

Abstract
Epidemiologic studies have shown that diabetes mellitus is associated positively with increased risk of pancreatic ductal adenocarcinoma (PDAC), and recent meta-analysis studies showed that metformin, reduces the risk of pancreatic cancer (PC). We tested the effects of metformin on pancreatic intraepithelial neoplasia (PanIN) and their progression to PDAC in p48Cre/+.LSL-KrasG12D/+ transgenic mice. Mice fed control diet showed 80% and 62% incidence of PDAC in males and females, respectively. Male mice showed 20% and 26%, and female mice showed 7% and 0% PDAC incidence with 1000- and 2000-ppm metformin treatments, respectively. Both doses of metformin decreased pancreatic tumor weights by 34% to 49% (P < 0.03–0.001). The drug treatment caused suppression of PanIN 3 (carcinoma in situ) lesions by 28% to 39% (P < .002) and significant inhibition of carcinoma spread in the pancreas. The pancreatic tissue and/or serum of mice fed metformin showed a significant inhibition of mammalian target of rapamycin (mTOR), extracellular signal-regulated kinases (ERK), phosphorylated extracellular signal-regulated kinases (pErk), and insulin-like growth factor 1 (IGF-1) with an increase in phosphorylated 5′ adenosine monophosphate kinase (pAMPK), tuberous sclerosis complex 1 (TSC1, TSC2), C-protein and an autophagy related protein 2 (ATG2). The cancer stem cell (CSC) markers were significantly decreased (P < 0.04–0.0002) in the pancreatic tissue. These results suggest that biologic effects of metformin are mediated through decreased CSC markers cluster of differentiation 44 (CD44 and CD133), aldehyde dehydrogenase isoform 1 (ALDH1), and epithelial cell adhesion molecule (EPCAM) and modulation of the mTOR signaling pathway. Our preclinical data indicate that metformin has significant potential for use in clinical trials for PC chemoprevention.
Introduction
Pancreatic cancer (PC) is a devastating disease almost uniformly lethal, with a <6% 5-year survival. The estimated incidence of PC in the United States has increased to 42,220 new cases with more than 38,460 deaths in 2013 and is now the fourth leading cause of cancer mortality in both men and women [1]. The Pancreatic Action Network estimates that, by 2020, PC will become the second leading cause of cancer-related deaths in the United States. Lack of early diagnosis and effective interventions are major factors in the poor prognosis and dismal survival rates [2–4]. So far, a range of targeted therapies against epidermal growth factor receptor, RAS/MAP kinase effector kinase, and vascular endothelial growth factor has failed to improve survival significantly in many clinical trials. The PC precursors, pancreatic intraepithelial neoplasia (PanIN), progress slowly over many years to the development of invasive PC [2–4]. Hence, developing preventive strategies to delay progression of PC is currently of intense interest.
Several epidemiological studies have shown that type 2 diabetes mellitus is positively associated with increased risk for developing PC and worse clinical outcome [5–8]. A recent analysis of a large pooled set of studies included in the National Cancer Institute Pancreatic Cancer Cohort Consortium has provided strong support for a positive association between obesity and increased risk of PC [9]. More than 80% of patients with PC have diabetes [5–9]. The interrelationship between PC and diabetes and how diabetes affects the clinical outcome of PC have yet to be fully elucidated. Improved understanding of the disease mechanisms shared by diabetes and PC may be a key to development of novel preventive strategies. Obesity, metabolic syndrome, new-onset diabetes, and chronic pancreatitis may increase risk of PC [10–13]; thus, patients with new-onset diabetes and/or chronic pancreatitis may constitute a population in whom PC can be detected early. Metformin (1,1-dimethylbiguanide hydrochloride), the most widely prescribed drug for treatment of type 2 diabetes worldwide, is emerging as a potential anticancer agent. Recent meta-analysis studies showed that metformin, an insulin-lowering agent, reduces risk of PC. A recent study of 973 patients demonstrated that metformin treatment was associated with a 62% reduction of risk for PC [10].
Several cancers, including PC, are characterized by aberrant activation of mammalian target of rapamycin (mTOR) [11]. Genetic and molecular epidemiologic data strongly suggest that use of metformin for lowering blood insulin levels and reversal of insulin resistance targets insulin-like growth factor (IGF) signaling and modulates AMP kinase (AMPK), which could be a useful strategy for PC prevention. In vitro and preclinical animal models confirmed that metformin induces AMPK activation, inhibits the Akt/mTOR pathway, and also eliminates cancer stem cells (CSCs) and inhibits tumor growth [12,13]. Metformin activation of phosphorylated AMPK (pAMPK) can inhibit mTOR activity in several ways, including directly targeting raptor, a component of rapamycin-sensitive mTOR complex 1 [14]. Both mTOR and AMPK control the energy status of a cell (ATP/AMP ratio) and regulate key aspects of cell growth. Insulin/IGF receptor signaling is mediated through the phosphatidylinositol 3-kinase/Akt/mTOR signaling pathway [15], a key pathway in PC proliferation [16]. Although epidemiological associations do not establish causation, they provide an important line of evidence that supports the need of further preclinical and clinical studies. Studies have shown clearly that metformin administration inhibits the growth of human pancreatic adenocarcinoma PANC-1 and MiaPaCa-2 tumor xenografts in vivo in a dose-dependent manner [17]. Studies have also demonstrated that metformin elicits a protective effect by targeting CSCs and mTOR signaling. However, efficacy and systematic analysis of metformin in a preclinical model that recapitulates human PC has not yet been evaluated.
In this study, we investigated the effects of the antidiabetic drug metformin on PanIN and their progression to pancreatic ductal adenocarcinoma (PDAC) in p48Cre/+.LSL-KrasG12D/+ transgenic mice. We examined the impact of metformin on tumor incidence and spread and the direct effects of metformin on mTOR signaling and on CSC markers. Our results provide key experimental evidence to guide future clinical investigations to establish metformin therapy in patients with PC.
Materials and Methods
Mouse Model, Diet, and Handling
The generation of p48Cre/+;LSL-KrasG12D/+ mice expressing activated KrasG12D oncogene has been described previously [18]. All animal research was performed under the animal protocols approved by the University of Oklahoma Health Sciences Center Institutional Animal Care and Use Committee. Animals were housed in ventilated cages under standardized conditions (21°C, 60% humidity, 12-hour light/12-hour dark cycle, 20 air changes per hour) in the University Rodent Barrier Facility. Semi-purified modified AIN-76A diet ingredients were purchased from Bio-Serv, Inc (Frenchtown, NJ). Metformin was procured from the Division of Cancer Prevention, National Cancer Institute chemoprevention drug repository. Metformin (1000 and 2000 ppm) was premixed with small quantities of casein and then blended into the diet using a Hobart Mixer. Both control and experimental diets were prepared weekly and stored in the cold room. Mice were allowed ad libitum access to the respective diets and to automated tap water purified by reverse osmosis. The primary antibodies used were pAMPK, annexin V, cluster of differentiation 133 (CD133), and C-protein from Abcam (Cambridge, MA); mTOR, CD24, CD44 (HCAM), extracellular signal-regulated kinases (ERK), phosphorylated extracellular signal-regulated kinases (pErk), epithelial cell adhesion molecule (EPCAM), α-tubulin, p70-S6K (serine/threonine kinase) and insulin-like growth factor 1 (IGF-1) from Santa Cruz Biotechnology (SantaCruz, CA); and c-Myc and β-actin from Cell Signaling Technology (Danvers, MA).
Breeding and Genotyping Analysis
LSL-KrasG12D/+ and p48Cre/+ mice were maintained in a C57BL/6 heterozygous genetic background. LSL-KrasG12D/+ and p48cre/+ mice were bred, and the offspring of activated p48Cre/+.LSL-KrasG12D/+ and C5BL/6 wild-type mice were generated at required quantities. Briefly, genomic DNA was isolated from tail tissue samples using the mini-prep kit (Invitrogen, Carlsbad, CA). Polymerase chain reaction (PCR) was performed for Kras and Cre genes using the following conditions: denaturation at 95°C for 5 minutes, followed by 35 cycles at 95°C for 1 minute, 60°C for 1 minute, and 72°C for 1 minute. Oligonucleotide primer sequences used were given as follows: Kras, 5′-CCTTTACAAGCGCACGCAGAG-3′ (sense) and 5′-AGCTAGCCACCATGGCTTGAGTAAGTCTGCA-3′ (antisense); p48Cre, 5′-ACCGTCAGTACGTGAGATATCTT-3′ (sense) and 5-ACCTGAAGATGTTCGCGATTATCT-3′ (antisense). PCR products were separated on a 2% agarose gel. Successful recombination yields were 550- and 350-bp products for Kras and Cre genes, respectively. The genotype of each pup was confirmed by tail DNA extraction and PCR.
Preclinical Efficacy Assay
Male and female p48Cre/+-LSL-KrasG12D/+ transgenic mice were used in the efficacy study (Figure W1). The experimental protocol is summarized in Figure W1. Briefly, 5-week-old mice were selected and randomized so that average body weights in each group were equal (n = 20–35 per group, p48Cre/+-LSL-KrasG12D/+ mice and n = 30 per group, C57BL/6 wild-type mice) and were fed AIN-76A diet for 1 week. At 6 weeks of age, mice were fed AIN-76A experimental diets containing 0-, 1000-, or 2000-ppm metformin until termination of the study (Figure W1). Mice were checked routinely for signs of weight loss or any signs of toxicity or any abnormalities. Body weight of each animal was measured once weekly for the first 6 weeks and then once a month until termination (Figure W2). After 265 days (38 weeks or ∼10 months) on experimental diets, all mice at 44 weeks (∼300 days) of age were killed by CO2 asphyxiation and necropsied; pancreata were collected from all groups, weighed, and snap frozen in liquid nitrogen for further analysis. Pancreata (head to tail) required for histopathologic and immunohistochemical (IHC) evaluations to identify PanIN lesions and PDAC and for evaluation of various molecular markers were fixed in 10% neutral buffered formalin as previously described.
Analysis of PanIN Lesions and PDAC
Normal and tumor pancreatic tissues were fixed in 10% neutral buffered formalin for 24 to 48 hours and then processed and embedded in paraffin according to standard protocols. Tissue sections (4 µm) of each pancreas stained with hematoxylin and eosin (H&E) were evaluated histologically by a pathologist blinded to the experimental groups. PanIN lesions and carcinomas were classified according to histopathology criteria as previously described [18]. To quantify the progression of PanIN lesions, the total number of ductal lesions and their grades were determined. The relative proportion of each PanIN lesion grade to the overall number of analyzed ducts was recorded for each animal. Similarly, pancreatic carcinoma and normal appearing pancreatic tissue were evaluated for all the animals.
IHC and Immunofluorescence
Five-micrometer fixed sections were incubated with primary antibodies in a hybridization chamber for 1 hour at room temperature or overnight at 4°C. The primary antibodies used were for mTOR, pAMPK, CD24, CD44 (HCAM), EPCAM, and annexin V. Following incubations with primary antibody, sections were incubated for 1 hour with anti-mouse or anti-rabbit secondary antibodies, as appropriate for each primary, then visualized with DAB and counterstained with hematoxylin for IHC and counterstained with 4′,6-diamidino-2-phenylindole (DAPI) for immunohistofluorescence (IHF). Slides were observed under an Olympus microscope 1X701, and digital computer images were recorded with an Olympus DP70 camera.
Protein Extraction and Western Blot Assay
Pancreata harvested from mice fed diets with or without metformin were homogenized and lysed in ice-cold lysis buffer. After a brief vortexing, the lysates were collected by centrifugation at 12,000g for 15 minutes at 4°C, and protein concentrations were measured with the Bio-Rad Protein Assay reagent (Bio-Rad, Hercules, CA). An aliquot (50 µg of protein per lane) of the total protein was separated with 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. After blocking with 5% milk powder, membranes were probed for expression of ERK, pERK, c-Myc, CD133, p70-S6, IGF-1, C-protein, β-actin, and ?-tubulin in hybridizing solution (1:500, in TBS-Tween 20 solution) using the respective primary antibodies and then probed with the appropriate HRP-conjugated secondary antibodies. Detection was performed using the SuperSignal West Pico Chemiluminescence procedure (Pierce, Rockford, IL). All the Western blot experiments were repeated two times with six untreated samples, six 1000-ppm metformin-treated samples, and six 2000-ppm metformin-treated samples. ImageJ software was used for quantification of the blots.
Quantitative Real-Time PCR Analysis
Total RNA from tumor samples was extracted using RNA Kit for isolation of total cellular RNA (Invitrogen) as per the manufacturer's instructions. Equal quantities of DNA-free RNA were used for reverse transcription (RT) reactions for making cDNA using SuperScript reverse transcriptase (Invitrogen). The real-time PCR was carried out in a 25-ml reaction volume using 3 ml of a 1:10 cDNA dilution containing SYBR Green master mix (BioRad) and primers for mTOR, Rheb, tuberous sclerosis complex 1 (TSC1), TSC2, cyclin D1, autophagy related protein 2 (ATG2), caspase-3, Bax, Bcl-2, CD44, EPCAM, CD133, aldehyde dehydrogenase 1 (ALDH1), and Dclk1 as previously described [13,19–21]. All PCRs were done in a Bio-Rad iCycler iQ real-time PCR detection system. The fluorescence threshold values (Ct) were calculated. Relative mRNA levels were assessed by standardization to glyceraldehyde phosphate dehydrogenase. Results are expressed as a fold difference in gene expression.
Statistical Analysis
The data are presented as means ± SE. Differences in body weights were analyzed by analysis of variance. Statistical differences between control and treated groups were evaluated using Fisher's exact test for PDAC incidence, and unpaired t test with Welch's correction was used for PanIN and PDAC lesions. Differences between groups are considered significant at P < .05.
Results
General Health of Animals
Transgenic p48Cre/+;LSL-KrasG12D/+ mice fed AIN-76A or metformin diets had steady body weight gains. As shown in Figure W2, there was no significant difference in body weight in the mice fed either AIN-76A or AIN-76A diet supplemented with either 1000- or 2000-ppm metformin. None of the animals fed the metformin diets exhibited any observable toxicity or any gross morphologic changes to liver, spleen, kidney, or lung.
Effect of Metformin Diet on Pancreas Weight and PDAC Incidence in Transgenic Mice
Pancreatic weight is a simple marker to determine the progression of tumor. In the C57BL/6J wild-type mice, the average pancreas weight was 0.29 ± 0.01 g in male mice and 0.28 ± 0.05 g in female mice. A significant increase in pancreas weight was observed in p48Cre/+; LSL-KrasG12D/+ mice compared with wild-type mice (1.45 ± 0.13 g in male mice and 1.1 ± 0.13 g in female mice, P < .0001). As summarized in Figure 1, A and B, the effect of metformin on the weight of pancreatic tumor revealed a significant difference between treatment groups in p48Cre/+;LSL-KrasG12D/+ mice. In transgenic mice fed 1000- or 2000-ppm metformin, the pancreas weights were 0.88 ± 0.14 and 0.95 ± 0.09 g, respectively, in males and 0.61 ± 0.04 and 0.56 ± 0.05 in females. In addition, no significant differences in the body weights were observed between wild-type mice and p48Cre/+;LSL-KrasG12D/+ mice fed with either AIN-76A or metformin-supplemented diets (Figure W2).
Figure 1.
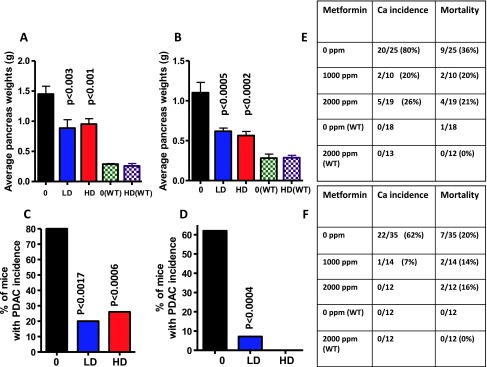
(A and B) Effect of metformin on pancreas weights at the termination of the experiment in male (A) and female (B) mice. Both doses ofmetformin significantly reduced the pancreatic tumor weights. (C and D) Effect of metformin on the incidence (percentage of mice with carcinomas) of PDAC in male (C) and female (D) mice. Effect of metformin on survival and mortality of male (E) and female (F) mice.
Extensive histopathologic analysis of the pancreas using H&E-stained slides revealed no microscopic pathologic alterations in wild-type mice fed either AIN-76A or metformin-supplemented diets (Figure 1, E and F). In contrast, AIN-76A diet-fed male and female p48Cre/+;LSL-KrasG12D/+ mice demonstrated 80% and 62% incidence of PDAC (percentage of mice with PDAC), respectively (Figure 1, C–F, and W3). Metformin treatment at 1000 and 2000 ppm significantly inhibited PDAC incidence by 67% to 80% in males and by up to 93% to 100% in female transgenic mice (Figure 1, C–F).
Inhibition of PanIN Lesion Progression and Carcinoma Spread by Metformin
Histologic analysis showed that none of the wild-type mice had PanIN lesions, whereas 100% penetrance of PanIN lesions was observed in the transgenic mice fed AIN-76A or metformin-supplemented diets. The numbers of PanIN 1, 2, and 3 lesions in male transgenic mice fed AIN-76A diet were (mean ± SE) 177 ± 36, 155 ± 15, and 169 ± 17, respectively; in the mice fed 1000-ppm metformin, PanIN 1, 2, and 3 numbers were 368 ± 59, 193 ± 28, and 103 ± 13, respectively; and in mice fed 2000-ppm metformin, they were 375 ± 13, 182 ± 18 and 122 ± 37, respectively (Figure 2A). The numbers of PanIN 1, 2, and 3 lesions in female transgenic mice fed AIN-76A diet were 193 ± 33, 150 ± 20, and 124 ± 18, respectively; in the mice fed 1000-ppm metformin, the numbers were 357 ± 36, 155 ± 20, and 79 ± 11; and in mice fed 2000-ppm metformin, they were 289 ± 47, 169 ± 15, and 185 ± 22, respectively (Figure 2B). The number of PanIN 3 lesions or carcinoma in situ was suppressed by 28% to 39% in the metformin-treated groups (Figure 2, A and B). A statistically significant increase in the number of PanIN 1 and 2 lesions was observed in metformin-supplemented mouse pancreas, suggesting a potential blockade of further progression of these lesions to carcinoma in situ and PDAC. Pancreata of transgenic mice fed AIN-76A diet showed a 43.6 ± 7.5% (male; Figure 2C) and 41 ± 7.5% (female; Figure 2D) spread of PDAC within the pancreas. Most importantly, the carcinoma spread within the pancreas was significantly inhibited (>95%, P < .0001; Figure 2, C and D) by both doses of metformin in both genders of transgenic mice. In support of the above, we observed that up to 29% of the pancreas from mice treated with metformin was normal appearing (i.e., free from PanIN lesions and carcinoma), whereas up to 9% was found to be normally appearing in the untreated transgenic mice (Figure 2, E and F).
Figure 2.

(A and B) Effect of metformin on the PanIN multiplicity (means ± SE; A—male, B—female). (C and D) Percentage of carcinoma spread per pancreas (C—male, D—female; 0—0 ppm, LD—1000 ppm, HD—2000 ppm). (E and F) Effect of metformin on normal pancreas (E—male, F—female). The data in the panels were analyzed by unpaired t test with Welch's correction; values are considered statistically significant at P < .05.
Modulation of mTOR/pAMPK Signaling by Metformin
Activation of the mTOR/pAMPK pathway was measured by IHC and IHF in PanIN lesions and PDAC. As shown in Figure 3, markedly decreased mTOR staining with a dramatic increase in pAMPK was observed in PanIN lesions in p48Cre/+;LSL-KrasG12D/+ mice fed metformin-supplemented diets compared with the pancreatic tissues from mice fed AIN-76A diet alone. The expression of mTOR and its related signaling molecules in the pancreas was further analyzed using real-time quantitative PCR and/or Western blot assays. Snap frozen pancreatic tissues were analyzed for the fold change in mRNA expression of mTOR, Rheb, cyclin D1, ATG2, and the tumor suppressors TSC1 and TSC2. Total RNA was isolated and reverse transcribed into cDNA and quantified with a real-time PCR assay. As shown in Figure 4, A to F, compared with AIN-76A-fed p48Cre/+; LSL-KrasG12D/+ mice, the pancreatic tissues of metformin-fed transgenic mice showed a significantly decreased expression of mTOR, Rheb, and cyclin D1 (Figure 4, A, B, and E) with an increase in TSC1, TSC2, and ATG2 (Figure 4, C, D, and F; P < .05). More than a two-fold decrease of mTOR, Rheb, and cyclin D1 mRNA expression was found in mice fed the diet containing metformin in comparison with AIN-76A diet-fed mice (P < .05). More than a three-fold increase in mRNA expression was found for TSC1, TSC2, and ATG2 in mice fed a diet containing metformin.
Figure 3.

Effect of metformin on mTOR and pAMPK in pancreatic tumors. IHC and immunofluorescence analyses were performed with paraffin-embedded and microsectioned pancreatic tissues as described in the Materials and Methods section. A significantly decreased expression of mTOR and an increased pAMPK expression were seen with metformin treatment.
Figure 4.
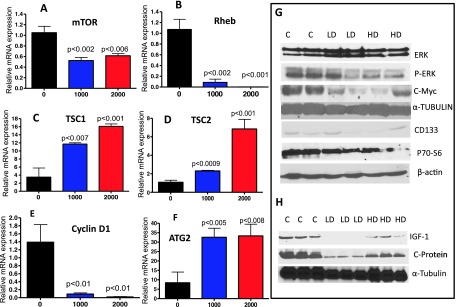
(A–F) Effect of metformin on the expression of mRNA for mTOR, Rheb, TSC1, TSC2, cyclin D1, and ATG2, as determined with quantitative real-time PCR. (G) Effect of metformin on the expression of ERK, pERK, cMYC, CD133, and p70-S6 proteins in PDAC. (H) Effect of metformin on IGF-1 and C-protein in the serum. Protein expression was analyzed by Western blot analysis as described in the text.
Western blot assays were performed to estimate the expression of ERK, pERK, c-Myc, p70-S6, and CD133 proteins in the pancreatic tissues and of IGF-1 and C-protein in serum. As shown in Figure 4G, there was an increase in the ERK levels with a significant decrease in pERK and its downstream markers in pancreatic tissues of mice treated with metformin-supplemented diets as compared with mice fed AIN-76A diet alone. The levels of IGF-1 and C-proteins in serum were decreased in mice treated with lower doses of metformin-supplemented diets (Figure 4H). These results are consistent with the effects of metformin on molecular signaling in earlier reports.
Inhibition of Cancer Stem Cell Markers by Metformin
To determine the expression of CSC markers in the pancreatic tissues of the transgenic mice, CD24, CD44, EPCAM, CD133, ALDH1, and DclK1 were analyzed using both IHF and real-time PCR approaches. IHF staining showed that PanIN lesions were positively labeled by both CD24 and CD44, which were co-localized in the pancreas of p48Cre/+;LSL-KrasG12D/+ mice fed AIN-76A diet alone (Figure 5A). Markedly decreased numbers of CD24 and CD44 and their co-localization were observed in lesions of mice fed a metformin-supplemented diet (Figure 5A). To examine the effect of metformin on the mRNA expression of CSC marker genes in pancreatic tissues, we measured the relative mRNA levels of the markers by quantitative RT-PCR. We found that metformin significantly decreased the expressions of CD24, CD44, EPCAM, CD133, and ALDH1 mRNA in pancreatic tissues of transgenic mice; there was a nonsignificant decrease in DclK1. These results suggest that the inhibition of tumor progression may be associated with the down-regulation of CSC markers (Figure 5, B–F) and are consistent with our in vivo tumor inhibition findings.
Figure 5.
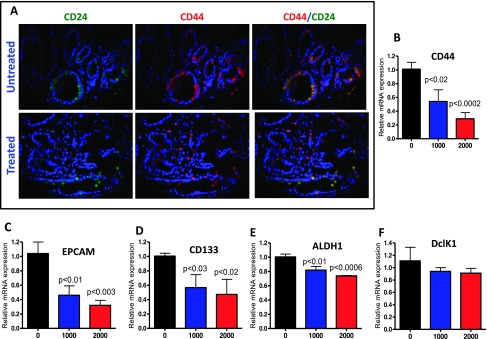
Effect of metformin on CSC markers in pancreatic tumors. (A) Immunofluorescence analysis for CD24 and CD44 was performed on paraffin-embedded and microsectioned pancreatic tissues as described in the Materials and Methods section. A significantly decreased expression of CD24, CD44, and CD44/CD244 ratio was seen with metformin treatment. (B-F) Effect of metformin on the expression of mRNA for the CSC markers CD44, EPCAM, CD133, ALDH1, and DclK1, as determined with quantitative real-time PCR.
Metformin Induces Apoptosis
We conducted fluorescence imaging microscopy to examine the effect of metformin on the expression of CSC surface biomarkers along with the apoptosis marker annexin V. As shown in Figure 6, the staining intensity of annexin V (green) was very low, whereas CD44 (red) was significantly higher in the PanIN lesions of mice fed AIN-76A diet. In contrast, we found a substantial increase in annexin V in lesions of treated pancreas, suggesting that metformin induces apoptosis in the lesions. Moreover, the CD44- and EPCAM-stained cells showed greater annexin V staining intensity in the metformin-treated pancreatic lesions compared with those in mice fed AIN-76A diet. These results demonstrate that metformin induces apoptosis and greatly decreases CSC markers CD44 and EPCAM (Figure 6). To further determine the effects of metformin on mRNA of apoptotic markers in pancreatic tissues, we measured relative mRNA levels of markers by quantitative RT-PCR. We found that metformin significantly increased the expressions of caspase-3 and Bax mRNA in pancreatic tissues of lower dose-treated transgenic mice; there was a nonsignificant increase in Bax with the higher dose (Figure 6). We observed a significant decrease in anti-apoptotic marker Bcl-2 on treatment with the lower dose of metformin (Figure 6). These results suggest that the inhibition of tumor progression may be associated with the increase in apoptosis and are consistent with our in vivo tumor inhibition findings.
Figure 6.

Effect of metformin on apoptosis and CSC markers. Dual immunofluorescence analysis for annexin V/CD44 (green/red) and annexin V/EPCAM (green/red) was performed with paraffin-embedded and microsectioned pancreatic tissues as described in the Materials and Methods section. A significant decrease in CSC markers (CD44 and EPCAM) was associated with an increased apoptosis (annexin V—green) on metformin treatment. Effect of metformin on the expression of mRNA for the apoptotic markers caspase-3 and Bax and anti-apoptotic marker Bcl-2 as determined with quantitative real-time PCR.
Discussion
Recent meta-analysis determined that the antidiabetic drug metformin reduces the incidence of overall cancers including liver, pancreatic, colorectal, and breast cancers as well as the mortality from these cancers [7,9]. Several observational and preclinical studies have shown that metformin may modify the risk for PC. The anticancer effects of metformin have also been demonstrated in many in vitro studies. In a retrospective study of patients with PC and preexisting diabetes, Sadeghi et al. observed that the overall survival duration was 4.1 months longer and the 1-year survival rate was 18.8% higher in patients treated with metformin than in those not treated with metformin. The beneficial effects of metformin were seen in all disease stages but were statistically significant only in patients with nonmetastatic disease. Metformin use was also associated with a 32% reduction in the risk of death [22]. These data provide strong supporting evidence that metformin has the potential to be used as a chemopreventive agent for treating high-risk individuals and patients with initial stages of PC. Studies conducted in various preclinical animal tumor models [13,23–25] and in vitro cancer cell lines [26–30] have shown that metformin inhibits tumor development and growth of cancer cells. While most previous in vitro studies used very high concentrations of metformin, the clinical significance of the inhibitory effects obtained using higher doses of metformin has been questioned. In PC studies, metformin has been shown to inhibit the growth of xenografts in nude mice through disruption of cross-talk between the insulin receptor and G protein-coupled receptors [15–17,31,32]. In a high-fat diet and carcinogen-induced pancreatic tumor model, metformin completely prevented the development of tumors [33].
To understand the mechanism for the anticancer effects of metformin, it is important to elucidate whether this antidiabetic drug can exert direct effects on in vivo transgenic animal models of PC that recapitulate the stepwise progression of precursor lesions to carcinoma as seen in humans. We have used a dose of metformin slightly higher than that used clinically to treat diabetes and a dose double that to evaluate its potential against prevention of PC progression in the p48Cre/+;LSL-KrasG12D/+ mouse model. This model is well established and extensively studied by us and others for chemoprevention of PC [4,18–20,34–37]. In the present study, we have demonstrated the chemopreventive effects of metformin against formation and progression of PanIN lesions to carcinoma. Recently, pancreatic xenograft studies have shown that metformin at 200 mg/kg body weight (∼1000 ppm in the diet) had maximal tumor suppression effects and caused a pronounced decrease in S6 and ERK [17]. We observed that metformin at 1000 ppm, i.e., ∼200 mg/kg body weight, showed 80% and 93% inhibition of PDAC incidence in male and female transgenic mice, respectively. However, we failed to observe a dose response effect with 2000-ppm treatment in male mice. A 74% suppression of PDAC incidence was seen in male mice treated with the higher dose of metformin, whereas no carcinoma was observed in female mice treated with this dose. These results are in line with the recent reports on the effective doses of metformin for PC. Further, Quinn et al. recently showed that metformin at 5 mg/ml in drinking water (equivalent to ∼5000 ppm in the diet) significantly inhibited lung tumorigenesis [38].
Consistent with the above PDAC incidence data, metformin-treated mice had a significant delay in progression of PanIN lesions versus untreated animals. A significant accumulation in the number of PanIN 1 and 2 lesions was observed in the metformin-treated pancreas compared with untreated, suggesting that many of the PanIN 1 and 2 lesions did not further progress into carcinoma in situ and PDAC. PanIN 3 lesions were reduced by 29% to 38% in the metformin-treated groups, clearly suggesting a blockade of the progression of precursor lesions to carcinoma. More than 95% inhibition of carcinoma spread was observed in male and female transgenic mice treated with metformin. Notably, the response rate is better in female mice compared to male mice because the transgenic male mice but not female mice with Kras activation develop much more aggressive PDAC (Figure 1, C and D). To our knowledge, this is the first report showing that the genetically predetermined progression of PanIN lesions to carcinoma in the conditional KrasG12D/+ mouse model can be attenuated by the antidiabetic drug metformin.
The ability of mutant oncogenic Kras to induce malignant transformation probably depends on its ability to persistently activate downstream effectors. We therefore reasoned that the ability of metformin to delay PanIN progression and inhibit carcinoma may be mediated, at least in part, by interfering with the Kras oncogenic pathways. Mechanistically, the metabolic regulation and antitumor activity of metformin are mediated mainly through activation of the AMPK signaling pathway. Metformin reduces mitochondrial ATP production by inhibiting complex I of oxidative phosphorylation [8], which results in the activation of the AMPK signaling pathway and, in turn, downregulates the mTOR pathway. Accumulating evidence from in vitro and in vivo studies has shown that metformin exerts its antitumor effect by targeting multiple signaling pathways such as AMPK/mTOR, anti-inflammatory pathways, cell cycle/apoptosis, insulin/IGF-1R, and angiogenesis and by targeting CSCs in cancers. Some studies also found that metformin could inhibit directly the mTOR pathway, independent of AMPK activation [39,40]. We determined the effects of metformin on mTOR and pAMPK signaling, CSC markers, and apoptosis. Our data suggest that metformin significantly regulates mTOR and pAMPK in pancreatic carcinogenesis. Our observation is consistent with other recent reports in which metformin disrupts mTOR, IGF-1, and CSC signaling pathways and prevents tumor growth [8,12,13,16,17,31,32,38]. We observed that metformin significantly reduces mTOR and its signaling molecules Rheb, pERK, c-Myc, p70-S6, and cyclin D1. We also found that metformin increases pAMPK and the tumor suppressors TSC1 and TSC2.
Recently, metformin has been reported to target CSCs selectively and to enhance the efficacy of chemotherapeutic drugs in blocking tumor growth [12]. CSCs are believed to be responsible for initiation of the malignant growth and metastasis and possess the ability to self-renew and give rise to differentiated tumor cells, which contribute to resistance to conventional therapeutics. Therefore, targeting CSCs may provide a novel strategy for cancer inhibition with better outcome. A detailed evaluation revealed that metformin did inhibit CSC markers in the PanIN lesions of the pancreas compared with nontreatment. Metformin inhibited the CSC markers CD24, CD44, EPCAM, CD133, ALDH1, and DclK1 in the present study. These markers have been identified as CSC markers in PC, and metformin was found to target these CSC markers in other cancers as well, thereby preventing tumor growth [8,12,13,20]. Metformin may also exert some of its antitumor activity through regulating lipid metabolism, endothelial function, immune functions [41–44], apoptosis, and insulin signaling. The analysis of serum revealed that metformin, at the lower dose, profoundly inhibits IGF-1 and C-protein, suggesting that these proteins could serve as biomarkers in individuals treated with metformin for chemoprevention of PC.
A previously reported case-controlled study of PC showed that a 2-year duration of metformin use was associated with reduced risk of PC [10], which suggests that metformin may not only prevent tumor development by inhibiting cell transformation and proliferation during the early stages of tumorigenesis but that it may also delay cancer progression [44]. Findings from the current study are consistent with other previous observations that support the use of metformin as a potential chemoprevention agent for PC and as a supplement for chemotherapy. Further transgenic mouse xenograft studies involving human PC cells along with improved imaging technologies [36,45,46] would strengthen the need for use of metformin for chemoprevention and treatment. Considering the high prevalence of diabetes in patients with PC and the lack of effective treatment strategies for this deadly disease, prospective studies should be conducted quickly to confirm the chemopreventive benefits of metformin use in patients with diabetes and PC.
In conclusion, our study shows that dietary metformin delayed the progression of PanIN lesions and ultimately decreased the incidence of invasive PC in p48Cre/+;LSL-KrasG12D/+ mice, suggesting that the anticancer action could be mediated, at least in part, through a direct effect on mTOR signaling and CSCs. One of the major concerns in chemoprevention strategies is potential toxicity associated with prolonged treatment. We show here that the widely used antidiabetic drug metformin targets PC cells along with cells positive for the markers of CSCs and mTOR signaling in oncogenic Kras-transformed pancreatic cells, inducing apoptosis without toxicity, and thus, its use may provide a new chemoprevention approach. Translational studies in early-phase clinical trials will validate the biomarkers of metformin that were observed in this study.
Supplementary Material
Acknowledgments
The authors thank the University of Oklahoma Health Sciences Center Rodent Barrier Facility staff. We also thank Julie Sando for valuable suggestions and editorial help.
Footnotes
We acknowledge support from the National Cancer Institute (N01CN-53300) and Kerley Cade Endowed Chair.
This article refers to supplementary materials, which are designated by Figures W1 to W3 and are available online at www.transonc.com.
References
- 1.American Cancer Society, author. Cancer Facts and Figures 2013. Atlanta, GA: American Cancer Society; 2013. [Google Scholar]
- 2.Mazur PK, Siveke JT. Genetically engineered mouse models of pancreatic cancer: unravelling tumour biology and progressing translational oncology. Gut. 2011;61:1488–1500. doi: 10.1136/gutjnl-2011-300756. [DOI] [PubMed] [Google Scholar]
- 3.Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2011;467:1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mohammed A, Janakiram NB, Lightfoot S, Gali H, Vibhudutta A, Rao CV. Early detection and prevention of pancreatic cancer: use of genetically engineered mouse models and advanced imaging technologies. Curr Med Chem. 2012;19:3701–3713. doi: 10.2174/092986712801661095. [DOI] [PubMed] [Google Scholar]
- 5.Gumbs AA. Obesity, pancreatitis, and pancreatic cancer. Obes Surg. 2008;18:1183–1187. doi: 10.1007/s11695-008-9599-3. [DOI] [PubMed] [Google Scholar]
- 6.Li D, Abbruzzese JL. New strategies in pancreatic cancer: emerging epidemiologic and therapeutic concepts. Clin Cancer Res. 2010;16:4313–4318. doi: 10.1158/1078-0432.CCR-09-1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeCensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B, Gandini S. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res (Phila) 2010;3:1451–1461. doi: 10.1158/1940-6207.CAPR-10-0157. [DOI] [PubMed] [Google Scholar]
- 8.Bao B, Wang Z, Li Y, Kong D, Ali S, Banerjee S, Ahmad A, Sarkar FH. The complexities of obesity and diabetes with the development and progression of pancreatic cancer. Biochim Biophys Acta. 2011;1815:135–146. doi: 10.1016/j.bbcan.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noto H, Goto A, Tsujimoto T, Noda M. Cancer risk in diabetic patients treated with metformin: a systematic review and meta-analysis. PLoS One. 2012;7:e33411. doi: 10.1371/journal.pone.0033411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li D, Yeung S-CJ, Hassan MM, Konopleva M, Abbruzzese JL. Antidiabetic therapies affect risk of pancreatic cancer. Gastroenterology. 2009;137:482–488. doi: 10.1053/j.gastro.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guertin DA, Sabatini DM. An expanding role for mTOR in cancer. Trends Mol Med. 2005;11:353–361. doi: 10.1016/j.molmed.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 12.Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009;69:7507–7511. doi: 10.1158/0008-5472.CAN-09-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bao B, Wang Z, Ali S, Ahmad A, Azmi AS, Sarkar SH, Banerjee S, Kong D, Li Y, Thakur S, et al. Metformin inhibits cell proliferation, migration and invasion by attenuating CSC function mediated by deregulating miRNAs in pancreatic cancer cells. Cancer Prev Res (Phila) 2012;5:355–364. doi: 10.1158/1940-6207.CAPR-11-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kisfalvi K, Rey O, Young SH, Sinnett-Smith J, Rozengurt E. Insulin potentiates Ca2+ signaling and phosphatidylinositol 4,5-bisphosphate hydrolysis induced by Gq protein-coupled receptor agonists through an mTOR-dependent pathway. Endocrinology. 2007;148:3246–3257. doi: 10.1210/en.2006-1711. [DOI] [PubMed] [Google Scholar]
- 16.Rozengurt E, Sinnett-Smith J, Kisfalvi K. Crosstalk between insulin/insulin-like growth factor-1 receptors and G protein-coupled receptor signaling systems: a novel target for the antidiabetic drug metformin in pancreatic cancer. Clin Cancer Res. 2010;16:2505–2511. doi: 10.1158/1078-0432.CCR-09-2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kisfalvi K, Moro A, Sinnett-Smith J, Eibl G, Rozengurt E. Metformin inhibits the growth of human pancreatic cancer xenografts. Pancreas. 2013;42:781–785. doi: 10.1097/MPA.0b013e31827aec40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mohammed A, Janakiram NB, Li Q, Madka V, Ely M, Lightfot S, Crawford H, Steele VE, Rao CV. The epidermal growth factor receptor inhibitor gefitinib prevents the progression of pancreatic lesions to carcinoma in a conditional LSL-KrasG12D/+ transgenic mouse model. Cancer Prev Res (Phila) 2010;11:1417–1426. doi: 10.1158/1940-6207.CAPR-10-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mohammed A, Janakiram NB, Ely M, Lightfoot S, Steele VE, Rao CV. Licofelone, a novel dual COX-LOX inhibitor prevents progression of PanIN lesions to pancreatic carcinoma by targeting miRNAs and cancer stem cells in p48Cre/+-LSL-KrasG12D/+ transgenic mice. Cancer Res; Proceedings of the 102nd Annual Meeting of the American Association for Cancer Research, Orlando, FL; AACR, Philadelphia, PA. 2011. p. 2839. [Google Scholar]
- 20.Mohammed A, Brewer M, Ritchie RL, Marya A, Lightfoot S, Janakiram NB, Steele VE, Rao CV. Metformin prevents progression of pancreatic intraepithelial neoplasia to ductal adenocarcinoma by targeting cancer stem cells and mTOR signaling; Proceedings of the 104th Annual Meeting of the American Association for Cancer Research, Washington, DC; AACR, Philadelphia. 2011. PAAbstract No. 2268. [Google Scholar]
- 21.Sureban SM, May R, Lightfoot SA, Hoskins AB, Lerner M, Brackett DJ, Postier RG, Ramanujam R, Mohammed A, Rao CV, et al. DCAMKL-1 regulates epithelial-mesenchymal transition in human pancreatic cells through a miR-200a-dependent mechanism. Cancer Res. 2013;71:2328–2338. doi: 10.1158/0008-5472.CAN-10-2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sadeghi N, Abbruzzese JL, Yeung SJ, Hassan M, Li D. Metformin use is associated with better survival of diabetic patients with pancreatic cancer. Clin Cancer Res. 2012;18:2905–2912. doi: 10.1158/1078-0432.CCR-11-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JW, Park S, Takahashi Y, Wang HG. The association of AMPK with ULK1 regulates autophagy. PLoS One. 2010;5:e15394. doi: 10.1371/journal.pone.0015394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhuang Y, Miskimins WK. Metformin induces both caspase-dependent and poly(ADP-ribose) polymerase-dependent cell death in breast cancer cells. Mol Cancer Res. 2011;9:603–615. doi: 10.1158/1541-7786.MCR-10-0343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sahra IB, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Y, Bost MF. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27:3576–3586. doi: 10.1038/sj.onc.1211024. [DOI] [PubMed] [Google Scholar]
- 26.Imamura H, Nhat KPH, Togawa H, Saito K, Iino R, Kato-Yamada Y, Nagai T, Noji H. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc Natl Acad Sci USA. 2009;106:15651–15656. doi: 10.1073/pnas.0904764106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heiden MGV, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vazquez A, Liu J, Zhou Y, Oltvai ZN. Catabolic efficiency of aerobic glycolysis: the Warburg effect revisited. BMC Syst Biol. 2010;4:58. doi: 10.1186/1752-0509-4-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quaile MP, Melich DH, Jordan HL, Nold JB, Chism JP, Polli JW, Smith GA, Rhodes MC. Toxicity and toxicokinetics of metformin in rats. Toxicol Appl Pharmacol. 2010;243:340–347. doi: 10.1016/j.taap.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 30.Sum CF, Webster JM, Johnson AB, Catalano C, Cooper BG, Taylor R. The effect of intravenous metformin on glucose metabolism during hyperglycaemia in type 2 diabetes. Diabet Med. 1992;9:61–65. doi: 10.1111/j.1464-5491.1992.tb01716.x. [DOI] [PubMed] [Google Scholar]
- 31.Kisfalvi K, Eibl G, Sinnett-Smith J, Rozengurt E. Metformin disrupts crosstalk between G protein-coupled receptor and insulin receptor signaling systems and inhibits pancreatic cancer growth. Cancer Res. 2009;69:6539–6545. doi: 10.1158/0008-5472.CAN-09-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006;66:10269–10273. doi: 10.1158/0008-5472.CAN-06-1500. [DOI] [PubMed] [Google Scholar]
- 33.Schneider MB, Matsuzaki H, Haorah J, Ulrich A, Standop J, Ding XZ, Adrian TE, Pour PM. Prevention of pancreatic cancer induction in hamsters by metformin. Gastroenterology. 2001;120:1263–1270. doi: 10.1053/gast.2001.23258. [DOI] [PubMed] [Google Scholar]
- 34.Mohammed A, Qian L, Janakiram NB, Lightfoot S, Steele VE, Rao CV. Atorvastatin delays progression of pancreatic lesions to carcinoma by regulating PI3/AKT signaling in p48Cre/+.LSL-KrasG12D/+ mice. Int J Cancer. 2012;131:1951–1962. doi: 10.1002/ijc.27456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mohammed A, Janakiram NB, Brewer M, Duff A, Lightfoot S, Brush RS, Anderson RE, Rao CV. Endogenous n-3 polyunsaturated fatty acids delay progression of pancreatic ductal adenocarcinoma in Fat-1-p48Cre/+-LSL-KrasG12D/+ mice. Neoplasia. 2012;14:1249–1259. doi: 10.1593/neo.121508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rao CV, Mohammed A, Janakiram NB, Qian L, Ritchie RL, Lightfoot S, Vibhudutta A, Steele VE. Inhibition of pancreatic intraepithelial neoplasia progression to carcinoma by nitric oxide-releasing aspirin in p48Cre/+-LSL-KrasG12D/+ mice. Neoplasia. 2012;14:778–787. doi: 10.1593/neo.121026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hocker JR, Mohammed A, Aston CE, Brewer M, Lightfoot SA, Rao CV, Hanas JS. Mass profiling of serum to distinguish mice with pancreatic cancer induced by a transgenic Kras mutation. Int J Cancer. 2013;133:2662–2671. doi: 10.1002/ijc.28285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quinn BJ, Dallos M, Kitagawa H, Kunnumakkara AB, Memmott RM, Hollander MC, Gills JJ, Dennis PA. Inhibition of lung tumorigenesis by metformin is associated with decreased plasma IGF-I and diminished receptor tyrosine kinase signaling. Cancer Prev Res (Phila) 2013;6:801–810. doi: 10.1158/1940-6207.CAPR-13-0058-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sahra BI, Regazzetti C, Robert G, Laurent K, Marchand-Brustel LY, Auberger P, Tanti JF, Giorgetti-Peraldi S, Bost F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011;71:4366–4372. doi: 10.1158/0008-5472.CAN-10-1769. [DOI] [PubMed] [Google Scholar]
- 40.Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B, Kemp BE, Bardeesy N, Dennis P, Schlager JJ, et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010;11:390–401. doi: 10.1016/j.cmet.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pollak M. Metformin and other biguanides in oncology: advancing the research agenda. Cancer Prev Res (Phila) 2010;3:1060–1065. doi: 10.1158/1940-6207.CAPR-10-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sahra BI, Marchand-Brustel LY, Tanti JF, Bost F. Metformin in cancer therapy: a new perspective for an old antidiabetic drug? Mol Cancer Ther. 2010;9:1092–1099. doi: 10.1158/1535-7163.MCT-09-1186. [DOI] [PubMed] [Google Scholar]
- 43.Li D. Metformin as an antitumor agent in cancer prevention and treatment. J Diabetes. 2011;3:320–327. doi: 10.1111/j.1753-0407.2011.00119.x. [DOI] [PubMed] [Google Scholar]
- 44.Yang YX. Do diabetes drugs modify the risk of pancreatic cancer? Gastroenterology. 2009;137:412–415. doi: 10.1053/j.gastro.2009.06.022. [DOI] [PubMed] [Google Scholar]
- 45.Tuli R, Surmak A, Reyes J, Hacker-Prietz A, Armour M, Leubner A, Blackford A, Tryggestad E, Jaffee EM, Wong J, et al. Development of a novel preclinical pancreatic cancer research model: bioluminescence image guided focal irradiation and tumor monitoring of orthotopic xenografts. Transl Oncol. 2012;5:77–84. doi: 10.1593/tlo.11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kayed H, Meyer P, He Y, Kraenzlin B, Fink C, Gretz N, Schoenberg SO, Sadick M. Evaluation of the metabolic response to cyclopamine therapy in pancreatic cancer xenografts using a clinical PET-CT system. Transl Oncol. 2012;5:335–343. doi: 10.1593/tlo.12166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.