

Abstract
The water channel aquaporin‐4 (AQP4) is the target of the immunoglobulin G autoantibody (AQP4‐IgG) in neuromyelitis optica (NMO). AQP4 is expressed in foot processes of astrocytes throughout the central nervous system, as well as in skeletal muscle and epithelial cells in kidney, lung and gastrointestinal organs. Phenotype analysis of AQP4 knockout mice indicates the involvement of AQP4 in water movement into and out of the brain, astrocyte migration, glial scar formation and neuroexcitatory phenomena. AQP4 monomers form tetramers in membranes, which further aggregate to form supramolecular assemblies called orthogonal arrays of particles. AQP4‐IgG is pathogenic in NMO by a mechanism involving complement‐ and cell‐mediated astrocyte cytotoxicity, which produces an inflammatory response with oligodendrocyte injury and demyelination. AQP4 orthogonal arrays are crucial in NMO pathogenesis, as they increase AQP4‐IgG binding to AQP4 and greatly enhance complement‐dependent cytotoxicity. Novel NMO therapeutics are under development that target AQP4‐IgG or AQP4, including aquaporumab monoclonal antibodies and small molecules that block AQP4‐IgG binding to AQP4, and enzymatic inactivation strategies to neutralize AQP4‐IgG pathogenicity.
Keywords: aquaporin, astrocyte, autoimmunity, complement, neuromyelitis optica, orthogonal array
Structure and Water Transport Function of Aquaporin‐4 (AQP4)
The aquaporins (AQPs) are a family of membrane water‐transporting proteins, some of which also transport glycerol and other small solutes 7. There are at least 12 related AQP proteins expressed in mammalian tissues with many more homologues in plants and lower organisms. AQP4, the target of neuromyelitis optica (NMO) autoantibodies, was originally identified in 1994 in rat lung by homology cloning 18. Figure 1A shows the amino acid sequence of human AQP4, with eight membrane‐embedded domains, six of which are membrane spanning, and cytoplasmically facing N‐ and C‐termini. High‐resolution X‐ray crystallography indicated that each AQP4 monomer consists of six helical, membrane‐spanning domains and two short helical segments surrounding a narrow aqueous pore (Figure 1B) 23, similar to other AQPs whose structures have been determined 81. AQP4 is an efficient water‐selective transport protein compared with other AQPs, as it has as relatively high single‐channel water permeability and excludes ions and small solutes such as urea 83. AQP4 structure analysis and molecular dynamics simulations provide a physical basis for its water transport selectivity 13.
Figure 1.
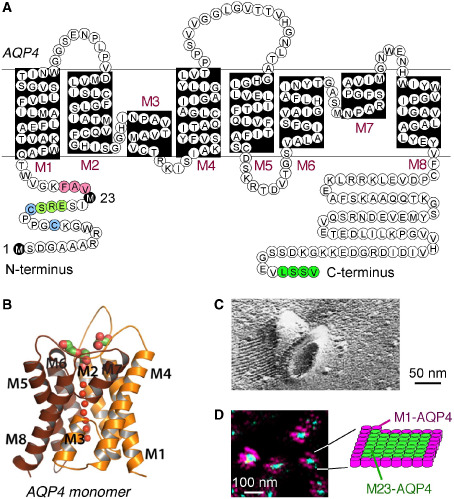
Structure and supramolecular assembly of AQP4. A. Amino acid sequence of human AQP4 showing Met‐1 and Met‐23 translation initiation sites (black), residues involved in intermolecular N‐terminus associations to form OAPs (pink); residues preventing OAP formation by M1‐AQP4 (light green), cysteine residues involved in palmitoylation‐regulated OAP assembly (blue), and C‐terminus PDZ domain (dark green). B. Crystal structure of human AQP4 monomer (PDB, 3GD8). C. Freeze‐fracture electron micrograph of M23‐AQP4‐expressing CHO cells showing AQP4 OAPs. D. Super‐resolution micrograph (dSTORM) in cells co‐expressing a green fluorescent M23‐AQP4 and red fluorescent M1‐AQP4. Diagram shows an OAP with M23‐AQP4‐enriched core and M1‐AQP4 periphery. Adapted from Rossi et al 60 and Yang et al 85.
Human AQP4 is expressed as two major isoforms produced by alternative splicing: a long isoform (called “M1”) with translation initiation at Met‐1 and a short isoform (called “M23”) with translation initiation at Met‐23 36, 84. Both isoforms are expressed in all AQP4‐containing cells including astrocytes, the relevant cell type in NMO. AQPs generally form homotetramers in cell membranes. In the case of AQP4, the M1‐ and M23‐AQP4 isoforms form heterotetramers 26, 41, 71. However, unlike most other AQPs, AQP4 tetramers further aggregate in the cell plasma membrane in supramolecular crystalline assemblies called orthogonal arrays of particles (OAPs). OAPs were originally defined by their regular, cobblestone‐like appearance as seen by freeze‐fracture electron microscopy (Figure 1C) 31, 82. AQP4 was discovered as the major OAP protein from the appearance of OAPs in AQP4‐transfected cells 85 and the absence of OAPs in AQP4 knockout mice 76, as confirmed by label‐fracture electron microscopy 53. OAP formation by AQP4 is essential in NMO pathogenesis, as described further below.
Our laboratory has extensively studied the structure and dynamics of OAPs in live cells, utilizing biophysical methods including quantum dot single‐particle tracking, photo‐bleaching and super‐resolution imaging. OAPs assemble in a post‐Golgi compartment and likely require plasma membrane‐specific factor(s) 59. OAP formation is stabilized by intermolecular N‐terminus interactions between M23‐AQP4 monomers in adjacent tetramers involving residues just downstream of Met‐23 10. M23‐AQP4 OAPs are very slowly diffusing compared to M1‐AQP4 tetramers 12 and rearrange over tens of minutes 11, 71. OAP size, shape and composition depend on the relative amounts of M1‐ vs. M23‐AQP4, with larger OAPs formed at higher M23 : M1 ratio 8, 17. Stochastic optical reconstruction microscopy with ∼20 nm spatial resolution showed individual OAPs containing both M1‐ and M23‐AQP4, with an M23‐enriched core and M1‐enriched periphery (Figure 1D) 60. Mathematical modeling, assuming random M1‐M23 association in heterotetramers and intertetrameric M23‐M23 association, provided a biophysical explanation for the M23 : M1 ratio dependence of OAP size, shape and composition 26.
However, despite a considerable body of data on OAP assembly, the biological importance of OAPs remains unclear, as does the expression of the two AQP4 isoforms. Speculated biological roles of OAPs include enhanced water permeability 14, 68, astrocyte‐to‐astrocyte adhesion 22 and AQP4 polarization to astrocyte end‐feet 15, 44; however, direct measurements of water permeability and cell‐to‐cell adhesion provided evidence against these proposed roles 61, 88. Recent data from our laboratory suggest distinct cellular localization and functions of AQP4 tetramers/small OAPs vs. large AQP4 OAPs (Smith and Verkman, unpublished results). Spatial segregation of small, M1‐enriched AQP4 tetramers/OAPs in lamellipodia and large, M23‐enriched OAPs in non‐lamellipodial cell membrane provided evidence for a dynamic, dual role of AQP4 in cell migration and substrate adhesion.
Cellular Expression Pattern of AQP4
AQP4 is expressed in astrocytes throughout the central nervous system (CNS). AQP4 is concentrated in astrocyte end‐feet that make contact with microcapillary endothelia forming the blood‐brain barrier and in ependymal cells at brain‐cerebrospinal interfaces 16, 43, 53 (Figure 2A). AQP4 expression in brain is altered in a variety of pathological conditions, generally increasing in reactive astrocytes and during glia scar formation 47; AQP4 expression is notably reduced in NMO lesions 37. AQP4 is also expressed in astrocyte‐like “supportive cells” in sensory organs including retinal Müller cells, Hensen's and Claudius' cells in inner ear and support cells in olfactory epithelium. AQP4 is absent in electrically excitable cells in contact with the support cells including neurons in brain and spinal cord, bipolar cells in retina, hair cells in inner ear and olfactory receptor neurons. AQP4 expression is particularly strong in optic nerve and spinal cord, the major tissues affected in NMO. Outside of the CNS, AQP4 is expressed at the basolateral membrane in epithelial cells in kidney, airways, gastrointestinal organs and, at a low level, in skeletal muscle. Interestingly, NMO pathology is not seen in these peripheral, AQP4‐expressing tissues.
Figure 2.
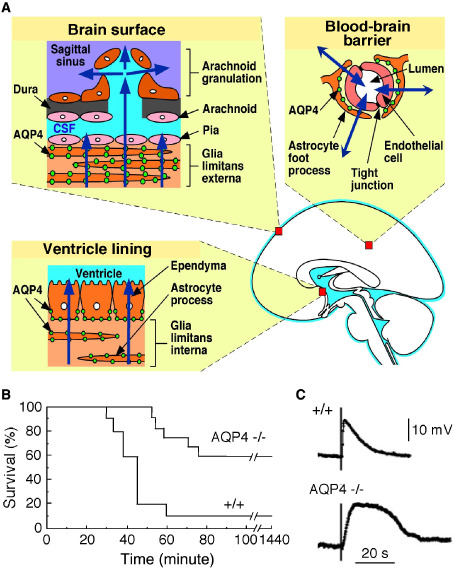
Role of AQP4 in brain water balance and extracellular space K + dynamics. A. Schematic showing site of AQP4 expression in brain and pathways for water movement out of brain. B. Improved survival of AQP4 knockout (−/−) mice following acute water intoxication. C. Delayed K+ clearance in brain following electrically induced seizure‐like neuroexcitation. Measurements done using K+‐sensitive microelectrodes inserted into brain cortex in live mice. Adapted from Binder et al 3 and Manley et al 38.
Biological Functions of AQP4
A substantial body of data on the physiological functions of AQPs has come from phenotype analysis of knockout mice lacking individual AQPs (reviewed in Verkman 77). Water‐selective AQPs are involved in transepithelial fluid transport and cell migration; AQPs that transport both water and glycerol (called “aquaglyceroporins”) are involved in cell proliferation, adipocyte metabolism and epidermal water retention. Loss‐of‐function mutations in human AQPs cause nephrogenic diabetes insipidus (AQP2) and congenital cataracts (AQP0). Many tumors express AQPs, whose roles may include facilitated tumor angiogenesis, tumor growth and invasion/metastasis 24, 79. AQP4 is strongly expressed in astrocytomas and glioblastomas, with AQP4 expression correlating with tumor grade 62. The reader is referred to recent reviews for discussion of the many interesting roles of AQPs and their cellular mechanisms 57, 78.
Studies on knockout mice have indicated several distinct roles for AQP4 (reviewed in Papadopoulos and Verkman 47). As might be anticipated from its expression pattern, AQP4 facilitates the movement of water between blood and brain and between brain and cerebrospinal fluid (CSF) compartments. AQP4 is the major water‐transporting pathway in astrocytes 69. AQP4 knockout mice show reduced cytotoxic (cell‐swelling) brain edema following water intoxication (Figure 2B) or ischemic stroke 38 and increased vasogenic (leaky‐vessel) brain edema in brain tumor 48 and brain abscess 4. AQP4 knockout mice also show increased ventricular enlargement in obstructive hydrocephalus 5. Transgenic AQP4 overexpressing mice show accelerated brain edema following water intoxication 86. AQP4 thus provides an important route for water movement into brain in cytotoxic edema, where the blood‐brain barrier is intact, and, apparently, for the removal of excess water in vasogenic edema, where water enters the brain through a disrupted blood‐brain barrier. Various theories of AQP4‐dependent edema fluid clearance from the CNS have been proposed; these theories typically involve AQP4‐dependent transcellular water flow. In one theory, water elimination occurs across AQP4‐rich interfaces, driven by hydrostatic pressure 48. Another theory postulates that excess water flows through brain parenchyma, transcellularly through AQP4 and then into perivenous spaces and cervical lymphatics 25. However, neither theory accounts for hydrostatically driven flow of solute‐free water through AQP4 being opposed by the osmotic gradients. These theories lack a rigorous description of AQP4‐dependent fluid clearance in vasogenic edema.
As mentioned above, AQPs facilitate cell migration 49, which includes AQP4 in astrocytes. AQP4 is polarized to the leading edge of migrating astrocytes in culture, and the migration of astrocytes from AQP4 knockout mice is slowed compared with astrocytes from wild‐type mice 64. In vivo, glial scar formation is impaired in AQP4 knockout mice following stab injury. Furthermore, the migration in brain (of wild‐type mice) of injected, fluorescently labeled AQP4‐expressing astrocytes was greatly slowed compared with AQP4‐deficient astrocytes 1. Mechanistic studies suggest that AQP‐facilitated water transport at the leading edge of migrating cells increases their lamellipodial extension 63. Actin depolymerization and/or active solute influx in lamellipodia creates an osmotic gradient driving water influx across the cell plasma membrane that facilitates plasma membrane expansion. In addition to its involvement in astrocyte migration and glial scar formation, AQP4 is probably an important determinant of the local invasiveness in glioblastomas.
The third distinct role of AQP4 is in neural signal transduction. AQP4 knockout mice show increased seizure duration and intensity following chemical or electrical stimulation 3, and prolonged, spreading depression in the cortex following mechanical stimulation 45. Neurosensory signal transduction is also impaired in AQP4 knockout mice, as demonstrated by evoked potential measurements and/or behavioral studies of vision 33, hearing 32 and olfaction 35. AQP4‐facilitated neural signal transduction probably involves effects on potassium (K+) dynamics in brain extracellular space (ECS) during neuroexcitation. Measurements in brain in vivo and in brain slices of AQP4‐deficient mice show slowed clearance of K+ from the ECS following neuroexcitation 3, 70 (Figure 2C). Mathematical modeling of ECS K+/water coupling 27 provided evidence that reduced astrocyte water permeability in AQP4 deficiency may be responsible for the slowed K+ clearance. The proposed mechanism involves reduced ECS shrinkage and consequent decrease in the electrochemical driving force for uptake of excess ECS K+ into astrocytes. A possible caveat, however, is that interpretation of data from knockout mice could be confounded by altered gene expression or other secondary effects. Neuroexcitatory phenomena following acute inhibition of AQP4 water transport should be measured if and when bona fide AQP4 inhibitors become available.
The fourth distinct role for AQP4, distinct from its role in NMO pathogenesis, is neuroinflammation. AQP4 knockout mice manifest an attenuated course of experimental autoimmune encephalomyelitis (EAE) following active immunization with myelin oligodendrocyte glycoprotein (MOG) peptide or adoptive transfer of MOG‐sensitized T‐lymphocytes 34. Mechanistic studies suggested a pro‐inflammatory role for AQP4. Intracerebral injection of lipopolysaccharide produced greater neuroinflammation in wild‐type than in AQP4 knockout mice. Secretion of the major cytokines tumor necrosis factor‐alpha (TNF‐α) and interleukin‐6 (IL‐6) was reduced in astrocyte cultures from AQP4 knockout mice. AQP4‐dependent neuroinflammation may involve astrocyte water permeability and consequent cell swelling and cytokine release, as amplified by a positive‐feedback cycle of cytokine secretion and local cytotoxic brain swelling.
AQP4‐IgG Binding to AQP4
AQP4‐IgG binds to the extracellular surface of AQP4, likely to three‐dimensional conformations involving all three extracellular loops of AQP4. Binding to three‐dimensional rather than linear epitopes is typical for autoantibodies in human autoimmune disorders. Some reports have shown that mutations or other alterations in AQP4 extracellular loops reduce NMO‐IgG binding 52, 87; however, because the reports studied polyclonal AQP4‐IgG and AQP4 mutation often interferes with its cellular processing and surface expression, such data are difficult to interpret. It is not unexpected that mutations at multiple positions in AQP4 extracellular loops reduce binding of an antibody that recognizes a precise, three‐dimensional conformation of its target.
Several studies have reported that binding of AQP4‐IgG in human NMO serum is greater to cells expressing M23‐AQP4 than to cells expressing M1‐AQP4 9, 42, 58. Because M23‐AQP4 forms OAPs and M1‐AQP4 does not, this suggests a preference for AQP4‐IgG binding to OAPs. Our laboratory measured NMO antibody‐binding affinity and specificity using a fluorescence ratio‐imaging assay 9. Figure 3A shows AQP4 immunofluorescence and immunoblot analysis of cells expressing M1‐ or M23‐AQP4. M1‐AQP4, which does not form OAPs, shows a smooth pattern of cellular fluorescence and a single (tetramer) band by native gel electrophoresis; M23‐AQP4, which forms OAPs, shows a punctate pattern of fluorescence and multiple higher order bands. Measurements used NMO patient serum, as well as monoclonal recombinant AQP4‐IgGs derived from clonally expanded plasma cells from the CSF of seropositive NMO patients, generated as described 2. AQP4‐IgG in NMO serum is polyclonal, consisting of many monoclonal antibodies with different AQP4‐binding characteristics. Binding measurements show wide variation in the absolute and relative affinities for AQP4‐IgG binding to M1 vs. M23‐AQP4, from nearly comparable binding to exclusive binding to M23‐AQP4 (Figure 3B). Of more than 30 monoclonal AQP4‐IgGs tested to date, the affinity of the tightest binding antibody was ∼15 nM. Most NMO sera and monoclonal AQP4‐IgGs tested showed substantially greater affinity to M23‐ vs. M1‐AQP4. Measurements in cells expressing M23‐AQP4 mutants with OAP‐disrupting mutations indicated that the differential binding of NMO‐IgG to M1‐ vs. M23‐AQP4 is due to OAP assembly rather than to differences in the M1 vs. M23 N‐termini. Measurements using purified Fab fragments derived from NMO‐rAbs suggested that a structural change in the AQP4 epitope upon array assembly, rather than bivalent NMO‐IgG binding, accounts for the greater binding affinity to OAPs.
Figure 3.
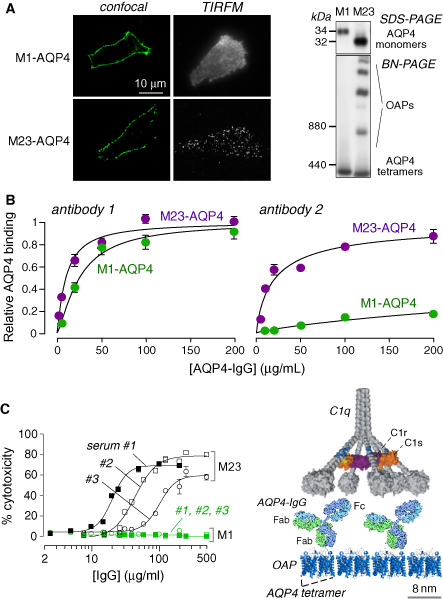
AQP4‐IgG binding to AQP4 and complement‐dependent cytotoxicity. A. (Left) Confocal and TIRFM of M1‐ and M23‐AQP4 transfected CHO cells immunostained for AQP4 using a C‐terminus anti‐AQP4 antibody. (Right) SDS‐PAGE (top) and BN‐PAGE (bottom) of cell homogenates. B. Binding of two monoclonal recombinant NMO autoantibodies to cells expressing M1‐AQP4 or M23‐AQP4. C. Cells expressing M1‐AQP4 are resistant to CDC caused by AQP4‐IgG. (Left) Cytotoxicity plotted as a function of total IgG concentration (from NMO patient serum) in the presence of human complement. (Right) Multivalent binding of C1q to Fc regions of clustered AQP4‐IgG bound to OAP‐assembled AQP4. Adapted from Crane et al 9 and Phuan et al 51.
Cellular Consequences of AQP4‐IgG Binding to AQP4
Binding of antibodies to their cellular target can alter target function (AQP4 water permeability), cause target internalization (AQP4 endocytosis) and/or damage target cells. Cytotoxicity can be caused by complement (complement‐dependent cytotoxicity, CDC) and cellular mechanisms (antibody‐dependent cellular cytotoxicity, ADCC), the latter involving various leukocytes including natural killer (NK) cells, granulocytes (neutrophils and eosinophils) and macrophages. Knowledge of the cellular consequences of AQP4‐IgG binding to AQP4 is important to understanding both NMO pathogenesis and the development of targeted therapeutics.
AQP4‐IgG is an IgG1 subclass antibody and, therefore, has CDC and ADCC effector functions. As expected when using different cellular systems and cytotoxicity readouts, AQP4‐IgG causes CDC when complement is present and ADCC when effector cells are present 19, 80. CDC is initiated by binding of complement protein C1q to the IgG1 Fc region, and ADCC is initiated by binding of effector cell Fcγ receptors to the IgG1 Fc region. As discussed further below, both CDC and ADCC are involved in NMO pathogenesis. CDCC (complement‐dependent cell‐mediated cytotoxicity) is important as well. Whereas ADCC involves direct effector‐cell binding to AQP4‐IgG (without the need for complement), CDCC involves complement‐dependent enhanced effector‐cell recruitment and AQP4‐IgG binding.
An important observation was that efficient CDC (but not ADCC) requires AQP4 assembly in OAPs 51. CDC was substantial for M23‐AQP4‐expressing cells but minimal or absent for M1‐AQP4‐expressing cells (Figure 3C). Measurements of complement protein binding suggested that the greatly enhanced CDC involves C1q binding to AQP4‐IgG when clustered on AQP4 OAPs. C1q is a large, multivalent protein whose binding affinity to IgG is greatly enhanced with increased binding valency. OAP formation by AQP4 thus enhances CDC at two levels: AQP4‐IgG binding affinity to AQP4 and C1q binding to clustered AQP4‐IgG.
Our data suggest that AQP4‐IgG does not inhibit AQP4 water permeability nor does it alter the cellular distribution or OAP assembly of AQP4 61. Figure 4A shows high water permeability in plasma membrane vesicles isolated from AQP4‐expressing cells compared to control null cells, as measured by stopped‐flow light scattering, a sensitive, quantitative method capable of resolving small differences in water permeability. High concentrations of NMO serum or monoclonal NMO‐IgGs did not inhibit AQP4 water permeability. The absence of effect of AQP4‐IgG on AQP4 water permeability is not unexpected, as AQP4‐IgG is large compared to AQP4 such that it is not sterically possible to accommodate more than one AQP4‐IgG per AQP4 tetramer, which contains four monomers and, thus, four separate water pores. The inability of AQP4‐IgG to inhibit AQP4 water permeability was confirmed by two other laboratories 40, 42, although one study that reported AQP4‐IgG inhibition of AQP4 water permeability 21 utilized a time‐to‐burst oocyte swelling assay, which is an inaccurate surrogate of osmotic water permeability.
Figure 4.
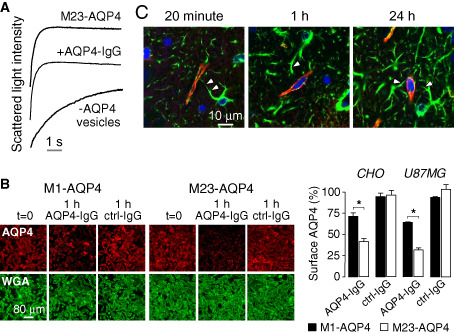
AQP4‐IgG does not inhibit AQP4 water permeability or cause AQP4 internalization in astrocytes in vivo. A. Stopped‐flow light scattering measurement of osmotic water permeability in plasma membrane vesicles from AQP4‐expressing and control cells. Vesicles were pre‐incubated with a high concentration of AQP4‐IgG where indicated. B. AQP4‐IgG‐induced internalization of M23‐AQP4 is more rapid than M1‐AQP4 in transfected cell lines. (Left) CHO cells expressing M1‐ or M23‐AQP4 stained for surface AQP4 (red) and plasma membrane (WGA, green) before (t = 0) and after 1 h incubation at 37°C with NMO‐IgG or control‐IgG. (Right) Percentage of AQP4 remaining at the cell surface for CHO and U87MG cells. C. Perivascular astrocyte surface localization of fluorescently labeled AQP4‐IgG in vivo in mice following intracerebral injection. Confocal microscopy showing AQP4‐IgG (red) in brain stained for GFAP (green) and nuclei (blue) at indicated times after intracerebral injection. Arrowheads indicate astrocytes stained for GFAP whose processes extend to the perivascular space. Adapted from Ratelade et al 54 and Rossi et al 61.
Super‐resolution imaging of OAPs showed that AQP4‐IgG binding to AQP4 does not affect OAP size 60, 61. One study, which reported that AQP4‐IgG causes AQP4 clustering at the plasma membrane 21, was likely observing general cellular toxicity, as addition of cytotoxic agents reproduced the clustering phenomena 60. An initial study demonstrated that adding AQP4‐IgG to cells stably transfected with a green fluorescent protein‐AQP4 chimera caused rapid internalization and degradation of AQP4 20. The same group subsequently reported AQP4‐IgG‐induced preferential internalization of M1‐AQP4 in astrocytes 21. However, cellular internalization of AQP4 and AQP4‐IgG, if it occurs in vivo, would protect astrocytes from CDC and ADCC. Using a dark quencher to resolve extracellular vs. intracellular localization of fluorescent AQP4‐IgG, our lab found rapid, selective internalization of AQP4‐IgG and AQP4 in transfected cell cultures. Internalization of M23‐AQP4 was more rapid than M1‐AQP4 in cells expressing these isoforms separately (Figure 4B), although identical rates of M23‐ and M1‐AQP4 were found in cells co‐expressing both isoforms 61. Importantly, there was little or no internalization of AQP4‐IgG or AQP4 in primary cultures of mouse astrocytes or in brain in vivo 54. Following injection of a fluorescent AQP4‐IgG in mouse brain, AQP4‐IgG and AQP4 were localized to perivascular astrocyte end‐feet for up to 24 h after injection, indicating minimal or no AQP4‐IgG internalization (Figure 4C). The polarized distribution of AQP4 may be responsible for the differences in cell culture vs. in vivo measurements.
Together, current evidence supports the conclusion that AQP4‐IgG is cytotoxic to astrocytes by CDC and ADCC mechanisms. We conclude that AQP4‐IgG binding does not affect AQP4 water permeability, plasma membrane aggregation or AQP4 internalization in vivo. Another proposed effect of AQP4‐IgG is internalization of glutamate transporter EAAT2 involving a direct AQP4‐EAAT2 interaction 20, which was proposed to cause glutamate excitotoxicity in vivo. However, follow‐up work showed no significant internalization of EAAT2 in astrocytes exposed to high concentrations of AQP4‐IgG or impairment in astrocyte glutamate uptake 54.
In Vivo Consequences of AQP4‐IgG Binding to AQP4
A central question in NMO pathogenesis is how the initial cellular effects of AQP4‐IgG binding to AQP4, and complement‐ and cell‐dependent astrocyte cytotoxicity produce NMO lesions. Human NMO lesions show loss of AQP4 and glial fibrillary acidic protein (GFAP) immunoreactivity, which indicates astrocyte damage, infiltration by neutrophils, eosinophils and macrophages, disruption of the blood‐brain barrier, perivascular deposition of activated complement, and demyelination with oligodendrocyte and neuron loss 28, 39, 46. A working model of NMO pathogenesis is diagrammed in Figure 5. Binding of AQP4‐IgG to AQP4 on astrocyte end‐feet activates complement, producing primary astrocyte injury. Astrocyte injury is followed by recruitment of inflammatory cells, first granulocytes and later macrophages, which further disrupt the blood‐brain barrier. Astrocyte loss and inflammation, with degranulation of neutrophils and eosinophils, and cytokine release secondarily damage oligodendrocytes, causing demyelination and neuron loss. Details of this mechanism are being elucidated.
Figure 5.
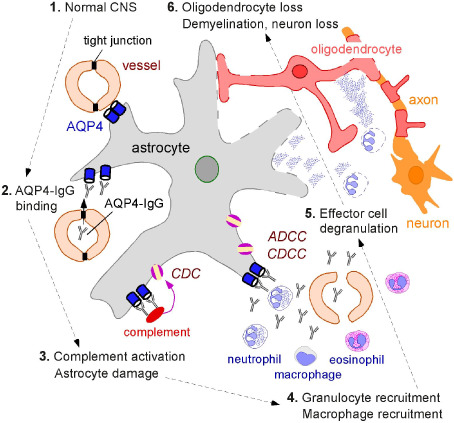
Proposed mechanisms of NMO pathogenesis. AQP4 is expressed at astrocyte end‐feet facing the blood‐brain barrier formed by endothelial cells connected by tight junctions (labeled “1”). In NMO, by unknown mechanisms, circulating AQP4‐IgG crosses the blood‐brain barrier and binds AQP4 on astrocytes (labeled “2”). This leads to recruitment and activation of complement and deposition of the membrane attack complex (MAC), producing astrocyte damage (labeled “3”). Complement activation and cytokine secretion by astrocytes recruit inflammatory cells (eosinophils, neutrophils and macrophages), which further disrupt the blood‐brain barrier, allowing more entry of AQP4‐IgG (labeled “4”). Degranulating inflammatory cells (labeled “5”) and astrocyte damage secondarily cause oligodendrocyte injury, myelin loss and axon damage (labeled “6”).
Rodent models support the conclusion that AQP4‐IgG is pathogenic in NMO and have provided information about the involvement of specific leukocyte types. The original studies showed that peripheral administration of AQP4‐IgG exacerbates CNS lesions in rats with preexisting neuroinflammation created by experimental autoimmune EAE 2, 6, 29 or pretreatment with complete Freund's adjuvant 30. The major caveat in these studies, however, is the background hyper‐inflammatory environment and presence of myelin protein‐reactive T cells. More compelling evidence for a pathogenic role of AQP4‐IgG came from a model involving intracerebral injection of AQP4‐IgG and human complement in mice, which produced lesions with loss of AQP4 and GFAP, neutrophil and macrophage infiltration, loss of myelin, and perivascular deposition of activated complement 56, 65. A recent variation of this model, involving continuous 3‐day intracerebral infusion of AQP4‐IgG by minipump, produced lesions that closely recapitulate human NMO including prominent eosinophil infiltration 89.
More relevant animal models of NMO are needed—ideally, models of AQP4‐IgG seropositivity that spontaneously develop NMO pathology in optic nerve and spinal cord. Mice made seropositive by intravenous administration of human AQP4‐IgG show rapid binding of AQP4‐IgG in AQP4‐expressing peripheral organs (kidney, stomach, trachea and skeletal muscle) and in circumventricular organs in brain that have a leaky blood‐brain barrier 55. However, NMO pathology was not seen, even following maneuvers to permeabilize the blood‐brain barrier. Mice are unlikely to be suitable second‐generation NMO models because of the very weak intrinsic activity of mouse complement and the presence in mouse serum of factor(s) that inhibit complement from multiple species.
Ex vivo mouse organ culture models of NMO spinal cord and optic nerve pathology have been developed 90, which have been useful in studying the role of inflammatory cells and soluble factors 89 and potential therapeutics 50, 72, 73, 75. Organ culture models allow exposure of NMO‐relevant tissues to defined conditions and factors. The ex vivo spinal cord model involves culture of 300‐μm‐thick vibratome‐cut transverse slices of mouse spinal cord on transwell porous supports. After 7 days in culture, exposure to AQP4‐IgG and complement produced marked loss of GFAP, AQP4 and myelin, deposition of activated complement, and microglial cell activation.
Notwithstanding the limitations of existing in vivo and ex vivo models, important information has been obtained on the role of specific leukocytes in NMO pathogenesis. Although CDC is a major mechanism in NMO pathogenesis, the importance of ADCC has become apparent. Intracerebral injection of AQP4‐IgG and NK cells in mice produced NMO‐like lesions with astrocyte injury but without myelin loss 56. Although NK cells and cytotoxic T cells are rarely seen in human NMO lesions 66, neutrophils, eosinophils and macrophages, which can produce ADCC and CDCC, are abundant in NMO lesions. Studies using the mouse intracerebral‐injection model have implicated important roles of neutrophils and eosinophils in NMO 67, 89, as demonstrated by reduced lesion size in neutropenic or eosinopenic mice and increased lesion size in neutrophilic or hypereosinophilic mice. Secreted neutrophil elastases and eosinophil granule toxins worsened NMO lesions. The neutrophil elastase inhibitor Sivelastat and the second‐generation antihistamine Cetirizine, which has eosinophil‐stabilizing action, reduced NMO pathology in the in vivo and ex vivo mouse models 89, suggesting the potential utility of neutrophil‐ and eosinophil‐targeted therapies in NMO.
NMO Therapeutics Targeting AQP4‐IgG and AQP4
Current NMO therapies include general immunosuppression (eg, corticosteroids, azathioprine, mycophenolate mofetil), immunomodulation (B‐cell depletion by rituximab) and plasma exchange. An open‐label clinical trial of a monoclonal antibody inhibitor of complement (eculizumab) is in progress. There is a need for new therapies with improved efficacy and without the potential long‐term side effects of general immunosuppressive drugs. The central role of AQP4‐IgG binding to AQP4 in NMO pathogenesis provides a unique opportunity to develop targeted therapeutics in NMO.
Several recently introduced therapeutic possibilities are based on blocking of AQP4‐IgG binding to AQP4 or inactivation of AQP4‐IgG to eliminate its CDC‐ and ADCC‐effector functions. In one strategy, a nonpathogenic, monoclonal antibody (“aquaporumab”) was generated from a recombinant monoclonal AQP4‐IgG that binds tightly to AQP4. Mutations were introduced in its Fc region to eliminate its CDC‐ and ADCC‐effector functions 73 (Figure 6A, left). Because an IgG1 antibody is significantly larger than an AQP4 tetramer as seen in Figure 3C (right), aquaporumab competitively displaces AQP4‐IgGs in NMO patient sera, preventing cytotoxicity (Figure 6A, right). Aquaporumab greatly reduced NMO lesions in spinal cord slice cultures and in mice receiving intracerebral AQP4‐IgG and complement. Because aquaporumab targeting of AQP4 is highly selective, minimal toxicity is anticipated. In an alternative blocker approach, high‐throughput screening identified several small‐molecule drugs and natural products, including the antiviral agent arbidol, that bind to AQP4 and sterically prevent AQP4‐IgG binding 72. Interestingly, small molecule screening also identified idiotype‐selective compounds that prevent antibody binding to AQP4 by interaction at the variable region of AQP4‐IgG 50. Further preclinical optimization of monoclonal antibody and small‐molecule blockers is in progress.
Figure 6.
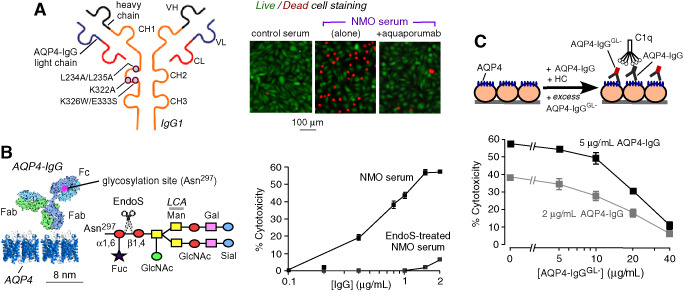
NMO therapies under development based on blocking of AQP4‐IgG binding to AQP4 and AQP4‐IgG inactivation. A. (Left) Schematic of AQP4‐IgG antibody showing heavy (VH) and light (VL) chain variable regions, light chain constant region (CL) and heavy chain constant regions (CH1‐CH3). Locations of amino acid mutations introduced in the CH2 domain of AQP4‐IgG to reduce CDC (K322A), ADCC (K326W/E333S) or both (L234A/L235A). (Right) Aquaporumab (AQmab) prevents CDC following exposure to AQP4‐IgG and complement as shown by live/dead cell assay. B. (Left) Schematic of IgG showing the Fc glycosylation at Asn‐297 and Fab binding to AQP4. Sugar moiety at Asn‐297 with EndoS cleavage site shown. Asn, asparagine; Fuc, fucose; GlcNac, N‐acetylglucosamine; Man, mannose; Gal, galactose; Sial, sialic acid. (Right) CDC in AQP4‐expressing CHO cells incubated with control of Endo S‐treated NMO serum and human complement. C. EndoS‐treated NMO‐IgG protects against CDC caused by (untreated) NMO‐IgG. LDH release assayed in CHO cells after 1 h incubation with indicated concentrations of NMO‐IgG and NMO‐IgGGL ‐, together with 2% human complement. Adapted from Tradtrantip et al 73 and Tradtrantip et al 75.
Antibody inactivation is another potential strategy for therapy of autoimmune disorders caused by pathogenic antibodies. This approach utilizes bacterial enzymes that target IgG‐class antibodies with very high selectivity, neutralizing their Fc effector functions and, hence, their pathogenicity. One such enzyme, Endoglycosidase S (EndoS) is a 108 kDa protein encoded by gene ndoS of Streptococcus pyogenes. EndoS selectively digests asparagine‐linked glycans on the heavy chain of all IgG subclasses without action on other immunoglobulin classes or other glycoproteins (Figure 6B, left). Glycosylation of a conserved asparagine (Asn‐297) on the CH2 domain of IgG heavy chains is essential for antibody‐effector functions. EndoS treatment of NMO patient serum prevented CDC (Figure 6B, right) and ADCC without impairing NMO‐IgG binding to AQP4 75. Cytotoxicity was also prevented by addition of EndoS after NMO‐IgG binding to AQP4. The EndoS‐treated, nonpathogenic NMO‐IgG competitively displaced pathogenic NMO‐IgG bound to AQP4 (Figure 6C) and prevented NMO pathology in spinal cord slice culture and mouse models of NMO. EndoS deglycosylation thus converts pathogenic NMO‐IgG into therapeutic blocking antibodies. An alternative enzymatic neutralization method utilizes the bacterial enzyme IdeS (IgG‐degrading enzyme of S. pyogenes), which selectively cleaves IgG antibodies to yield Fc and F(ab')2 fragments. IdeS treatment of monoclonal NMO‐IgGs and NMO patient serum abolished CDC and ADCC 74. Binding of NMO‐IgG to AQP4 was similar to that of the NMO‐F(ab')2 generated by IdeS cleavage. NMO‐F(ab')2 competitively displaced pathogenic NMO‐IgG, preventing cytotoxicity, and in addition, the Fc fragments generated by IdeS cleavage reduced ADCC. IdeS greatly reduced NMO lesions in mice. EndoS or IdeS treatment of blood may be beneficial in NMO, perhaps by therapeutic apheresis using surface‐immobilized enzyme.
Biology of AQP4 and AQP4‐IgG: Remaining Questions
Although much has been learned about the biology of AQP4, largely from knockout mice, many questions remain about cellular mechanisms as well as the biological significance of M1‐ vs. M23‐AQP4 expression and their assembly in OAPs. It remains unclear whether loss of AQP4 water‐transport function is directly responsible for defective clearance of excess brain water and for impaired neuroexcitation in knockout mice or whether these phenotypes represent secondary phenomena. AQP4‐selective inhibitors are needed both as research tools to study the physiological functions of AQP4 and as potential therapeutics, for example, to reduce cytotoxic brain swelling in ischemic stroke.
The binding of AQP4‐IgG to extracellular loops on AQP4 has been clearly demonstrated, although it is likely that different monoclonal AQP4‐IgGs have very different binding mechanisms. Co‐crystallization of purified AQP4 with monoclonal AQP4‐IgGs is a high priority. Resolution of the composition of polyclonal AQP4‐IgG in NMO patient sera is important as well, as it would be interesting to correlate AQP4‐IgG concentration and composition with clinical disease activity. Although substantial progress has been made in elucidating the pathogenic role of AQP4‐IgG in NMO and the cellular consequence of AQP4‐IgG binding to AQP4, major questions remain unresolved. It is not known why NMO lesions localize mainly to spinal cord and optic nerve without affecting peripheral, AQP4‐expressing organs. It is not known how peripheral AQP4‐IgG initially enters the CNS. While the roles of neutrophils, eosinophils and cytotoxic T cells in NMO have now been well studied, the role of macrophages is unclear, as is the involvement of AQP4‐sensitized T cells. Finally, the efficacy of the recently introduced AQP4‐IgG/AQP4 blocker and AQP4‐IgG inactivation strategies needs to be tested in human clinical trials.
Acknowledgments
We thank Drs Jeffrey Bennett (Univ. Colorado Denver) and Marios Papadopoulos (St. Georges Univ. London) for continued collaboration on NMO pathogenesis mechanisms and therapeutics, and for many productive discussions, as well as Drs Julien Ratelade, Hua Zhang and Andrea Rossi for their contributions to NMO research in the Verkman laboratory. This work was supported by grants from the Guthy‐Jackson Charitable Foundation and the National Institutes of Health (grants EY13574, EB00415, DK35124, HL73856, DK86125 and DK72517).
References
- 1. Auguste KI, Jin S, Uchida K, Yan D, Manley GT, Papadopoulos MC, Verkman AS (2007) Greatly impaired migration of implanted aquaporin‐4‐deficient astroglial cells in mouse brain toward a site of injury. FASEB J 21:108–116. [DOI] [PubMed] [Google Scholar]
- 2. Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C et al (2009) Intrathecal pathogenic anti‐aquaporin‐4 antibodies in early neuromyelitis optica. Ann Neurol 66:617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Binder DK, Yao X, Zador Z, Sick TJ, Verkman AS, Manley GT (2006) Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin‐4 water channels. Glia 53:631–636. [DOI] [PubMed] [Google Scholar]
- 4. Bloch O, Papadopoulos MC, Manley GT, Verkman AS (2005) Aquaporin‐4 gene deletion in mice increases focal edema associated with staphylococcal brain abscess. J Neurochem 95:254–262. [DOI] [PubMed] [Google Scholar]
- 5. Bloch O, Auguste KI, Manley GT, Verkman AS (2006) Accelerated progression of kaolin‐induced hydrocephalus in aquaporin‐4‐deficient mice. J Cereb Blood Flow Metab 26:1527–1537. [DOI] [PubMed] [Google Scholar]
- 6. Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M et al (2009) Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo . Ann Neurol 66:630–643. [DOI] [PubMed] [Google Scholar]
- 7. Carbrey JM, Agre P (2009) Discovery of the aquaporins and development of the field. Handb Exp Pharmacol 190:3–28. [DOI] [PubMed] [Google Scholar]
- 8. Crane JM, Bennett JL, Verkman AS (2009) Live cell analysis of aquaporin‐4 M1/M23 interactions and regulated orthogonal array assembly in glial cells. J Biol Chem 284:35850–35860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Crane JM, Lam C, Rossi A, Gupta T, Bennett JL, Verkman AS (2011) Binding affinity and specificity of neuromyelitis optica autoantibodies to aquaporin‐4 M1/M23 isoforms and orthogonal arrays. J Biol Chem 286:16516–16524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Crane JM, Verkman AS (2009) Determinants of aquaporin‐4 assembly in orthogonal arrays revealed by live‐cell single‐molecule fluorescence imaging. J Cell Sci 122:813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Crane JM, Verkman AS (2009) Reversible, temperature‐dependent supramolecular assembly of aquaporin‐4 orthogonal arrays in live cell membranes. Biophys J 97:3010–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Crane JM, Van Hoek AN, Skach WR, Verkman AS (2008) Aquaporin‐4 dynamics in orthogonal arrays in live cells visualized by quantum dot single particle tracking. Mol Biol Cell 19:3369–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cui Y, Bastien DA (2011) Water transport in human aquaporin‐4: molecular dynamics (MD) simulations. Biochem Biophys Res Commun 412:654–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fenton RA, Moeller HB, Zelenina M, Snaebjornsson MT, Holen T, MacAulay N (2010) Differential water permeability and regulation of three aquaporin 4 isoforms. Cell Mol Life Sci 67:829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Frydenlund DS, Bhardwaj A, Otsuka T, Mylonakou MN, Yasumura T, Davidson KG et al (2006) Temporary loss of perivascular aquaporin‐4 in neocortex after transient middle cerebral artery occlusion in mice. Proc Natl Acad Sci USA 103:13532–13536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Frigeri A, Gropper MA, Umenishi F, Kawashima M, Brown D, Verkman AS (1995) Localization of MIWC and GLIP water channel homologs in neuromuscular, epithelial and glandular tissues. J Cell Sci 108:2993–3002. [DOI] [PubMed] [Google Scholar]
- 17. Furman CS, Gorelick‐Feldman DA, Davidson KG, Yasumura T, Neely JD, Agre P, Rash JE (2003) Aquaporin‐4 square array assembly: opposing actions of M1 and M23 isoforms. Proc Natl Acad Sci U S A 100:13609–13614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hasegawa H, Ma T, Skach W, Matthay MA, Verkman AS (1994) Molecular cloning of a mercurial‐insensitive water channel expressed in selected water‐transporting tissues. J Biol Chem 269:5497–5500. [PubMed] [Google Scholar]
- 19. Hinson SR, Pittock SJ, Lucchinetti CF, Roemer SF, Fryer JP, Kryzer TJ, Lennon VA (2007) Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology 69:2221–2231. [DOI] [PubMed] [Google Scholar]
- 20. Hinson SR, Roemer SF, Lucchinetti CF, Fryer JP, Kryzer TJ, Chamberlain JL et al (2008) Aquaporin‐4‐binding autoantibodies in patients with neuromyelitis optica impair glutamate transport by down‐regulating EAAT2. J Exp Med 205:2473–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hinson SR, Romero MF, Popescu BF, Lucchinetti CF, Fryer JP, Wolburg H et al (2012) Molecular outcomes of neuromyelitis optica (NMO)‐IgG binding to aquaporin‐4 in astrocytes. Proc Natl Acad Sci U S A 109:1245–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hiroaki Y, Tani K, Kamegawa A, Gyobu N, Nishikawa K, Suzuki H et al (2006) Implications of the aquaporin‐4 structure on array formation and cell adhesion. J Mol Biol 355:628–639. [DOI] [PubMed] [Google Scholar]
- 23. Ho JD, Yeh R, Sandstrom A, Chorny I, Harries WE, Robbins RA et al (2009) Crystal structure of human aquaporin 4 at 1.8 A and its mechanism of conductance. Proc Natl Acad Sci U S A 106:7437–7442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hu J, Verkman AS (2006) Increased migration and metastatic potential of tumor cells expressing aquaporin water channels. FASEB J 20:1892–1894. [DOI] [PubMed] [Google Scholar]
- 25. Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA et al (2012) A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med 4:147ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jin BJ, Rossi A, Verkman AS (2011) Model of aquaporin‐4 supramolecular assembly in orthogonal arrays based on heterotetrameric association of M1‐M23 isoforms. Biophys J 100:2936–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jin BJ, Zhang H, Binder DK, Verkman AS (2013) Aquaporin‐4‐dependent K+ and water transport modeled in brain extracellular space following neuroexcitation. J Gen Physiol 141:119–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim W, Kim SH, Kim HJ (2011) New insights into neuromyelitis optica. J Clin Neurol 7:115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kinoshita M, Nakatsuji Y, Kimura T, Moriya M, Takata K, Okuno T et al (2009) Neuromyelitis optica: passive transfer to rats by human immunoglobulin. Biochem Biophys Res Commun 386:623–627. [DOI] [PubMed] [Google Scholar]
- 30. Kinoshita M, Nakatsuji Y, Kimura T, Moriya M, Takata K, Okuno T et al (2010) Anti‐aquaporin‐4 antibody induces astrocytic cytotoxicity in the absence of CNS antigen‐specific T cells. Biochem Biophys Res Commun 394:205–210. [DOI] [PubMed] [Google Scholar]
- 31. Landis DM, Reese TS (1974) Arrays of particles in freeze‐fractured astrocytic membranes. J Cell Biol 60:316–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li J, Verkman AS (2001) Impaired hearing in mice lacking aquaporin‐4 water channels. J Biol Chem 276:31233–31237. [DOI] [PubMed] [Google Scholar]
- 33. Li J, Patil RV, Verkman AS (2002) Mildly abnormal retinal function in transgenic mice without Muller cell aquaporin‐4 water channels. Invest Ophthalmol Vis Sci 43:573–579. [PubMed] [Google Scholar]
- 34. Li L, Zhang H, Varrin‐Doyer M, Zamvil SS, Verkman AS (2011) Proinflammatory role of aquaporin‐4 in autoimmune neuroinflammation. FASEB J 25:1556–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lu DC, Zhang H, Zador Z, Verkman AS (2008) Impaired olfaction in mice lacking aquaporin‐4 water channels. FASEB J 22:3216–3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lu M, Lee MD, Smith BL, Jung JS, Agre P, Verdijk MA et al (1996) The human AQP4 gene: definition of the locus encoding two water channel polypeptides in brain. Proc Natl Acad Sci U S A 93:10908–10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lucchinetti CF, Mandler RN, McGavern D, Bruck W, Gleich G, Ransohoff RM et al (2002) A role for humoral mechanisms in the pathogenesis of Devic's neuromyelitis optica. Brain 125:1450–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW et al (2000) Aquaporin‐4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med 6:159–163. [DOI] [PubMed] [Google Scholar]
- 39. Marignier R, Nicolle A, Watrin C, Touret M, Cavagna S, Varrin‐Doyer M et al (2010) Oligodendrocytes are damaged by neuromyelitis optica immunoglobulin G via astrocyte injury. Brain 133:2578–2591. [DOI] [PubMed] [Google Scholar]
- 40. Melamud L, Fernandez JM, Rivarola V, Di Giusto G, Ford P, Villa A, Capurro C (2012) Neuromyelitis optica immunoglobulin G present in sera from neuromyelitis optica patients affects aquaporin‐4 expression and water permeability of the astrocyte plasma membrane. J Neurosci Res 90:1240–1248. [DOI] [PubMed] [Google Scholar]
- 41. Neely JD, Christensen BM, Nielsen S, Agre P (1999) Heterotetrameric composition of aquaporin‐4 water channels. Biochemistry 38:11156–11163. [DOI] [PubMed] [Google Scholar]
- 42. Nicchia GP, Mastrototaro M, Rossi A, Pisani F, Tortorella C, Ruggieri M et al (2009) Aquaporin‐4 orthogonal arrays of particles are the target for neuromyelitis optica autoantibodies. Glia 57:1363–1373. [DOI] [PubMed] [Google Scholar]
- 43. Nielsen S, Nagelhus EA, Amiry‐Moghaddam M, Bourque C, Agre P, Ottersen OP (1997) Specialized membrane domains for water transport in glial cells: high‐resolution immunogold cytochemistry of aquaporin‐4 in rat brain. J Neurosci 17:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Noell S, Fallier‐Becker P, Deutsch U, Mack AF, Wolburg H (2009) Agrin defines polarized distribution of orthogonal arrays of particles in astrocytes. Cell Tissue Res 337:185–195. [DOI] [PubMed] [Google Scholar]
- 45. Padmawar P, Yao X, Bloch O, Manley GT, Verkman AS (2005) K+ waves in brain cortex visualized using a long‐wavelength K+‐sensing fluorescent indicator. Nat Methods 2:825–827. [DOI] [PubMed] [Google Scholar]
- 46. Papadopoulos MC, Verkman AS (2012) Aquaporin 4 and neuromyelitis optica. Lancet Neurol 11:535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Papadopoulos MC, Verkman AS (2013) Aquaporin water channels in the nervous system. Nature Rev Neurosci 14:265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Papadopoulos MC, Manley GT, Krishna S, Verkman AS (2004) Aquaporin‐4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J 18:1291–1293. [DOI] [PubMed] [Google Scholar]
- 49. Papadopoulos MC, Saadoun MC, Verkman AS (2008) Aquaporins and cell migration. Pflugers Arch 456:693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Phuan PW, Anderson MO, Tradtrantip L, Zhang H, Tan J, Bennett JL, Verkman AS (2012) A small molecule screen yields idiotype‐specific blockers of neuromyelitis optica immunoglobulin G binding to aquaporin‐4. J Biol Chem 287:36837–36844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Phuan PW, Ratelade J, Rossi A, Tradtrantip L, Verkman AS (2012) Complement‐dependent cytotoxicity in neuromyelitis optica requires aquaporin‐4 assembly in orthogonal arrays. J Biol Chem 287:13829–13839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pisani F, Mastrototaro M, Rossi A, Nicchia GP, Tortorella C, Ruggieri M et al (2011) Identification of two major conformational aquaporin‐4 epitopes for neuromyelitis optica autoantibody binding. J Biol Chem 286:9216–9224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rash JE, Yasumura T, Hudson CS, Agre P, Nielsen S (1998) Direct immunogold labeling of aquaporin‐4 in square arrays of astrocyte and ependymocyte plasma membranes in rat brain and spinal cord. Proc Natl Acad Sci U S A 95:11981–11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ratelade J, Bennett JL, Verkman AS (2011) Evidence against cellular internalization in vivo of NMO‐IgG, aquaporin‐4, and excitatory amino acid transporter 2 in neuromyelitis optica. J Biol Chem 286:45156–45164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ratelade J, Bennett JL, Verkman AS (2011) Intravenous neuromyelitis optica autoantibody in mice targets aquaporin‐4 in peripheral organs and area postrema. PLoS One 6:e27412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ratelade J, Zhang H, Saadoun S, Bennett JL, Papadopoulos MC, Verkman AS (2012) Neuromyelitis optica IgG and natural killer cells produce NMO lesions in mice without myelin loss. Acta Neuropathol 123:861–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rojek A, Praetorius J, Frøkiaer J, Nielsen S, Fenton RA (2008) A current view of the mammalian aquaglyeroporins. Ann Rev Physiol 70:301–327. [DOI] [PubMed] [Google Scholar]
- 58. Rossi A, Crane JM, Verkman AS (2011) Aquaporin‐4 Mz isoform: brain expression, supramolecular assembly and neuromyelitis optica antibody binding. Glia 59:1056–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rossi A, Baumgart F, van Hoek AN, Verkman AS (2012) Post‐Golgi supramolecular assembly of aquaporin‐4 in orthogonal arrays. Traffic 13:43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rossi A, Moritz TJ, Ratelade J, Verkman AS (2012) Super‐resolution imaging of aquaporin‐4 orthogonal arrays of particles in cell membranes. J Cell Sci 125:4405–4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rossi A, Ratelade J, Papadopoulos MC, Bennett JL, Verkman AS (2012) Neuromyelitis optica (NMO) IgG does not alter aquaporin‐4 water permeability, plasma membrane M1/M23 isoform content or supramolecular assembly. Glia 60:2027–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Saadoun S, Papadopoulos MC, Davies DC, Krisha S, Bell BA (2002) Aquaporin‐4 expression is increased in oedematous human brain tumours. J Neurol Neurosurg Psychiatry 72:262–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Saadoun S, Papadopoulos MC, Hara‐Chikuma M, Verkman AS (2005) Impairment of angiogenesis and cell migration by targeted aquaporin‐1 gene disruption. Nature 434:786–792. [DOI] [PubMed] [Google Scholar]
- 64. Saadoun S, Papadopoulos MC, Watanabe H, Yan D, Manley GT, Verkman AS (2005) Involvement of aquaporin‐4 in astroglial cell migration and glial scar formation. J Cell Sci 118:5691–5698. [DOI] [PubMed] [Google Scholar]
- 65. Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC (2010) Intra‐cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain 133:349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Saadoun S, Bridges LR, Verkman AS, Papadopoulos MC (2012) Paucity of natural killer and cytotoxic T cells in human neuromyelitis optica lesions. Neuroreport 23:1044–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Saadoun S, Waters P, Macdonald C, Bell BA, Vincent A, Verkman AS, Papadopoulos MC (2012) Neutrophil protease inhibition reduces neuromyelitis optica‐immunoglobulin G‐induced damage in mouse brain. Ann Neurol 71:323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Silberstein C, Bouley R, Huang Y, Fang P, Pastor‐Soler N, Brown D, Van Hoek AN (2004) Membrane organization and function of M1 and M23 isoforms of aquaporin‐4 in epithelial cells. Am J Physiol 287:F501–F511. [DOI] [PubMed] [Google Scholar]
- 69. Solenov E, Watanabe H, Manley GT, Verkman AS (2004) Sevenfold‐reduced osmotic water permeability in primary astrocyte cultures from AQP‐4‐deficient mice, measured by a fluorescence quenching method. Am J Physiol Cell Physiol 286:C426–C432. [DOI] [PubMed] [Google Scholar]
- 70. Strohschein S, Huttmann K, Gabriel S, Binder DK, Heinemann U, Steinhauser C (2011) Impact of aquaporin‐4 channels on K+ buffering and gap junction coupling in the hippocampus. Glia 59:973–980. [DOI] [PubMed] [Google Scholar]
- 71. Tajima M, Crane JM, Verkman AS (2010) Aquaporin‐4 (AQP4) associations and array dynamics probed by photobleaching and single‐molecule analysis of green fluorescent protein‐AQP4 chimeras. J Biol Chem 285:8163–8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tradtrantip L, Zhang H, Anderson MO, Saadoun S, Phuan PW, Papadopoulos MC et al (2012) Small‐molecule inhibitors of NMO‐IgG binding to aquaporin‐4 reduce astrocyte cytotoxicity in neuromyelitis optica. FASEB J 26:2197–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tradtrantip L, Zhang H, Saadoun S, Phuan PW, Lam C, Papadopoulos MC et al (2012) Anti‐aquaporin‐4 monoclonal antibody blocker therapy for neuromyelitis optica. Ann Neurol 71:314–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tradtrantip L, Asavapanumas N, Verkman AS (2013) Therapeutic cleavage of anti‐aquaporin‐4 autoantibodies in neuromyelitis optica by an IgG‐selective proteinase. Mol Pharmacol 83:1268–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tradtrantip L, Ratelade J, Zhang H, Verkman AS (2013) Enzymatic deglycosylation converts neuromyelitis optica anti‐AQP4 IgG into a therapeutic blocking antibody. Ann Neurol 73:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Verbavatz JM, Ma T, Gobin R, Verkman AS (1997) Absence of orthogonal arrays in kidney, brain and muscle from transgenic knockout mice lacking water channel aquaporin‐4. J Cell Sci 110:2855–3860. [DOI] [PubMed] [Google Scholar]
- 77. Verkman AS (2012) Aquaporins in clinical medicine. Annu Rev Med 63:303–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Verkman AS (2005) More than just water channels: unexpected cellular roles of aquaporins. J Cell Sci 118:3225–3232. [DOI] [PubMed] [Google Scholar]
- 79. Verkman AS, Hara‐Chikuma M, Papadopoulos MC (2008) Aquaporins—new players in cancer biology. J Mol Med (Berl) 86:523–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Vincent T, Saikali P, Cayrol R, Roth AD, Bar‐Or A, Prat A, Antel JP (2008) Functional consequences of neuromyelitis optica‐IgG astrocyte interactions on blood‐brain barrier permeability and granulocyte recruitment. J Immunol 181:5730–5737. [DOI] [PubMed] [Google Scholar]
- 81. Walz T, Fujiyoshi Y, Engel A (2009) The AQP structure and functional implications. Handb Exp Pharmacol 190:31–56. [DOI] [PubMed] [Google Scholar]
- 82. Wolburg H, Wolburg‐Buchholz K, Fallier‐Becker P, Noell S, Mack AF (2011) Structure and functions of aquaporin‐4‐based orthogonal arrays of particles. Int Rev Cell Mol Biol 287:1–41. [DOI] [PubMed] [Google Scholar]
- 83. Yang B, Verkman AS (1997) Water and glycerol permeability of aquaporins 1‐5 and MIP determined quantitatively by expression of epitope‐tagged constructs in Xenopus oocytes. J Biol Chem 272:16140–16146. [DOI] [PubMed] [Google Scholar]
- 84. Yang B, Ma T, Verkman AS (1995) cDNA cloning, gene organization, and chromosomal localization of a human mercurial insensitive water channel. Evidence for distinct transcriptional units. J Biol Chem 270:22907–22913. [DOI] [PubMed] [Google Scholar]
- 85. Yang B, Brown D, Verkman AS (1996) The mercurial insensitive water channel (AQP‐4) forms orthogonal arrays in stably transfected Chinese hamster ovary cells. J Biol Chem 271:4577–4580. [PubMed] [Google Scholar]
- 86. Yang B, Zador Z, Verkman AS (2008) Glial cell aquaporin‐4 overexpression in transgenic mice accelerates cytotoxic brain swelling. J Biol Chem 283:15280–15286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yu X, Green M, Gilden D, Lam C, Bautista K, Bennett JL (2011) Identification of peptide targets in neuromyelitis optica. J Neuroimmunol 236:65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhang H, Verkman AS (2008) Evidence against involvement of aquaporin‐4 in cell‐cell adhesion. J Mol Biol 382:1136–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zhang H, Verkman AS (2013) Eosinophil pathogenicity mechanisms and therapeutics in neuromyelitis optica. J Clin Invest 123:2306–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Zhang H, Bennett JL, Verkman AS (2011) Ex vivo spinal cord slice model of neuromyelitis optica reveals novel immunopathogenic mechanisms. Ann Neurol 70:943–954. [DOI] [PMC free article] [PubMed] [Google Scholar]