

Significance
The nuclear factor kappa B (NF-κB) family of transcription factors regulates hundreds of genes in response to stress. Stress responses must be robust when needed and then completely and rapidly turned off. A family of inhibitor proteins, the IκBs, is responsible for turning off NF-κBs. Our study shows that IκBα turns off NF-κB(RelA/p50) by rapidly binding to the DNA-bound NF-κB, forming a transient ternary complex. The ternary complex lasts for only milliseconds before the DNA dissociates from the complex. The resulting IκBα–NF-κB complex is extremely stable and cannot be dissociated. Our study shows how the macromolecular interactions of IκBα are precisely tuned to efficiently and irreversibly turn off NF-κB transcriptional activity.
Keywords: transcription activation, DNA binding, NF-κB transcription, NF-κB post-induction repression, stopped-flow fluorescence kinetics
Abstract
We previously demonstrated that IκBα markedly increases the dissociation rate of DNA from NF-κB. The mechanism of this process remained a puzzle because no ternary complex was observed, and structures show that the DNA and IκBα binding sites on NF-κB are overlapping. The kinetics of interaction of IκBα with NF-κB and its complex with DNA were analyzed by using stopped-flow experiments in which fluorescence changes in pyrene-labeled DNA or the native tryptophan in IκBα were monitored. Rate constants governing the individual steps in the reaction were obtained from analysis of the measured rate vs. concentration profiles. The NF-κB association with DNA is extremely rapid with a rate constant of 1.5 × 108 M−1⋅s−1. The NF-κB–DNA complex dissociates with a rate constant of 0.41 s−1, yielding a KD of 2.8 nM. When IκBα is added to the NF-κB–DNA complex, we observe the formation of a transient ternary complex in the first few milliseconds of the fluorescence trace, which rapidly rearranges to release DNA. The rate constant of this IκBα-mediated dissociation is nearly equal to the rate constant of association of IκBα with the NF-κB–DNA complex, showing that IκBα is optimized to repress transcription. The rate constants for the individual steps of a more folded mutant IκBα were also measured. This mutant associates with NF-κB more rapidly than wild-type IκBα, but it associates with the NF-κB–DNA complex more slowly and also is less efficient at mediating dissociation of the NF-κB–DNA complex.
The nuclear factor κB (NF-κB) signaling system regulates cell growth, immune responses, inflammatory viral responses, and apoptotic death (1–5) and is often misregulated in cancer, arthritis, asthma, diabetes, AIDS, and viral infections (6–8). The NF-κB family of transcription factors consists of different homodimers or heterodimers of RelA (p65), p50, p52, c-Rel, and RelB (4), with the RelA/p50 heterodimer being the most abundant one in many cell types (9). NF-κBs are distinguished by a Rel homology domain, within which is an N-terminal domain, a dimerization domain, and a nuclear localization signal (NLS). The dimerization and N-terminal domains bind to the κB elements in the promoter and enhancer regions of target genes (10), whereas the dimerization domain and the NLS interact with the inhibitor proteins, IκBs (11, 12) (Fig. 1).
Fig. 1.

(A) Structure of the NF-κB–IκBα complex to the κB DNA [Protein Data Bank (PDB) ID code 1VKX (10)]. The RelA subunit of NF-κB is shown in green, the p50 subunit is shown in cyan, and the κB DNA is shown in (orange). Highlighted in bright pink is the AmC6–pyrene fluorophore, bound to the κB sequence. (B) Structure of the NF-κB–IκBα complex (subunits, coloring, and orientation are the same as in A bound and IκBα (magenta) [PDB ID code 1NFI (12)]. The naturally occurring Trp-258 used as the fluorophore to monitor IκBα binding is highlighted in bright pink. (C) Schematic diagram of the NF-κB signaling pathway showing the feedback loop that produces newly synthesized IκBα in purple.
IκBs are a family of homologous proteins that have an N-terminal regulatory domain, followed by six or more ankyrin repeats (ARs) and sometimes a PEST domain (rich in proline, glutamic acid, serine, and threonine) (13). Although the classical IκB family members include IκBα, IκBβ, and IκBε, experiments in knockout cell lines identified IκBα as the main mediator of the rapid postinduction repression of NF-κB activity (14). In resting cells and in vitro, IκBα was shown to bind extremely tightly to NF-κB (KD = 40 pM) (15, 16). This stable complex can only be dissociated by proteasome-mediated degradation following stimulation-induced phosphorylation (by the IκB kinase IKK) and subsequent ubiquitination (17–20). Proteasomal degradation of IκBα exposes the NLS on NF-κB, resulting in rapid translocation to the nucleus, where it binds the DNA and regulates transcription of numerous κB-dependent genes (19). The IκBα gene itself has κB sites and is strongly activated by NF-κB (21–23). The newly synthesized IκBα enters the nucleus, binds NF-κB (thereby inhibiting its transcriptional activity), and returns it to the cytosol (14). The newly synthesized IκBα is rapidly degraded by an ubiquitin-independent mechanism unless it is stabilized by binding to NF-κB, so the cellular concentration of free IκBα is always extremely low (15) (Fig. 1C).
We previously showed that IκBα accelerates the dissociation of NF-κB from the DNA (24). By using surface plasmon resonance (SPR) experiments, a pseudo-second-order rate constant of 106 M−1⋅s−1 was measured for this accelerated dissociation (24). The mechanism by which IκBα mediates this accelerated dissociation should involve formation of a ternary complex (DNA–NF-κB–IκBα); however, none was observed in the SPR experiments (24). A ternary complex was observed by NMR when an excess of DNA was added to the NF-κB–IκBα complex (25). In the NMR experiments, even at high concentrations of DNA, the first four ARs of IκBα remained associated with NF-κB, and the DNA caused disappearance of the signals from the AR(5–6)PEST region of IκBα. Dissociation of IκBα from NF-κB was not observed, even with addition of a large excess of DNA. In the present work, we describe fluorescence kinetics measurements of the individual steps of binding of IκBα, NF-κB, and DNA (Scheme 1). Formation of a very transient ternary complex is observed along the way to the efficient IκBα-mediated dissociation of NF-κB from the DNA. Each of the rate constants in Scheme 1 have been determined. The fluorescence intensity of pyrene-labeled DNA (DNA*) was measured for k1, k−1, k3, k5, and k−5. Changes in the fluorescence intensity of W258 in IκBα were measured in separate experiments to determine k2, k−2, k4, and k−4. It is important to note that the only species in Scheme 1 that is transcriptionally active is the NF-κB–DNA complex (N-D). None of the NF-κB complexes containing IκBα are transcriptionally active.
Scheme 1.
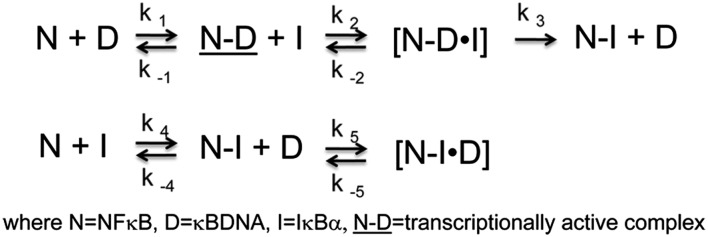
A stabilized mutant IκBαC186P/A220P (26), which we previously showed binds with the same affinity to NF-κB but was less efficient at mediating dissociation of the NF-κB–DNA complex in the SPR experiments (24), was also studied. Together, the results allow a full characterization of the formation of the transient ternary complex formed during IκBα-mediated dissociation of NF-κB from DNA.
Results and Discussion
DNA–NF-κB Binding Kinetics.
We measured DNA binding to a pyrene-labeled DNA hairpin (DNA*) containing the IFN-κB promoter sequence (27). The fluorescence of the pyrene attached to the DNA increased upon NF-κB binding, and the data fit best to a double exponential (Fig. 2 A and B) with the large amplitude phase being strongly dependent on NF-κB concentration and yielding an association rate constant of 1.5 × 108 M−1⋅s−1 (Fig. 2B). The smaller amplitude phase, with a rate constant of 5.0 × 107 M−1⋅s−1, was more weakly concentration-dependent. Similar biexponential behavior was also observed for the papillomavirus E2 protein with its specific DNA target (28, 29). As was observed for the E2 protein, extrapolation of the main amplitude phase results in an estimate of the kd that is faster than the measured one (see below), most likely because such extrapolated off rates represent the dissociation of an encounter complex and not the overall dissociation rate of a consolidated complex.
Fig. 2.

Stopped-flow fluorescence kinetic experiments in which a pyrene-labeled DNA hairpin (DNA*) (0.1 μM) was mixed with various concentrations of NF-κB. (A) Stopped-flow fluorescence trace for the association reaction (final concentrations were 500 nM NF-κB and 100 nM DNA*). Both the single-exponential fit (blue) and the double-exponential fit (red) are shown with the corresponding residuals below in the same colors. (B) Plot of the association rates at varying NF-κB concentrations. The main amplitude phase was linear with [NF-κB] yielding a k of 1.5 (± 0.2) × 108 M−1⋅s−1 (k1 in Scheme 1). The minor amplitude phase fit yielded a k of 5.0 (± 0.2) × 107 M−1⋅s−1. The y axis is the same for both plots.
Dissociation of NF-κB from DNA.
To determine the baseline rate constant for DNA dissociation from NF-κB, the DNA*–NF-κB complex was rapidly mixed with unlabeled DNA, and above a 20:1 molar excess of unlabeled DNA, full dissociation was observed with a kd of 0.41 s−1 (Fig. 3A). The KD for the hairpin DNA binding to NF-κB calculated from the association and dissociation rate constants was 2.8 nM, very similar to the reported value measured by SPR using longer DNA oligomers containing the same κB sequence (24).
Fig. 3.

(A) Stopped-flow fluorescence trace corresponding to dissociation of DNA* from NF-κB in the presence of unlabeled DNA. (B) Fluorescence traces corresponding to the dissociation of DNA* from NF-κB in the presence of a 5-fold (blue) or 20-fold (red) excess of IκBα or no IκBα (black). Upon binding of IκBα to the DNA*–NF-κB complex, the fluorescence increased in the first 100 ms and then decreased to the values expected for free DNA*. The first part of the curve represents the formation of the transient ternary complex DNA*–NF-κB–IκBα. The DNA* dissociation phase fit to a biexponential function with the larger amplitude phase becoming more rapid with increasing IκBα concentration. (C) The dissociation rates increased linearly with IκBα concentration, yielding a rate constant for IκBα-mediated dissociation of the NF-κB–DNA* complex of 3.68 (± 0.02) × 106 M−1⋅s−1 (k3 in Scheme 1). A horizontal line marks the NF-κB–DNA* dissociation rate (0.41 s−1) in the absence of IκBα.
IκBα-Mediated Dissociation of NF-κB from the DNA.
When even relatively low concentrations of IκBα were mixed with the same concentration of the DNA*–NF-κB complex instead of unlabeled DNA, the DNA* was rapidly displaced from the NF-κB. Within the first 100 ms, the fluorescence of the pyrene label in DNA*, which is highly sensitive to its environment, increased further upon association of the IκBα and then decreased back to the fluorescence expected for the free DNA* as IκBα-mediated dissociation of the DNA* occurred (Fig. 3B). The amplitude of the transient fluorescence increase was proportional to the concentration of IκBα and thus must be due to ternary complex formation. The second phase of each curve, corresponding to dissociation of the DNA*, could be fit to a biexponential process in which the larger amplitude phase became more rapid with increasing IκBα concentration. A smaller amplitude phase displayed a concentration-independent rate constant of 14 s−1. A plot of the rate of the main amplitude phase vs. concentration of IκBα yielded a second-order rate constant (k3 in Scheme 1), of 3.68 × 106 M−1⋅s−1 (Fig. 3C). Even at low concentrations of IκBα, the IκBα-mediated dissociation of NF-κB from the DNA was much faster than the rate constant for dissociation of DNA from NF-κB in the absence of IκBα, 0.41 s−1.
Association of IκBα with the NF-κB–DNA Complex.
The change in fluorescence of the native Trp, W258, in IκBα was used to measure the rate of association of IκBα with the NF-κB–DNA complex. Trp-258 was previously shown to be a sensitive probe for the folding of IκBα upon binding to NF-κB (30). This experiment would measure the formation of the ternary complex if it were stable; however, because it is transient, the product of this association experiment is expected to be the IκBα–NF-κB complex. The change in the IκBαW258 fluorescence upon binding the NF-κB–DNA complex fit to a single exponential (Fig. 4 A and B), and the rate vs. concentration data yielded a second-order rate constant (k2 in Scheme 1) of 5.65 × 106 M−1⋅s−1. For comparison, we measured the association rates of IκBα binding to NF-κB, which also fit to a single exponential (Fig. 4 C and D). The rate vs. concentration data yielded a rate constant (k4 in Scheme 1) of 1.44 × 107 M−1⋅s−1, 2.5-fold faster than the rate constant of IκBα association to the NF-κB–DNA complex. At first we were puzzled that the Trp fluorescence could be fit to a single exponential even though the IκBα-mediated dissociation process should involve two steps—the formation of the ternary complex and then the release of the DNA. However, the Trp fluorescence is expected to increase in both the ternary complex and the IκBα–NF-κB final complex. Given that the rate constant of IκBα association to the NF-κB–DNA complex (5.65 × 106 M−1⋅s−1; k2 in Scheme 1) is similar to the IκBα-mediated dissociation rate constant (3.68 × 106 M−1⋅s−1; k3 in Scheme 1), we think it is unlikely that we could have resolved two phases in the association reaction.
Fig. 4.

Stopped-flow fluorescence kinetic experiments in which IκBα was mixed with various concentrations of either the DNA–NF-κB complex (A and B) or NF-κB alone (C and D), and the change in intensity of the Trp fluorescence from the native W258 in IκBα was monitored. (A) Stopped-flow fluorescence trace for the association reaction (final concentrations were 0.1 μM IκBα and/or 0.4 μM DNA–NF-κB). The data fit to a single exponential, and the residuals are plotted below the graph. (B) Plot of the rates vs. DNA–NF-κB concentrations yielded a k of 5.65 (± 0.02) × 106 M−1⋅s−1 (k2 in Scheme 1). (C) Stopped-flow fluorescence trace for the association reaction (final concentrations were 0.1 μM IκBα and/or 0.4 μM NF-κB). The data fit to a single exponential, and the residuals are plotted below the graph. (D) Plot of the rates vs. NF-κB concentrations yielding a k of 1.44 (± 0.04) × 107 M−1⋅s−1 (k4 in Scheme 1).
Reverse Reactions.
Because W258 is the only Trp in IκBα, mutation of this residue to Phe provided a spectroscopically silent IκBα for monitoring the dissociation kinetics of IκBα. However, even when the IκBαW258F mutant was mixed in large excess with the NF-κB–IκBα complex, no fluorescence change (i.e., no dissociation) was observed over 10 min, in agreement with the very high stability of the complex (16).
We recently showed that by adding an excess of DNA to the NF-κB–IκBα complex, a “backwards ternary complex” could be formed (k5, Scheme 1) (25). To measure the rate of backwards ternary complex formation, we mixed DNA* with the NF-κB–IκBα complex. These association curves could be fit with a single exponential, and a plot of the rate vs. concentration of NF-κB–IκBα complex yielded a second-order rate constant of 8.6 × 107 M−1⋅s−1 (Table 1). We attempted to measure the dissociation of the DNA* from this backwards ternary complex by mixing a large excess of unlabeled DNA with a 5:1:1 mixture of DNA*–NF-κB–IκBα (conditions under which at least some of the DNA* is in the ternary complex). The rate constant of dissociation of DNA from this putative backwards ternary complex (k−5, Scheme 1) was 0.96 s−1 (Table 1). We were unable to observe dissociation of IκBα from the DNA–NF-κB–IκBα ternary complex, even when a large excess of IκBαW258F was added. This “irreversibility” of IκBα binding was also observed in NMR experiments (25).
Table 1.
Rate constants for the individual steps in the IκBα–NF-κB binding reactions of wild-type and the C186PA220P mutant IκBα
| Syringe 1 (label) | Syringe 2 (varying concentrations) | Reaction step | Rate constant | Rate constant value (fold difference) |
| DNA* | NF-κB | N+D→ND | k1 (M−1⋅s−1) | 1.5 (± 0.2) × 108 |
| NF-κB/DNA* | Unlabeled DNA | ND→N+D | k−1 (s−1) | 0.41 ± 0.01 |
| IκBα | NF-κB/DNA | ND+I→NDI | k2 (M−1⋅s−1) | 5.65 (± 0.02) × 106 |
| NF-κB/DNA/IκBα(W258) | IκBα(W258F) | NDI→ND+I | k−2 (M−1⋅s−1) | <0.01 |
| NF-κB/DNA* | IκBα | ND+I→NI + D | k3 (M−1⋅s−1) | 3.68 (± 0.02) × 106 |
| IκBα(W258) | NF-κB | N+I→NI | k4 (M−1⋅s−1) | 1.44 (± 0.04) × 107 |
| IκBα(W258)/NF-κB | IκBα(W258F) | NI→N+I | k−4 (s−1) | <0.01 |
| DNA* | NF-κB/IκBα | NI+D→ NDI | k5 (M−1⋅s−1) | 8.6 (± 0.4) × 107 |
| NF-κB/ IκBα/DNA* | Unlabeled DNA | NDI→NI+D | k−5 (s−1) | 0.96 ± 0.01 |
| IκBα(CPAP)(W258) | NF-κB/DNA | ND+I→NDI | k2 (M−1⋅s−1) | 5.0 (± 0.3) × 106 (1.15 times more slowly) |
| NF-κB/DNA/IκBα(CPAP)(W258) | IκBα(W258F) | NDI→ND+I | k−2 (M−1⋅s−1) | <0.01 |
| NF-κB/DNA* | IκBα(CPAP) | ND+I→NI + D | k3 (M−1⋅s−1) | 2.48 (± 0.08) × 106 (1.5 times more slowly) |
| IκBα(CPAP)(W258) | NF-κB | N+I→ NI | k4 (M−1⋅s−1) | 4.1 (± 0.2) × 107 (2.8 times faster) |
| IκBα(CPAP)(W258)/NF-κB | IκBα(W258F) | NI→N+I | k−4 (s−1) | <0.01 |
| DNA* | NF-κB/IκBα(CPAP) | NI+D→NDI | k5 (M−1⋅s−1) | 1.3 (± 0.1) × 107 (6.7 times more slowly) |
| NF-κB/ IκBα(CPAP)/DNA* | Unlabeled DNA | NDI→NI+D | k−5 (s−1) | 0.66 ± 0.01 (0.69) |
Effects of the Stabilizing C186P/A220P Mutation.
All of the experiments just described were repeated with IκBαC186P/A220P because previous SPR experiments showed that this mutant IκBα mediates dissociation of NF-κB off the DNA 1.5-fold more slowly than wild-type IκBα (Fig. 5A). IκBαC186P/A220P also formed a transient ternary complex upon binding to the DNA*–NF-κB complex (Fig. 5B) 1.15-fold slower than wild type (k2 in Scheme 1) (Table 1). The rate constant of IκBαC186P/A220P-mediated dissociation (k3 in Scheme 1) of NF-κB from the DNA was 1.5-fold slower, similar to the previous SPR (24). The binding of IκBαC186P/A220P to NF-κB in the absence of DNA (k4 in Scheme 1) was 2.8-fold faster than wild type (Fig. 5C), but association of IκBαC186P/A220P with the NF-κB–DNA complex was 1.15-fold slower, and DNA binding to form the backwards ternary IκBαC186P/A220P–NF-κB–DNA complex was 6.7-fold slower, all consistent with the increased “foldedness” of this mutant.
Fig. 5.
Comparison of the kinetics of IκBαC186P/A220P with wild type. (A) Plot of the rate vs. IκBαC186P/A220P (●) or wild-type IκBα (■) concentration at constant DNA*–NF-κB complex yielding a rate constant (k3 in Scheme 1) of 2.48 (± 0.08) × 106 M−1⋅s−1 compared with 3.68 (± 0.02) × 106 M−1⋅s−1 for wild-type IκBα (data replotted from Fig. 3C). (B) Fluorescence traces corresponding to the dissociation of DNA* in the presence of a 5-fold (blue) and 20-fold (red) excess of IκBαC186P/A220P showing ternary complex formation. (C) Plot of the rate of IκBαC186P/A220P (●) or wild-type IκBα (■) association vs. NF-κB concentration yielding an association rate constant (k4 in Scheme 1) of 4.1 (± 0.2) × 107 M−1⋅s−1 for IκBαC186P/A220P, 2.8-fold faster than wild-type 1.44 (± 0.04) × 107 M−1⋅s−1 (data replotted from Fig. 4D).
Conclusion
Postinduction repression of NF-κB signaling by IκBα is surprisingly rapid, considering that it occurs by a typical feedback mechanism in which NF-κB activates transcription of the IκBα gene, resulting in newly synthesized IκBα, which must travel back to the nucleus and compete with DNA for NF-κB binding. We recently showed that IκBα appears to do more than just compete with DNA for NF-κB binding; rather, it accelerates the dissociation of NF-κB from κB target sites (24). Here we followed changes in fluorescence of pyrene-labeled DNA and of the naturally occurring Trp in IκBα to measure the rate constants of all of the kinetic steps in IκBα-mediated dissociation of NF-κB from the DNA (Scheme 2).
Scheme 2.
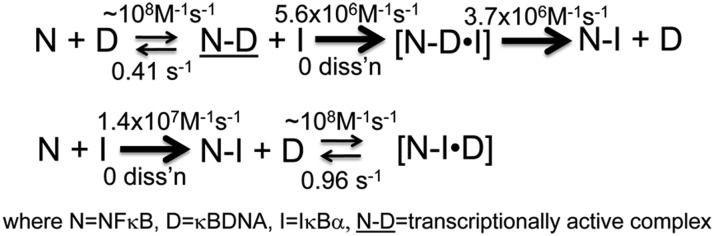
DNA binding to NF-κB is extremely rapid, almost diffusion controlled. Dissociation is also rapid, consistent with the observation of rapid binding and unbinding of NF-κB from transcription sites in cells (31) and lending credence to speculations that NF-κB–responsive transcription likely occurs in rapid bursts and that NF-κB can readily move among many sites in actively transcribed DNA (32).
The association rate constant of IκBα binding to the NF-κB–DNA complex was 2.5-fold slower than the rate constant for IκBα binding to NF-κB. In both cases, IκBα association probably involves initial binding of AR(1–4), for which we do not have a fluorescence probe, and then binding and folding of AR(5–6)PEST, probed by Trp-258 fluorescence. The 2.5-fold slower association in the presence of DNA is likely due to the fact that the AR(5–6)PEST binding site is occupied by DNA in the DNA–NF-κB complex.
When IκBα associates with the DNA*–NF-κB complex, the fluorescence of the pyrene–DNA first increases, indicative of formation of a transient ternary complex, and then decreases to the fluorescence of free pyrene–DNA. Indeed, the rate constant for IκBα binding to the NF-κB–DNA complex followed by IκBα Trp-258 fluorescence is nearly the same as the rate constant for IκBα-mediated dissociation of the NF-κB–DNA complex followed by pyrene fluorescence (Scheme 2), explaining why the ternary complex is so transient and had never been observed before. Almost instantly, when IκBα encounters the NF-κB–DNA complex, the DNA is released, resulting in formation of the extremely high-affinity NF-κB–IκBα complex, which does not dissociate. We could not, under any circumstances, observe IκBα dissociation—either from the IκBα–NF-κB complex or from the IκBα–NF-κB–DNA ternary complex. Thus, we write the steps subsequent to the ternary complex formation as irreversible in Scheme 2. In the classic case of the lac operon, addition of the effector molecules, allolactose or isopropyl β-d-thiogalactoside (IPTG), also dramatically increase the dissociation rate of the operator DNA (33). Binding of the effector molecule, at a site distant from the DNA binding site, is reversible and alters the conformation of the LacI protein to an ensemble of structures that more weakly bind DNA. In contrast, we have no evidence that IκBα alters the conformational ensemble of NF-κB; rather, parts of the IκBα molecule, notably AR (5, 6) PEST, occupy part of the DNA binding site in a mutually exclusive manner (25) (Fig. 1 A and B). In fact, the mutually exclusive binding of NF-κB by IκBα or DNA is a paradigm of the NF-κB field (34). Whereas addition of excess DNA to lacI weakens binding of IPTG (35), addition of very large excesses of DNA did not dissociate the IκBα–NF-κB complex.
Our results reveal how precisely tuned the macromolecular interactions of IκBα are to so efficiently terminate transcription. Once IκBα associates with the NF-κB–DNA complex, the captured NF-κB molecule is essentially irreversibly dissociated from the DNA. The resulting IκBα–NF-κB complex is then exported from the nucleus (36), and the transcription activity of that NF-κB molecule is irreversibly terminated. Indeed, the half-life of the IκBα–NF-κB complex in IKK knockout cells has been reported at >12 h (15). Our results predict that every molecule of newly synthesized IκBα that manages to enter the nucleus will be critical for determining the dynamics of NF-κB signaling (37).
Materials and Methods
Protein Expression and Purification.
Human wild-type IκBα(67-287)—referred to simply as IκBα—and mutants (C186P/A220P and W258F) were expressed in Escherichia coli BL21 DE3 cells and purified by using a Hi-Load Q Sepharose (GE Healthcare) followed by a Superdex 75 column (GE Healthcare) as described (16, 38). The protein concentrations were determined by spectrophotometry (εIκBα, εIκBαC186P/A220P = 12,950 M−1cm−1, and εIκBαW258F = 7,450 M−1cm−1).
N-terminal hexahistidine–NF-κB (His6–p50(39-350)/RelA(19-321)) heterodimer was coexpressed by using the method described (39) and purified by nickel-affinity chromatography (Ni-NTA Agarose; Qiagen), cation exchange chromatography (Mono S column; GE Healthcare), and, finally, size-exclusion chromatography (Superdex 200; GE Healthcare). The protein concentration was determined by spectrophotometry (εNF-κB = 43,760 M−1cm−1).
Stopped-Flow Kinetics.
A pyrene (N-hydroxyl succinimide ester)-labeled hairpin DNA 5′-AmMC6/GGGAAATTCCTCCCCCAGGAATTTCCC-3′ (IDT Technologies) sequence corresponding to the IFN-κB site was used (DNA*). The labeling procedures were described (40). The excitation wavelength was 343 nm, and the fluorescence emission was measured at 376 nm with a cutoff filter of 350 nm. Intrinsic fluorescence of the naturally occurring W258 in IκBα was followed by using an excitation wavelength of 280 nm and an emission wavelength of 345–355 nm, using a cutoff filter of 320 nm. All stopped-flow kinetic experiments were performed at 25 °C by using an SX-20 stop-flow apparatus (Applied Photophysics). The mixing volume was 200 μL, and the sampling time was linear (2,000 points).
Association Kinetics Experiments.
The association kinetics for the DNA* (constant concentration, 0.1 μM) were measured by using 10 different NF-κB concentrations (0.2, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1, 1.2, and 1.4 μM). Twenty traces were collected, and on average seven were selected and averaged for each NF-κB concentration. The data were fit with a double-exponential equation by using the program pro Fit (Version 6.1.14; QuantumSoft). The association rates were plotted against the NF-κB concentration to obtain the association rate constant by using pro Fit to implement a Levenberg–Marquardt algorithm. The errors are the errors of the fitted line. Given that the concentrations of DNA* and NF-κB are all well above the KD of 2.8 nM for the interaction, the condition for pseudo-first-order reaction was met at ratios lower than 1:5.
To measure the association of IκBα to the NF-κB–DNA complex and the association of IκBα with NF-κB alone, we took advantage of the native Trp fluorescence of W258. Eight different concentrations of the NF-κB–DNA complex or the NF-κB alone (0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.8, and 1.0 μM) were mixed with IκBα (0.1 μM). Twenty traces were collected, and 10 (on average) were selected and averaged for each concentration of the NF-κB–DNA complex or NF-κB. The data were processed by using pro Fit as described above, except that these data fit to a single exponential.
To measure the association of DNA with the NF-κB–IκBα complex (formation of the backwards ternary complex), a constant concentration of DNA* (0.1 μM) was mixed with five different concentrations (0.25, 0,5, 0.75, 1.0, and 1.5 μM) of the NF-κB–IκBα (1:1) complex. The data were processed as described for the association of DNA* to NF-κB.
Dissociation Kinetics Experiments.
The dissociation of DNA* from the NF-κB–DNA* complex was measured by adding different concentrations (0.5, 1.0, 1.5, 2.0, 3.0, 5.0, and 10 μM) of unlabeled DNA to 0.1 μM of the NF-κB–DNA* complex (1:1) to achieve ratios between 5:1 and 100:1. The data were fit with a single-exponential model. Traces of DNA* mixed with unlabeled DNA and NF-κB mixed with unlabeled DNA showed no fluorescence change and demonstrated that the kinetic traces encompass the entire amplitude of the dissociation.
The rate of dissociation of DNA* from the ternary DNA*–NF-κB–IκBα complex was measured by adding different concentrations of unlabeled DNA to the ternary complex DNA*–NF-κB–IκBα (0.5:0.1:0.1 μM). The data were analyzed in the same manner as for the NF-κB–DNA dissociation. An excess of DNA* was required in these experiments to form the backwards ternary complex.
We attempted to measure the dissociation rate of IκBα from the ternary complex by monitoring the change in Trp fluorescence of IκBα as different concentrations of the mutant IκBα(W258F) (0.5, 1.0, 2.0, 3.0, and 4.0 μM) were added to the ternary complex of DNA–NF-κB–IκBα (0.5:0.1:0.1 μM). No dissociation of IκBα was observed even at the highest ratio of 40:1.
Measurement of IκBα-Mediated Dissociation of the NF-κB–DNA Complex.
The rate of IκBα-mediated dissociation of the NF-κB–DNA complex was measured adding different concentrations of IκBα (0.2, 0.4, 0.5, 0.6, 0.8, 1.0, 1.5, and 2.0 μM) to the NF-κB–DNA* (0.1:0.1 μM) complex. The dissociation phase of each curve was fit using pro Fit implementing a double-exponential dissociation model. The observed dissociation rates of the main amplitude phase were plotted against the IκBα concentration and fit using a linear model to obtain the rate constant.
Measurement of the Rate of Each Step Using the C186P/A220P Mutant IκBα.
All of the experiments reported above were repeated by using the IκBαC186P/A220P in place of the wild-type protein.
Acknowledgments
We thank Diego Ferreiro for helpful discussions. This work was supported by Grant GM71862 from the National Institutes of Health.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
References
- 1.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132(3):344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 2.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 3.Martone R, et al. Distribution of NF-kappaB-binding sites across human chromosome 22. Proc Natl Acad Sci USA. 2003;100(21):12247–12252. doi: 10.1073/pnas.2135255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoffmann A, Natoli G, Ghosh G. Transcriptional regulation via the NF-kappaB signaling module. Oncogene. 2006;25(51):6706–6716. doi: 10.1038/sj.onc.1209933. [DOI] [PubMed] [Google Scholar]
- 5.Schreiber J, et al. Coordinated binding of NF-kappaB family members in the response of human cells to lipopolysaccharide. Proc Natl Acad Sci USA. 2006;103(15):5899–5904. doi: 10.1073/pnas.0510996103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baldwin AS., Jr Series introduction: The transcription factor NF-kappaB and human disease. J Clin Invest. 2001;107(1):3–6. doi: 10.1172/JCI11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar A, Takada Y, Boriek AM, Aggarwal BB. Nuclear factor-kappaB: Its role in health and disease. J Mol Med (Berl) 2004;82(7):434–448. doi: 10.1007/s00109-004-0555-y. [DOI] [PubMed] [Google Scholar]
- 8.Lee CH, Jeon YT, Kim SH, Song YS. NF-kappaB as a potential molecular target for cancer therapy. Biofactors. 2007;29(1):19–35. doi: 10.1002/biof.5520290103. [DOI] [PubMed] [Google Scholar]
- 9.Sen R, Baltimore D. Inducibility of kappa immunoglobulin enhancer-binding protein Nf-kappa B by a posttranslational mechanism. Cell. 1986;47(6):921–928. doi: 10.1016/0092-8674(86)90807-x. [DOI] [PubMed] [Google Scholar]
- 10.Chen FE, Huang DB, Chen YQ, Ghosh G. Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature. 1998;391(6665):410–413. doi: 10.1038/34956. [DOI] [PubMed] [Google Scholar]
- 11.Huxford T, Huang DB, Malek S, Ghosh G. The crystal structure of the IkappaBalpha/NF-kappaB complex reveals mechanisms of NF-kappaB inactivation. Cell. 1998;95(6):759–770. doi: 10.1016/s0092-8674(00)81699-2. [DOI] [PubMed] [Google Scholar]
- 12.Jacobs MD, Harrison SC. Structure of an IkappaBalpha/NF-kappaB complex. Cell. 1998;95(6):749–758. doi: 10.1016/s0092-8674(00)81698-0. [DOI] [PubMed] [Google Scholar]
- 13.Baeuerle PA, Baltimore D. NF-kappa B: Ten years after. Cell. 1996;87(1):13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 14.Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: Temporal control and selective gene activation. Science. 2002;298(5596):1241–1245. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- 15.O’Dea EL, et al. A homeostatic model of IkappaB metabolism to control constitutive NF-kappaB activity. Mol Syst Biol. 2007;3:111. doi: 10.1038/msb4100148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergqvist S, et al. Thermodynamics reveal that helix four in the NLS of NF-kappaB p65 anchors IkappaBalpha, forming a very stable complex. J Mol Biol. 2006;360(2):421–434. doi: 10.1016/j.jmb.2006.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Traenckner EB, et al. Phosphorylation of human I kappa B-alpha on serines 32 and 36 controls I kappa B-alpha proteolysis and NF-kappa B activation in response to diverse stimuli. EMBO J. 1995;14(12):2876–2883. doi: 10.1002/j.1460-2075.1995.tb07287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Traenckner EB, Baeuerle PA. Appearance of apparently ubiquitin-conjugated I kappa B-alpha during its phosphorylation-induced degradation in intact cells. J Cell Sci Suppl. 1995;19:79–84. doi: 10.1242/jcs.1995.supplement_19.11. [DOI] [PubMed] [Google Scholar]
- 19.Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18(49):6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 20.Chen ZJ, Parent L, Maniatis T. Site-specific phosphorylation of IkappaBalpha by a novel ubiquitination-dependent protein kinase activity. Cell. 1996;84(6):853–862. doi: 10.1016/s0092-8674(00)81064-8. [DOI] [PubMed] [Google Scholar]
- 21.Scott ML, Fujita T, Liou HC, Nolan GP, Baltimore D. The p65 subunit of NF-kappa B regulates I kappa B by two distinct mechanisms. Genes Dev. 1993;7(7A):1266–1276. doi: 10.1101/gad.7.7a.1266. [DOI] [PubMed] [Google Scholar]
- 22.Sun SC, Ganchi PA, Ballard DW, Greene WC. NF-kappa B controls expression of inhibitor I kappa B alpha: Evidence for an inducible autoregulatory pathway. Science. 1993;259(5103):1912–1915. doi: 10.1126/science.8096091. [DOI] [PubMed] [Google Scholar]
- 23.de Martin R, et al. Cytokine-inducible expression in endothelial cells of an I kappa B alpha-like gene is regulated by NF kappa B. EMBO J. 1993;12(7):2773–2779. doi: 10.1002/j.1460-2075.1993.tb05938.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bergqvist S, et al. Kinetic enhancement of NF-kappaB–DNA dissociation by IkappaBalpha. Proc Natl Acad Sci USA. 2009;106(46):19328–19333. doi: 10.1073/pnas.0908797106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sue SC, Alverdi V, Komives EA, Dyson HJ. Detection of a ternary complex of NF-kappaB and IkappaBalpha with DNA provides insights into how IkappaBalpha removes NF-kappaB from transcription sites. Proc Natl Acad Sci USA. 2011;108(4):1367–1372. doi: 10.1073/pnas.1014323108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferreiro DU, et al. Stabilizing IkappaBalpha by “consensus” design. J Mol Biol. 2007;365(4):1201–1216. doi: 10.1016/j.jmb.2006.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoffmann A, Leung TH, Baltimore D. Genetic analysis of NF-kappaB/Rel transcription factors defines functional specificities. EMBO J. 2003;22(20):5530–5539. doi: 10.1093/emboj/cdg534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferreiro DU, de Prat-Gay G. A protein-DNA binding mechanism proceeds through multi-state or two-state parallel pathways. J Mol Biol. 2003;331(1):89–99. doi: 10.1016/s0022-2836(03)00720-4. [DOI] [PubMed] [Google Scholar]
- 29.Ferreiro DU, Sánchez IE, de Prat Gay G. Transition state for protein-DNA recognition. Proc Natl Acad Sci USA. 2008;105(31):10797–10802. doi: 10.1073/pnas.0802383105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Truhlar SME, Torpey JW, Komives EA. Regions of IkappaBalpha that are critical for its inhibition of NF-kappaB.DNA interaction fold upon binding to NF-kappaB. Proc Natl Acad Sci USA. 2006;103:18951–18956. doi: 10.1073/pnas.0605794103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bosisio D, et al. A hyper-dynamic equilibrium between promoter-bound and nucleoplasmic dimers controls NF-kappaB-dependent gene activity. EMBO J. 2006;25(4):798–810. doi: 10.1038/sj.emboj.7600977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Turner DA, et al. Physiological levels of TNFalpha stimulation induce stochastic dynamics of NF-kappaB responses in single living cells. J Cell Sci. 2010;123(Pt 16):2834–2843. doi: 10.1242/jcs.069641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Riggs AD, Newby RF, Bourgeois S. lac repressor—operator interaction. II. Effect of galactosides and other ligands. J Mol Biol. 1970;51(2):303–314. doi: 10.1016/0022-2836(70)90144-0. [DOI] [PubMed] [Google Scholar]
- 34.Liou HC, Baltimore D. Regulation of the NF-kappa B/rel transcription factor and I kappa B inhibitor system. Curr Opin Cell Biol. 1993;5(3):477–487. doi: 10.1016/0955-0674(93)90014-h. [DOI] [PubMed] [Google Scholar]
- 35.Lewis M. The lac repressor. C R Biol. 2005;328(6):521–548. doi: 10.1016/j.crvi.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 36.Huang TT, Kudo N, Yoshida M, Miyamoto S. A nuclear export signal in the N-terminal regulatory domain of IkappaBalpha controls cytoplasmic localization of inactive NF-kappaB/IkappaBalpha complexes. Proc Natl Acad Sci USA. 2000;97(3):1014–1019. doi: 10.1073/pnas.97.3.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalita MK, et al. Sources of cell-to-cell variability in canonical nuclear factor-κB (NF-κB) signaling pathway inferred from single cell dynamic images. J Biol Chem. 2011;286(43):37741–37757. doi: 10.1074/jbc.M111.280925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Croy CH, Bergqvist S, Huxford T, Ghosh G, Komives EA. Biophysical characterization of the free IkappaBalpha ankyrin repeat domain in solution. Protein Sci. 2004;13(7):1767–1777. doi: 10.1110/ps.04731004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sue SC, Cervantes C, Komives EA, Dyson HJ. Transfer of flexibility between ankyrin repeats in IkappaB* upon formation of the NF-kappaB complex. J Mol Biol. 2008;380(5):917–931. doi: 10.1016/j.jmb.2008.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Studer SM, Joseph S. Binding of mRNA to the bacterial translation initiation complex. Methods Enzymol. 2007;430:31–44. doi: 10.1016/S0076-6879(07)30002-5. [DOI] [PubMed] [Google Scholar]