7-dehydrocholesterol in Smith-Lemli-Opitz syndrome. Am J Med Genet. 2000;93:360–365. doi: 10.1002/1096-8628(20000828)93:5<360::aid-ajmg4>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Lindegaard ML, Wassif CA, Vaisman B, Amar M, Wasmuth EV, Shamburek R, Nielsen LB, Remaley AT, Porter FD. Characterization of placental cholesterol transport: ABCA1 is a potential target for in utero therapy of Smith-Lemli-Opitz syndrome. Hum Mol Genet. 2008;17:3806–3813. doi: 10.1093/hmg/ddn278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A, Koenig K, Scahill L, Tierney E, Porter FD, Nwokoro NA. Smith-Lemli-Opitz syndrome. J Am Acad Child Adolesc Psychiatry. 2001;40:506–507. doi: 10.1097/00004583-200105000-00008. [DOI] [PubMed] [Google Scholar]
- Matabosch X, Ying L, Serra M, Wassif CA, Porter FD, Shackleton C, Watson G. Increasing cholesterol synthesis in 7-dehydrosterol reductase (DHCR7) deficient mouse models through gene transfer. J Steroid Biochem Mol Biol. 2010;122:303–309. doi: 10.1016/j.jsbmb.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPheeters ML, Warren Z, Sathe N, Bruzek JL, Krishnaswami S, Jerome RN, Veenstra-Vanderweele J. A systematic review of medical treatments for children with autism spectrum disorders. Pediatrics. 2011;127:e1312–21. doi: 10.1542/peds.2011-0427. [DOI] [PubMed] [Google Scholar]
- Mondelli M, Sicurelli F, Scarpini C, Dotti MT, Federico A. Cerebrotendinous xanthomatosis: 11-year treatment with chenodeoxycholic acid in five patients. an electrophysiological study. J Neurol Sci. 2001;190:29–33. doi: 10.1016/s0022-510x(01)00563-9. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Matsuzawa Y, Takemura K, Kubo M, Miki H, Tarui S. Combined treatment with chenodeoxycholic acid and pravastatin improves plasma cholestanol levels associated with marked regression of tendon xanthomas in cerebrotendinous xanthomatosis. Metabolism. 1991;40:741–746. doi: 10.1016/0026-0495(91)90094-d. [DOI] [PubMed] [Google Scholar]
- Natowicz MR, Evans JE. Abnormal bile acids in the Smith-Lemli-Opitz syndrome. Am J Med Genet. 1994;50:364–367. doi: 10.1002/ajmg.1320500413. [DOI] [PubMed] [Google Scholar]
- Neven B, Valayannopoulos V, Quartier P, Blanche S, Prieur AM, Debre M, Rolland MO, Rabier D, Cuisset L, Cavazzana-Calvo M, de Lonlay P, Fischer A. Allogeneic bone marrow transplantation in mevalonic aciduria. N Engl J Med. 2007;356:2700–2703. doi: 10.1056/NEJMoa070715. [DOI] [PubMed] [Google Scholar]
- Nwokoro NA, Mulvihill JJ. Cholesterol and bile acid replacement therapy in children and adults with Smith-Lemli-Opitz (SLO/RSH) syndrome. Am J Med Genet. 1997;68:315–321. doi: 10.1002/(sici)1096-8628(19970131)68:3<315::aid-ajmg13>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Opitz JM. RSH (so-called Smith-Lemli-Opitz) syndrome. Curr Opin Pediatr. 1999;11:353–362. doi: 10.1097/00008480-199908000-00015. [DOI] [PubMed] [Google Scholar]
- Pappu AS, Steiner RD, Connor SL, Flavell DP, Lin DS, Hatcher L, Illingworth DR, Connor WE. Feedback inhibition of the cholesterol biosynthetic pathway in patients with Smith-Lemli-Opitz syndrome as demonstrated by urinary mevalonate excretion. J Lipid Res. 2002;43:1661–1669. doi: 10.1194/jlr.m200163-jlr200. [DOI] [PubMed] [Google Scholar]
- Porter FD. Smith-Lemli-Opitz syndrome: Pathogenesis, diagnosis and management. Eur J Hum Genet. 2008;16:535–541. doi: 10.1038/ejhg.2008.10. [DOI] [PubMed] [Google Scholar]
- Porter FD, Herman GE. Malformation syndromes caused by disorders of cholesterol synthesis. J Lipid Res. 2011;52:6–34. doi: 10.1194/jlr.R009548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada IJ, Ramos A. Botulinum toxin-responsive oromandibular dystonia in cerebrotendinous xanthomatosis. Parkinsonism Relat Disord. 2011;17:570–572. doi: 10.1016/j.parkreldis.2011.04.006. [DOI] [PubMed] [Google Scholar]
- Quezado ZM, Veihmeyer J, Schwartz L, Nwokoro NA, Porter FD. Anesthesia and airway management of pediatric patients with Smith-Lemli-Opitz syndrome. Anesthesiology. 2002;97:1015–1019. doi: 10.1097/00000542-200210000-00041. [DOI] [PubMed] [Google Scholar]
- Rodrigues CM, Spellman SR, Sola S, Grande AW, Linehan-Stieers C, Low WC, Steer CJ. Neuroprotection by a bile acid in an acute stroke model in the rat. J Cereb Blood Flow Metab. 2002;22:463–471. doi: 10.1097/00004647-200204000-00010. [DOI] [PubMed] [Google Scholar]
- Ryan AK, Bartlett K, Clayton P, Eaton S, Mills L, Donnai D, Winter RM, Burn J. Smith-Lemli-Opitz syndrome: A variable clinical and biochemical phenotype. J Med Genet. 1998;35:558–565. doi: 10.1136/jmg.35.7.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salen G, Batta AK, Tint GS, Shefer S. Comparative effects of lovastatin and chenodeoxycholic acid on plasma cholestanol levels and abnormal bile acid metabolism in cerebrotendinous xanthomatosis. Metabolism. 1994;43:1018–1022. doi: 10.1016/0026-0495(94)90183-x. [DOI] [PubMed] [Google Scholar]
- Salen G, Meriwether TW, Nicolau G. Chenodeoxycholic acid inhibits increased cholesterol and cholestanol synthesis in patients with cerebrotendinous xanthomatosis. Biochem Med. 1975;14:57–74. doi: 10.1016/0006-2944(75)90020-4. [DOI] [PubMed] [Google Scholar]
- Salen G, Shefer S, Nguyen L, Ness GC, Tint GS, Shore V. Sitosterolemia. J Lipid Res. 1992;33:945–955. [PubMed] [Google Scholar]
- Schaaf CP, Koster J, Katsonis P, Kratz L, Shchelochkov OA, Scaglia F, Kelley RI, Lichtarge O, Waterham HR, Shinawi M. Desmosterolosis-phenotypic and molecular characterization of a third case and review of the literature. Am J Med Genet A. 2011;155A:1597–1604. doi: 10.1002/ajmg.a.34040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel S, Kasprian G, Prayer D, Krssak M, Sycha T, Auff E. Visualisation of treatment response in a case of cerebrotendinous xanthomatosis. J Neurol Neurosurg Psychiatry. 2011;82:703–704. doi: 10.1136/jnnp.2009.196444. [DOI] [PubMed] [Google Scholar]
- Selva-O'Callaghan A, Bardes I, Jacas C, Jubany L, Lorenzo-Bosquet C, Cuberas-Borros G, Vilardell-Tarres M. SPECT imaging for brain improvement quantification in a patient with cerebrotendinous xanthomatosis. Clin Nucl Med. 2011;36:38–39. doi: 10.1097/RLU.0b013e3181feed83. [DOI] [PubMed] [Google Scholar]
- Setchell KD, Heubi JE. Defects in bile acid biosynthesis--diagnosis and treatment. J Pediatr Gastroenterol Nutr. 2006;43:S17–22. doi: 10.1097/01.mpg.0000226386.79483.7b. [DOI] [PubMed] [Google Scholar]
- Shefer S, Salen G, Batta AK, Honda A, Tint GS, Irons M, Elias ER, Chen TC, Holick MF. Markedly inhibited 7-dehydrocholesterol-delta 7-reductase activity in liver microsomes from Smith-Lemli-Opitz homozygotes. J Clin Invest. 1995;96:1779–1785. doi: 10.1172/JCI118223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishehbor MH, Brennan ML, Aviles RJ, Fu X, Penn MS, Sprecher DL, Hazen SL. Statins promote potent systemic antioxidant effects through specific inflammatory pathways. Circulation. 2003;108:426–431. doi: 10.1161/01.CIR.0000080895.05158.8B. [DOI] [PubMed] [Google Scholar]
- Sikora DM, Pettit-Kekel K, Penfield J, Merkens LS, Steiner RD. The near universal presence of autism spectrum disorders in children with Smith-Lemli-Opitz syndrome. Am J Med Genet A. 2006;140:1511–1518. doi: 10.1002/ajmg.a.31294. [DOI] [PubMed] [Google Scholar]
- Sikora DM, Ruggiero M, Petit-Kekel K, Merkens LS, Connor WE, Steiner RD. Cholesterol supplementation does not improve developmental progress in Smith-Lemli-Opitz syndrome. J Pediatr. 2004;144:783–791. doi: 10.1016/j.jpeds.2004.02.036. [DOI] [PubMed] [Google Scholar]
- Smith DW, Lemli L, Opitz JM. A Newly Recognized Syndrome of Multiple Congenital Anomalies. J Pediatr. 1964;64:210–217. doi: 10.1016/s0022-3476(64)80264-x. [DOI] [PubMed] [Google Scholar]
- Starck L, Lovgren-Sandblom A, Bjorkhem I. Simvastatin treatment in the SLO syndrome: A safe approach? Am J Med Genet. 2002;113:183–189. doi: 10.1002/ajmg.10722. [DOI] [PubMed] [Google Scholar]
- Steiner RD, Linck LM, Flavell DP, Lin DS, Connor WE. Sterol balance in the Smith-Lemli-Opitz syndrome. reduction in whole body cholesterol synthesis and normal bile acid production. J Lipid Res. 2000;41:1437–1447. [PubMed] [Google Scholar]
- Takada K, Aksentijevich I, Mahadevan V, Dean JA, Kelley RI, Kastner DL. Favorable preliminary experience with etanercept in two patients with the hyperimmunoglobulinemia D and periodic fever syndrome. Arthritis & Rheumatism. 2003;48:2645–2651. doi: 10.1002/art.11218. [DOI] [PubMed] [Google Scholar]
- Tierney E, Nwokoro NA, Kelley RI. Behavioral phenotype of RSH/Smith-Lemli-Opitz syndrome. Ment Retard Dev Disabil Res Rev. 2000;6:131–134. doi: 10.1002/1098-2779(2000)6:2<131::AID-MRDD7>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Tierney E, Nwokoro NA, Porter FD, Freund LS, Ghuman JK, Kelley RI. Behavior phenotype in the RSH/Smith-Lemli-Opitz syndrome. Am J Med Genet. 2001;98:191–200. doi: 10.1002/1096-8628(20010115)98:2<191::aid-ajmg1030>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Tierney E, Conley SK, Goodwin H, Porter FD. Analysis of short-term behavioral effects of dietary cholesterol supplementation in Smith-Lemli-Opitz syndrome. American Journal of Medical Genetics Part A. 2010;152A:91–95. doi: 10.1002/ajmg.a.33148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tint GS, Irons M, Elias ER, Batta AK, Frieden R, Chen TS, Salen G. Defective cholesterol biosynthesis associated with the Smith-Lemli-Opitz syndrome. N Engl J Med. 1994;330:107–113. doi: 10.1056/NEJM199401133300205. [DOI] [PubMed] [Google Scholar]
- Tokimura Y, Kuriyama M, Arimura K, Fujiyama J, Osame M. Electrophysiological studies in cerebrotendinous xanthomatosis. J Neurol Neurosurg Psychiatry. 1992;55:52–55. doi: 10.1136/jnnp.55.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia A, Rajadurai A, Carle AB, Kochevar IE. 7-dehydrocholesterol enhances ultraviolet A-induced oxidative stress in keratinocytes: Roles of NADPH oxidase, mitochondria, and lipid rafts. Free Radic Biol Med. 2006;41:1704–1718. doi: 10.1016/j.freeradbiomed.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij A, Nijenhuis AA, Wijburg FA, Schutgens RB. Highly increased CSF concentrations of cholesterol precursors in Smith-Lemli-Opitz syndrome. J Inherit Metab Dis. 1997;20:578–580. doi: 10.1023/a:1005355026186. [DOI] [PubMed] [Google Scholar]
- Vaughan DK, Peachey NS, Richards MJ, Buchan B, Fliesler SJ. Light-induced exacerbation of retinal degeneration in a rat model of Smith-Lemli-Opitz syndrome. Exp Eye Res. 2006;82:496–504. doi: 10.1016/j.exer.2005.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrips A, Wevers RA, Van Engelen BG, Keyser A, Wolthers BG, Barkhof F, Stalenhoef A, De Graaf R, Janssen-Zijlstra F, Van Spreeken A, Gabreels FJ. Effect of simvastatin in addition to chenodeoxycholic acid in patients with cerebrotendinous xanthomatosis. Metabolism. 1999;48:233–238. doi: 10.1016/s0026-0495(99)90040-9. [DOI] [PubMed] [Google Scholar]
- Wassif CA, Krakowiak PA, Wright BS, Gewandter JS, Sterner AL, Javitt N, Yergey AL, Porter FD. Residual cholesterol synthesis and simvastatin induction of cholesterol synthesis in Smith-Lemli-Opitz syndrome fibroblasts. Mol Genet Metab. 2005;85:96–107. doi: 10.1016/j.ymgme.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Woollett LA. Review: Transport of maternal cholesterol to the fetal circulation. Placenta. 2011;32:S218–21. doi: 10.1016/j.placenta.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu LB, Sheflin LG, Porter NA, Fliesler SJ. 7-dehydrocholesterol-derived oxysterols and retinal degeneration in a rat model of Smith-Lemli-Opitz syndrome. Biochimica ET Biophysica Acta-Molecular and Cell Biology of Lipids. 2012;1821(6):877–883. doi: 10.1016/j.bbalip.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuhara H, Tonooka M, Kamei K, Sakamoto K. Membrane effects of various drugs on isolated rat hepatocytes and erythrocytes. Toxicol Appl Pharmacol. 1985;79:453–460. doi: 10.1016/0041-008x(85)90142-5. [DOI] [PubMed] [Google Scholar]
- Zarcone JR, Hellings JA, Crandall K, Reese RM, Marquis J, Fleming K, Shores R, Williams D, Schroeder SR. Effects of risperidone on aberrant behavior of persons with developmental disabilities: I. A double-blind crossover study using multiple measures. Am J Ment Retard. 2001;106:525–538. doi: 10.1352/0895-8017(2001)106<0525:EOROAB>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Zolotushko J, Flusser H, Markus B, Shelef I, Langer Y, Heverin M, Bjorkhem I, Sivan S, Birk OS. The desmosterolosis phenotype: Spasticity, microcephaly and micrognathia with agenesis of corpus callosum and loss of white matter. Eur J Hum Genet. 2011 doi: 10.1038/ejhg.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]