

Significance
The BK calcium-activated potassium channels are important regulators of electrical and calcium signaling in many types of tissue. BK channel activation involves complex conformational changes. We investigated the molecular details of such conformational changes with scanning mutagenesis and electrophysiological measurements. We found multiple pore residues that reorient their side chains during channel opening. These findings bridge the gap between our knowledge about the static X-ray structure of ion channel pores and knowledge about their functional dynamics.
Keywords: gating, structure–function, ion channel pore
Abstract
Three deep-pore locations, L312, A313, and A316, were identified in a scanning mutagenesis study of the BK (Ca2+-activated, large-conductance K+) channel S6 pore, where single aspartate substitutions led to constitutively open mutant channels (L312D, A313D, and A316D). To understand the mechanisms of the constitutive openness of these mutant channels, we individually mutated these three sites into the other 18 amino acids. We found that charged or polar side-chain substitutions at each of the sites resulted in constitutively open mutant BK channels, with high open probability at negative voltages, as well as a loss of voltage and Ca2+ dependence. Given the fact that multiple pore residues in BK displayed side-chain hydrophilicity-dependent constitutive openness, we propose that BK channel opening involves structural rearrangement of the deep-pore region, where multiple residues undergo conformational changes that may increase the exposure of their side chains to the polar environment of the pore.
Large-conductance, Ca2+-activated K+ (BK) channels regulate physiological processes such as neurotransmitter release, smooth muscle contraction, and hair cell frequency tuning (1–12). BK channel proteins are homotetramers formed by BK α-subunits, which then associate with different β- or γ-subunits in a tissue-specific manner (13–22). The α-subunit of the BK (KCa1.1) channel is encoded by the KCNMA1 gene, first discovered in Drosophila as the slowpoke mutation (dSlo) (23, 24), and later identified in mouse (mSlo1) and human (hSlo1) (25, 26). Each α-subunit has seven transmembrane segments (S0–S6), with the S6 segments lining the pore. A similar structural arrangement is found in other members of the K+ channel protein family.
From a functional point of view, the gating behavior of BK channels can be described as a central C⇔O (i.e., closed⇔open) transition, influenced by voltage sensor movement and/or Ca2+ binding (27–31). The structural basis for this C⇔O transition is believed to be conformational changes of the S6 segment and/or the selectivity filter (32, 33), controlling the passage of K+ ions across the membrane.
Molecular details of gating-related conformational changes have been probed with mutagenesis-based methods for BK and related channels. Although it has been demonstrated that voltage-gated (Kv) K+ channels open by a cytoplasmic S6 “bundle crossing” gate (34–37), evidence has accumulated that the opening conformational change of BK, like cyclic nucleotide-gated (CNG) channels, occurs deeper in the pore, closer to the selectivity filter (38–46). With the goal of further understanding the opening conformational change in BK channels, we used a histidine substitution/protonation strategy and identified a residue in BK S6 (M314 in hSlo1) whose side chain turns more toward the pore when the channel is open. The open conformation can be stabilized by the presence of side-chain charges at this location, with the aspartate mutant being the most effective in keeping the channel open in neutral pH (47).
To uncover more dynamic details at other pore residues during BK channel gating, we scanned the S6 segment cytoplasmic to the selectivity filter with single aspartate substitutions (I308D→N328D). Because the S6 residues of K+ channels reside on the interface between a polar (the water and ion filled pore) and a nonpolar (the rest of the protein in the membrane) environment, we expect the charged side chains of substituted aspartates to prefer the more aqueous environment of the pore, making the conformations with such side-chain orientation energetically favorable. If these conformations correspond to any functional states of the channel, such states may be stabilized, and will be functionally measurable. Our previous studies identified the M314D mutant channels as favoring the open state, consistent with greater exposure of this side chain to the pore upon opening.
In addition, the S6-formed pore is believed to host a “gate” that, when closed, prevents the passage of ions. The nature of such a gate, in its closed conformation, is proposed to be an occlusion structure formed by the side chains of amino acid residues at the gate location. In the structural model of the closed KcsA channel, at three residue locations (T107, A111, V115) to the intracellular end of its TM2 (equivalent of S6 in mammalian K+ channels) (Fig. 1), one could see the side chains at equivalent locations from the four subunits come very close to each other in the pore, occluding passage of K+ (48, 49). In the structural model of an open KcsA channel, pore diameters at the same locations are significantly larger (50, 51).
Fig. 1.

Sequence alignment and structural models. (A) Sequence alignment of KcsA (accession no. NP_631700), Shaker (accession no. CAA29917), and hSlo1 (accession no. AAB65837), starting from the threonine in the “TVGYGD” signature sequence of the K+ channel selectivity filter. The numbers by the channel name indicate the sequence number for the threonine in the corresponding channel. Highlighted in yellow are T107, A111, and V115 of KcsA, and P475, V478, and F481 of Shaker. Three residues of hSlo1 are highlighted as follows: L312, blue; A313, red; A316, green. The brown bar below the sequences indicates the selectivity filter, and the gray bar indicates residues scanned by aspartate substitution. (B) Side, top, and bottom views of two KcsA structure models: 1R3J, the closed conformation; and 3F5W, the open conformation. Only the P and S6 helices are shown. In the side views, one of the four subunits was taken away to better view the pore. The brown vertical lines indicate ion permeation through the selectivity filter. The equivalent residues to L312, A313, and A316 are colored the same way as in A.
One way to functionally locate such a tightly packed occlusion structure is to try to disrupt it with small, charged, or large side-chain amino acid substitutions. Application of this method on the Shaker K+ channels, together with prior evidence from cysteine accessibility experiments, identified V478 or F481 as candidate locations for the gate (52). With location P475, the aspartate mutant is constitutively open, the result of a shifted energetic balance between the closed and the open states (52, 53) (Fig. 1). If the BK channel gate is also formed by specific, intersubunit hydrophobic interactions between four equivalent residues in the tetramer, such as suggested by the KcsA structure, the charges on the substituted aspartate side chains may also prevent such interactions and reveal the location of the gate.
Results
Despite strong conservation of the selectivity filter sequence among different K+ channels, amino acid sequences along the S6 pore, cytoplasmic to the selectivity filter, are quite different. In Fig. 1, sequences are aligned for KcsA, Shaker, and hSlo1 (human BK), starting at the threonine of the signature “TVGYGD.”
Aspartate Scanning Mutagenesis Identified Multiple Constitutively Open Mutants in the Deep-Pore Region of BK.
We scanned BK S6 cytoplasmic to the selectivity filter (residues 308→328 in hSlo1) with single aspartate substitutions. For some positions, voltage- and Ca2+-dependent gating is largely preserved, with only small shifts of G–V curves compared with the wild-type BK channel (E324D, G327D, N328D) (Fig. 2 and Table S1). Channels with aspartate substitutions at other positions had significantly right-shifted G–V curves (L309D, G311D, E321D, I323D, and L325D), suggesting the closed states are energetically favored relative to wild type, whereas left-shifted G–V curves (I308D, G310D, S317D, Y318D, P320D) suggest the open states being energetically favored (Fig. 2 and Table S1). M314D, the mutation described in our previous study (47), had a constant G/Gmax of about 0.25 between −100 and 0 mV in 0 Ca2+ (Fig. 2). Three other mutants, L312D, A313D, and A316D, were constitutively open, with open probabilities close to 1 at −100 mV in 0 Ca2+, as well as a loss of voltage and Ca2+ dependence in channel gating (Fig. 2, highlighted number labels in blue, red, and green; Table S1) (Fig. 1, highlighted residues in blue, red, and green). Intracellular Ca2+, at a concentration of 85 µM, shifted the G–V curves in the negative direction, except for the three constitutively open mutants. Under these conditions, channels such as I308D, M314D, S317D, Y318D, and P320D also had high open probabilities at negative potentials (G/Gmax ≥ 0.5, −100 mV).
Fig. 2.
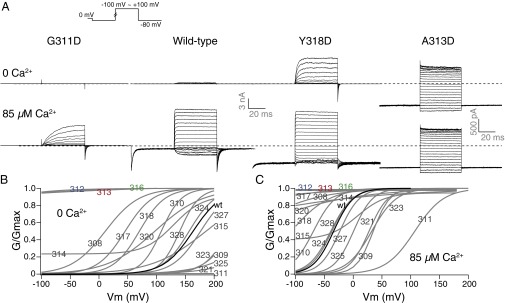
Single aspartate substitutions along BK S6 resulted in different gating phenotypes. (A) Trace examples of current recorded in response to families of voltage protocols (Inset), in 0 and 85 µM Ca2+ from the same patch. (B) Boltzmann fits of G–V curves for aspartate-substituted mutants (gray) compared with the wild-type (black) G–V, in 0 Ca2+. (C) Boltzmann fits of G–V curves for aspartate-substituted mutants (gray) compared with the wild-type (black) G–V, in 85 µM Ca2+. The numbers on the curves indicate the location of the mutation. The number labels for L312, A313, and A316 are colored the same way as in Fig. 1.
In comparison with similar studies on Shaker (the N-type inactivation removed Shaker-IR, specifically) channels, where aspartate substitution at a single location P475D (equivalent to BK P320D, which had a left-shifted G–V curve in 0 Ca2+ compared with wild type) led to constitutively open channels, we found three locations in BK with the same gating behavior. In addition, these residues are located in the deep-pore region of the BK channel, unlike P475 in Shaker, which is in the intracellular “bundle-crossing” region (52, 53). The three locations in BK, L312, A313, and A316, or their equivalents in related channels, have been individually identified in previous studies to contribute to aspects of gating (46, 54, 55). The L312 residue, for example, was previously identified in an alanine scanning mutagenesis study to yield a low-voltage–activated mutant (L312A) (54).
Most Amino Acid Substitutions at the L312 Location Produced Channels with High Open Probability in 0 Ca2+ and Low Voltage.
Consistent with these previous reports (54), replacement at the L312 location by cysteine (C), valine (V), serine (S), or glutamine (Q) led to channels with high open probability (G/Gmax > 0.5) in 0 Ca2+ and hyperpolarized voltage (−100 mV). We report additional replacements showing similar gating behavior, including glycine (G), threonine (T), aspartate (D), and asparagine (N). Besides a much reduced voltage dependence for the gating of these mutants, the Ca2+ sensitivity seems to be lost as well, because higher Ca2+ concentration did not further promote channel opening (Fig. 3 and Table S2). The methionine (M) substitution retained gating behavior of the wild-type channel, so did the phenylalanine (F) except for small rightward G–V shifts in both 0 and 85 µM Ca2+. Both the alanine (A) and isoleucine (I) substitutions led to significant leftward shifts of the G–V curves compared with wild type in 0 Ca2+. For the channels that are not constitutively open, higher intracellular Ca2+ concentration at 85 µM shifted the G–V curves further to the left and led to an apparent reduction of voltage dependence in the case of isoleucine (I) (note the shallow slope of the G–V curve for L312I) (Fig. 3). Because the mutations were made in the pore region, they most likely influenced the C⇔O equilibrium, which can result in constitutive opening, as well as apparent losses of voltage and Ca2+ dependence via allosteric mechanisms (31).
Fig. 3.
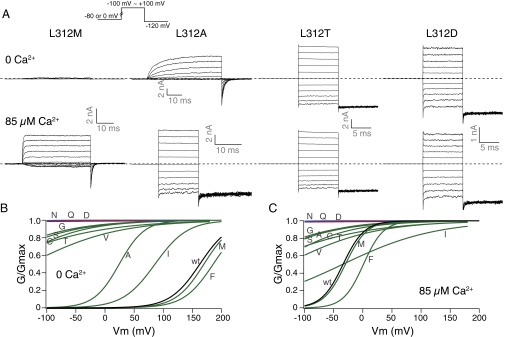
Gating of L312X mutants. (A) Trace examples of current recorded in response to families of voltage protocols (Inset), in 0 and 85 µM Ca2+ from the same patch. (B) Boltzmann fits of G–V curves for charged (red), strongly polar (blue), and other (green) side-chain substituted mutants, compared with the wild-type (black) G–V, in 0 Ca2+. (C) Comparison of G–V curves in 85 µM Ca2+. Colors are the same as in B. Letters on the curves indicate the mutant amino acids.
Charged and Polar Amino Acid Substitutions at the A313 Location Led to Constitutively Open Channels.
Previous implications of the A313 location in BK channel gating can be found in cysteine scanning mutagenesis and modification studies (46). The A313C channels can be modified by [2-(trimethylammonium)ethyl]methanethiosulfonate (MTSET) in both closed and open states. The modification rate, as measured by MTSET block of the current, is state dependent, with the open-state modification happening faster than the closed-state modification by 2–3 orders of magnitude. When modified by the positively charged MTSET, the A313C mutant is also constitutively open (46). We replaced A313 with the other amino acids. In 0 Ca2+, replacement of the endogenous alanine by phenylalanine (F), isoleucine (I), leucine (L), cysteine (C), valine (V), or methionine (M) caused the G–V curves to shift rightward; replacement by glycine (G), tyrosine (Y), threonine (T), tryptophan (W), or serine (S) led to left-shifted G–V curves. In addition to the aspartate (D) substitution, channels with asparagine (N), glutamine (Q), glutamate (E), histidine (H), lysine (K), arginine (R), or proline (P) at the A313 location were constitutively open. Except for proline (P), all of the side chains causing constitutive openness at position 313 are charged or polar. Higher intracellular Ca2+ concentration at 85 µM shifted the G–V curves to the left, except for the channels that were already constitutively open (Fig. 4 and Table S3). Inward rectification of steady-state currents was observed for A313K and A313R. Such rectification was absent from other constitutively active mutants (Fig. 4, trace examples), but was reminiscent of the MTSET block of the A313C channels (46). The lysine (K) and arginine (R) side chains are both large and positively charged, as is MTSET.
Fig. 4.
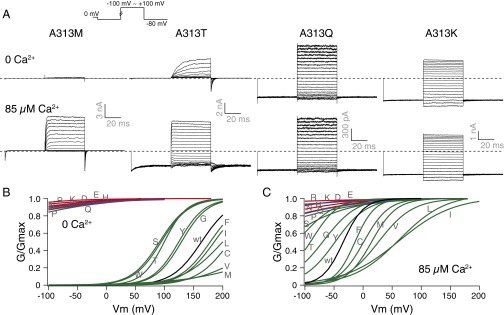
Gating of A313X mutants. (A) Trace examples of current recorded in response to families of voltage protocols (Inset), in 0 and 85 µM Ca2+ from the same patch. (B) Boltzmann fits of G–V curves for charged (red), strongly polar (blue), and other (green) side-chain substituted mutants, compared with the wild-type (black) G–V, in 0 Ca2+. (C) Comparison of G–V curves in 85 µM Ca2+. Colors are the same as in B. Letters on the curves indicate the mutant amino acids.
Charged and Polar Amino Acid Substitutions at the A316 Location Produced Constitutively Open Channels.
The previous cysteine scanning mutagenesis studies also reported state-dependent differences of MTSET modification rates for A316C, similar to those of A313C. In addition, the A316 location is unique in that the MTSET modification led to total loss of current, because of block at a positive membrane potential (46). Similar observation was made for the A88 (equivalent of A316 in hSlo1) location in the related bacterial channel MthK, where bulky side-chain substitutions led to reduced single-channel conductance. The A88D mutant of MthK was also constitutively open (55). We went on to replace A316 with all of the other amino acids.
In 0 Ca2+, replacement of the endogenous A316 with methionine (M), phenylalanine (F), tyrosine (Y), valine (V), cysteine (C), isoleucine (I), or leucine (L) led to rightward shifts of the G–V curves; whereas serine (S), glycine (G), threonine (T), or proline (P) substitutions caused leftward shifts of the G–V curves. At the A316 location, besides the aspartate (D) substitution, asparagine (N), glutamine (Q), glutamate (E), lysine (K), and arginine (R) all led to constitutively open channels. The histidine mutant (A316H) also had substantial open probability with 0 Ca2+ and hyperpolarization (−100 mV). The 85 µM Ca2+ on the intracellular side shifted the G–V curves to the left except for the channels that were already constitutively open (Fig. 5 and Table S4). An even larger degree of inward rectification of steady-state currents was observed for A316K and A316R, compared with those same mutants at the A313 location (Fig. 5, trace examples).
Fig. 5.
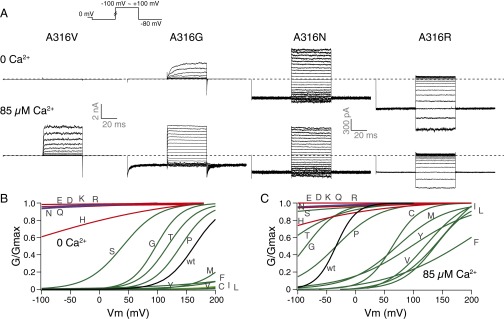
Gating of A316X mutants. (A) Trace examples of current recorded in response to families of voltage protocols (Inset), in 0 and 85 µM Ca2+ from the same patch. (B) Boltzmann fits of G–V curves for charged (red), strongly polar (blue), and other (green) side-chain substituted mutants, compared with the wild-type (black) G–V, in 0 Ca2+. (C) Comparison of G–V curves in 85 µM Ca2+. Colors are the same as in B. Letters on the curves indicate the mutant amino acids.
Side-Chain Solvation Energy of Substituted Amino Acids Correlates with the Propensity for Mutant Channels to Stay Open.
As shown in Figs. 2–5, although we used Boltzmann fits to idealize the G–V curves, the fitting parameters such as V1/2 and z do not necessarily describe the energetics of channel gating without a more comprehensive kinetic model, especially for the constitutively open mutants. To quantify the effects of the mutations on the G–V curves, we used another parameter, G/Gmax at the V1/2 of wild-type BK channel—(G/Gmax)mut(V1/2,wt). This parameter qualitatively describes the position of the G–V curves. The wild-type value is defined as 0.5; a number smaller than 0.5 means that the mutant has a G–V curve on the right side of the wild type and that it is more difficult for the channel to open; a number larger than 0.5 means that the mutant has a G–V curve on the left side of the wild type and that it is easier for the channel to open (Materials and Methods). The constitutively open channels have (G/Gmax)mut(V1/2,wt) values close to 1, in 0 Ca2+. We limited the analysis to 0 Ca2+ conditions to focus on the Ca2+-independent C⇔O transition. We plotted (G/Gmax)mut(V1/2,wt) against amino acid side-chain volume (Fig. 6, Upper) or solvation energy [free energies of transfer of amino acid side chains to water from cyclohexane (56)] (Fig. 6, Lower).
Fig. 6.
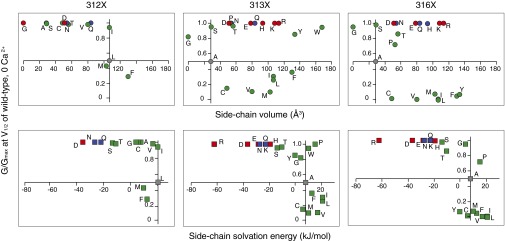
Correlation between side-chain properties and steady-state gating. Normalized macroscopic conductance at the V1/2 of wild type (in 0 Ca2+) is plotted against either side-chain volume (Upper) or side-chain solvation energy (Lower), for mutants at the three deep-pore locations: L312X (Left), A313X (Center), and A316X (Right). Charged residues are shown with red symbols; strongly polar residues are shown with blue symbols; other mutant residues are shown with green symbols. The endogenous wild-type residues are shown with gray symbols, where the axes intersect.
At L312, quite a few mutants did not produce currents that can be measured under the experimental conditions (neutral pH, 0 or 85 µM Ca2+, and voltage ranges from −100 to +200 mV). Among the mutants that did produce measurable currents, most of them were constitutively open, and they all have side chains that are both smaller and more hydrophilic than that of the endogenous leucine (Fig. 6).
For the A313 mutants, (G/Gmax)mut(V1/2,wt) had little correlation with the size of the side chains. However, side chains with negative solvation energies all resulted in high open probability channels. [The solvation energy of tyrosine (Y) at 25 °C and pH 7 is −0.59 kJ⋅mol−1 (56).] Among these substitutions, A313R, A313D, A313E, A313N, A313Q, A313K, and A313H were constitutively open, whereas A313S, A313T, and A313Y had significantly left-shifted G–V curves in comparison with wild type, resulting in large (G/Gmax)mut(V1/2,wt) values (Fig. 6). The A313W mutant also had a G–V curve that is left shifted, to an extent where the G/Gmax reached 0.95 at the V1/2 of wild type in 0 Ca2+ (Fig. 6), but the channel can still close with hyperpolarization (Fig. 4). The A313P mutant, however, was constitutively open, like the charged and the strongly polar substituted mutants (Figs. 4 and 6).
Side-chain size also did not correlate with (G/Gmax)mut(V1/2,wt) for the A316 mutants. However, charged and strongly polar residues led to constitutively open channels. Like the cases for L312 and A313, the sign of the substituted charges did not seem to create much difference in how likely the channels stay open. Other side chains with negative solvation energies such as serine (S) and threonine (T) also made it easier for the channel to open, evident from the leftward G–V shifts (Figs. 5 and 6). Unlike the A313 location, where A313Y had a left-shifted G–V curve in comparison with wild type, the G–V curve for A316Y was greatly right shifted. In addition, the proline substitution at A316 did not make the channel constitutively open (Figs. 5 and 6).
The Deep-Pore Location, as Well as Reduction in Side-Chain Solvation Energy, Are Important Factors in Rendering the Mutant Channels Constitutively Open.
Because side-chain solvation energy seemed to be an important factor in determining the constitutive openness of the L312X, A313X, and A316X mutant channels, we asked whether differences in side-chain solvation energy between the substituted aspartate and the endogenous residue are larger at these locations than at other locations along S6, hence the more significant gating effects. In Fig. 7A, we plotted ΔV1/2 (in 0 Ca2+, compared with wild type) of the aspartate mutants for all of the tested locations along S6. For the same locations, we plotted in Fig. 7B the difference in side-chain solvation energy between the substituted aspartate and the endogenous residue. With the exception of E321D, E324D, and N328D, reduction in solvation energy was larger than 36 kJ⋅mol−1 for all of the other locations, but only L312D, A313D, and A316D were constitutively open. [The slow kinetics and nonzero open probability at negative voltages (47) for M314D were not reflected by ΔV1/2.] Therefore, conformational changes involving these three deep-pore locations may have larger energetic contributions to the C⇔O equilibrium than those involving the other S6 residues.
Fig. 7.

Similar changes in side-chain solvation energy resulted in significant differences in ΔV1/2 at different locations along BK S6. (A) Differences in V1/2 between the aspartate mutants and wild type, ΔV1/2, plotted for residues 308→328 along BK S6, in 0 Ca2+. V319D, I322D, and I326D did not produce measurable currents. (B) Differences in solvation energy between the aspartate side chain and the side chain of the endogenous residue at locations along S6 in BK.
It is also noteworthy that the same amino acid switches produced different, even opposite gating effects at different locations along the pore. For example, the leucine-to-aspartate switch, although making L312D constitutively open, led to greatly right-shifted G–V curves at L309 and L325. The isoleucine-to-aspartate switch at I308 made the channels easier to open, but very high voltages and/or Ca2+ were needed to open I323D channels. The three glycines in the pore, G310, G311, and G327, displayed three different gating phenotypes when replaced by aspartates. The G–V curves of G327D were close to those of wild type; G310D had significantly left-shifted G–V curves; whereas G311D was among the most difficult to open. The glutamate-to-aspartate switch, despite a small change of the side chains, made the E321D channels hard to open, but E324D G–V curves overlapped with those of wild type (Figs. 2 and 7, and Table S1). These observations suggest diversity in the types of S6 side-chain movements during BK channel opening.
Discussion
Starting with the finding that the L312D, A313D, and A316D mutant BK channels were all constitutively open, we further investigated the mechanisms of such gating phenotypes by other amino acid substitutions. A salient similarity among all three locations is that not only aspartate but also other charged or polar residues, positive or negative, irrespective of side-chain size, led to constitutively open channels.
For the L312X mutants, besides side-chain hydrophilicity, amino acids with side-chain volume less than that of the endogenous leucine also made the mutant channels constitutively open. Our results are consistent with previous studies (54), the interpretation of which included a hypothetical, specific intersubunit hydrophobic interaction (L312 with F315 from neighboring subunits) to stabilize the closed conformation (54). In these previous studies, L312 was identified as a unique location where an alanine substitution led to low-voltage–activated BK channels (L312A). Because the 313 and 316 positions have alanines endogenously, they were not examined in the alanine scan mutagenesis. By scanning the BK S6 pore with other amino acid substitutions, we demonstrate that the constitutive activity was not a unique feature of the L312 location.
Specific, closed-state–stabilizing, intersubunit interactions, as well as side-chain dynamics, are proposed to explain why the A88D (equivalent of A316D in hSlo1) mutant in MthK is constitutively open (55). Analysis of other substitutions at the same location, together with reference to the open and closed structure of another channel, bacterial NaK, led to the hypothesis that the A88 side chain points to the center of the pore in the open conformation, but points tangential to the central pathway in the closed conformation, participating in intersubunit helical packing with hydrophobic chains of a few residues in the neighboring inner helix (equivalent of S5 in BK) (55). Because these studies were motivated by X-ray crystal structure models of MthK showing the unique position of A88, other pore-lining residues were not subjected to mutagenesis to investigate their potential roles in gating-related conformational changes.
Our aspartate scanning mutagenesis of the BK channel S6 revealed a couple of prominent features about the BK channel pore. First, unlike the Shaker channel, where aspartate substitution at the intracellular end of S6 resulted in channels being constitutively open, the constitutively open BK mutants had aspartate substitutions in the deep-pore region, immediately cytoplasmic to the selectivity filter and above the intracellular end of S6. In addition, we found three locations along BK S6 that, when individually replaced by aspartate, made the channel constitutively open.
If the aspartates made mutant channels constitutively open by eliminating a specific, intersubunit interaction at the BK channel gate (Fig. 8A), such a gate does not appear to be at the intracellular end of S6 in BK. It is also unlikely that three independent gates are present in the deep pore of BK, each formed by intersubunit interactions of equivalent L312s, A313s, or A316s, respectively, because disrupting only one of those interactions should still leave the other two intact, unless some other conformational changes happened to render the other two unable to close either. Therefore, if the constitutively open channels are just results of disruption of an intersubunit hydrophobic interaction, such interaction will need to involve all L312, A313, and A316 side chains, spanning two helical turns on S6.
Fig. 8.

Schematic cross-sections of the BK channel pore. (A) The “closed-state, side-chain interaction” model, stating that at the gate location in wild-type channels, side chains from four equivalent locations in S6 form hydrophobic interactions to occlude ionic passage. When aspartate is substituted into this location on S6, the gate can no longer close. (B) The “open-state, side-chain stabilization” model, stating that the side chains of certain residue(s) orient more toward the pore in the open state than in the closed state of the channel. Substitution with aspartate at these locations stabilizes the open state. These residues are not necessarily located at the gate.
Alternatively, although various specific interactions have been proposed to explain the open phenotypes of BK-related pore mutants, given the fact that multiple pore residues in BK displayed side-chain hydrophilicity-dependent constitutive openness, the simpler interpretation may be the energetic preference of the hydrophilic side chains to turn toward the polar environment of the pore. It is possible that L312, A313, and A316 side chains are more exposed to the polar environment of the pore when the channel is open, compared with when the channel is closed. When charged or polar residues are substituted into these locations, the pore-exposed orientation of the side chains should be more energetically favorable, leading to a stabilized open conformation (Fig. 8B). Our results do not exclude the possibility that one of the three locations could host the “gate” and the other two participate in channel opening by turning their side chains. In any case, the open-state stabilization mechanism (Fig. 8B) needs to be included to explain all of the observations.
Together with M314, we have now identified at least four residues in BK deep pore that may undergo side-chain reorientation during channel opening. The structure of the BK channel in this region is not known. When threading the hSlo1 sequence onto the structural models of the related Kv1.2 and MthK, we found this deep-pore region to be less resolved than the well-conserved parts of the helices (47). If the deep-pore region were also a well-defined α-helix, then at least two out of the four locations (L312, A313, M314, and A316) would be on the opposite sides of the helix (46). A rigid-body, rotational movement, as proposed for lower S6 of Shaker and CNG channels (57, 58), does not seem to be able to accommodate the fact that all four side chains go from more buried to more exposed conformations upon opening. However, even with α-helical structures, movements of side chains independent of the rigid-body motion were observed when comparing the open and closed structural models of KcsA (50, 51). Moreover, it is still possible that the structure of the deep-pore region in BK is not a rigid α-helix in all states related to opening, allowing for more flexible movements of the amino acid side chains during channel gating.
At the three identified deep-pore locations in this study, a reduction of ≥27 kJ⋅mol−1 in solvation energy per subunit, compared with the wild-type residue, was sufficient to render the mutant channels constitutively open (Fig. 6) [taking the difference between histidine, −19.5 kJ⋅mol−1; and alanine, 7.57 kJ⋅mol−1; both measured at 25 °C and pH 7 (56)]. Neither depolarization nor intracellular Ca2+ was necessary. However, the A313S and A316S channels, which have a reduction of about 22 kJ⋅mol−1 in side-chain solvation energy per subunit, were not constitutively open, despite considerable leftward G–V shifts relative to wild type (Figs. 4 and 5). In comparison, based on the Horrigan–Aldrich model of BK channel gating, the equilibrium energy difference between the open and closed states for wild-type BK channels in the absence of Ca2+ at zero voltage (Vm = 0 mV) is ∼34 kJ⋅mol−1 (31) (Materials and Methods), which is significantly smaller than the local maximum possible perturbation via the alanine-to-serine switch (4 × 22 kJ⋅mol−1). This comparison suggests that the side-chain reorientations, as part of the C⇔O pathway, may not be transitioning from completely buried to completely pore exposed, and that the mutations may not have achieved their maximum possible local perturbations. It is also evident that the proposed side-chain reorientations, if not directly perturbing the pore gate, must contribute to the C⇔O pathway together with other, yet-unknown conformational changes, the combined energetic effects of which will eventually determine the overall C⇔O equilibrium.
It is important to point out that L312 is different from A313 and A316, in that side-chain sizes and structural isomerism also seem to contribute to the gating phenotypes. Comparing A313 and A316, a possible structural break introduced by a proline (P) made A313P stay open but A316P could still close. Bulky substitutions such as tyrosine (Y) also resulted in different gating behavior between the two locations. In terms of permeation, lysine (K) and arginine (R) substitutions at both A313 and A316 resulted in inward rectification of the current, but the extent of the rectification is significantly larger in A316K and A316R.
Differences in the mutant phenotypes suggest that, although these side chains may all go through structural reorientation during channel opening, the degree of such movement, as well as local steric constraints, may be different. Some side chains may move from barely buried, pointing tangentially to the pore, to directly pore pointing, as suggested for A88 in MthK; some may start from deeply buried in the membrane and move to be just exposed to the pore. The movement of others may be of a lesser amount, from just buried to barely exposed, for example. Even with the same type and amount of side-chain movement, the effect on channel gating also depends on where such movement is along the C⇔O pathway, in an energetic sense (59). Therefore, amino acids at other S6 locations could also undergo similar conformational changes (notice left-shifted G–V curves with other aspartate mutants along S6, Figs. 2 and 7). However, the energetic effects of their movements were not as significant, the manipulation of which, with our methods, did not lead to channels being constitutively open. On the other hand, greatly right-shifted G–V curves were also observed for some S6 residues when substituted by aspartate (Figs. 2 and 7). One possibility is that these side chains may move in a way to go from a pore-exposed to a buried conformation upon opening, so that the charges on aspartate may stabilize the closed conformation. This hypothesis will need to be tested with further experiments.
Materials and Methods
Molecular Biology and Cell Culture.
Site-directed mutagenesis was conducted using the Stratagene QuikChange kit. Success of mutagenesis was confirmed by DNA sequencing. HEK293 cells were cultured following standard procedures. Wild-type and mutant BK channel cDNAs were transiently transfected into HEK cells with Lipofectamine 2000. Enhanced green fluorescent protein (EGFP) was cotransfected as a marker.
Electrophysiology.
Recordings of BK channel currents were performed 24–48 hours after transfection. Voltage-clamp recordings were performed at room temperature (22–24 °C) in the inside-out configuration on patches pulled off from HEK cells. The patch electrodes (1–3 MΩ in resistance) contained the following (in mM): 6 KCl, 136 KOH, 20 Hepes, 2 MgCl2, adjusted to pH 7.2 with MeSO3H. Bath (intracellular side) solution contained the following (in mM): 6 KCl, 136 KOH, 20 Hepes, adjusted to pH 7 with MeSO3H. For solutions with nominal 0 Ca2+, 5 mM EDTA was included. For solutions with 85 µM free Ca2+, 1.81 mM CaCl2 and 5 mM NTA were included. Free Ca2+ concentrations were measured with Ca2+-sensitive electrodes. The Axopatch 200A amplifier was used for voltage-clamp recordings.
Data Analysis.
Currents were recorded from the patch and digitized using the ITC-16 A/D converter. Data were analog filtered at 10 kHz, digitized at 25 kHz, collected with PULSE software, and analyzed off-line with custom software written in Igor Pro. G–V curves were obtained by tail-current measurements for most mutants. For mutants with very fast tail currents, the G–V curves were calculated with steady-state current measurement divided by driving force. Similar voltage commands were used in the presence of 0 Ca2+ at pH 7 to record “blank” current signals for subtracting linear leak and capacitive transients, scale as needed. Currents were normalized to the maximum current obtained in the same patch with saturating Ca2+ concentration, or at high voltages (up to +300 mV). Group G–V curves were obtained by fitting a Boltzmann function to averaged data points [G/Gmax = base + 1/(1 + ezF((V1/2−V)/RT)], with base = 0 for wild-type and most mutant BK channels. A nonzero base value was obtained during fitting for mutant channels with constant, nonzero G/Gmax values at negative voltages. To improve clarity, error bars were not included in figures where a number of G–V curves were presented. G–V curves with error bars (representing SEM) on individual mutants [except for the L312X mutants, where our data largely overlap with previously published results (54)] are presented in Fig. S1. Fitting of the Boltzmann function to G–V curves were performed with the Levenberg–Marquardt (a version of least-squares) algorithm in Igor Pro. Error estimates of the fitting parameters z and V1/2, as reported in Tables S1–S4, represent the SDs of the parameter distributions, calculated from surface plots of the χ2 values against the fitting parameters.
G/Gmax at Wild-Type V1/2 [(G/Gmax)mut(V1/2,wt)] as an Alternative, but Related Measure to ΔΔG.
The macroscopic conductance is related to the open probability (Popen), number of channels in the patch (n), and single-channel conductance (γ) by the following:
The maximum macroscopic conductance, Gmax, is determined by the following:
In wild-type BK channels, because Pmax is very close to 1, G/Gmax is an approximate measure of Popen (this may not be the case in some mutants):
In a simplified two-state model (all of the open states have the same energy; all of the closed states have the same energy) for a voltage-dependent channel (BK channels can be considered as purely voltage-dependent channels in 0 Ca2+):
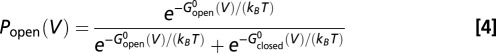 |
(Boltzmann distribution), where  and
and  are the voltage-dependent free energy of the channel in the open and closed states, respectively. By definition, the steady-state free energy difference between the closed and open states, ΔG0, is also a voltage-dependent parameter:
are the voltage-dependent free energy of the channel in the open and closed states, respectively. By definition, the steady-state free energy difference between the closed and open states, ΔG0, is also a voltage-dependent parameter:
substituting Eq. 5 into Eq. 4, we have the following:
For wild-type channels, at V1/2, substituting Popen = 0.5 into Eq. 6:
For any mutant channel, according to Eq. 6,
If we define ΔΔG as the difference between the mutant channel and wild type as follows:
then our measurement of G/Gmax at wild-type V1/2 can be considered as equivalent to the conventional ΔΔG measurement. The only difference is that we are measuring the same parameter at a different voltage, instead of at 0 mV:
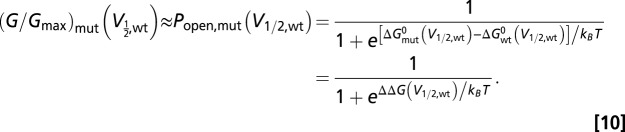 |
From Eq. 10, it is apparent that when (G/Gmax)mut(V1/2,wt) is less than 0.5, ΔΔG(V1/2,wt) is positive; therefore, it is more difficult for the mutant than wild type to open. When (G/Gmax)mut(V1/2,wt) is larger than 0.5, ΔΔG(V1/2,wt) is negative; therefore, the mutant made it easier for the channel to open. When (G/Gmax)mut(V1/2,wt) is close to 1, ΔΔG(V1/2,wt) should have a very large negative value, indicating no need for additional energy to open the channel, once the membrane voltage reached the V1/2 of wild type, in 0 Ca2+ (Fig. 6). We chose to report (G/Gmax)mut(V1/2,wt), instead of ΔΔG, because we are not confident our mutants could all be approximated as two-state channels. With this measurement, we also do not rely on accuracy of the fitting parameters (V1/2 and z in the first-order Boltzmann equation, for example) to arrive at qualitative conclusions about energetics. The fittings in this study mainly served as a way to idealize and extrapolate data.
Steady-State Energy Calculations.
R = 8.314 J⋅K−1⋅mol−1
T = 298 K
L0 = 10−6 [the C⇔O equilibrium constant in the Horrigan–Aldrich model (31) of BK channel gating in 0 Ca2+, V = 0 mV]
C = 7.4 (allosteric factor describing interaction between channel opening and Ca2+ binding)
D = 25 (allosteric factor describing interaction between channel opening and voltage sensor activation)
The steady-state free energy difference between the unliganded closed and open states, at 0 mV:
Perturbation in ΔG0 caused by one Ca2+ sensor binding:
Perturbation in ΔG0 caused by one voltage sensor activation:
Supplementary Material
Acknowledgments
We thank Amelia Hall, Travis Cox, and Dr. He Liu for making some of the mutants. We thank Dr. Jennifer Greeson-Bernier, Dr. D. Brent Halling, Dr. Thomas Middendorf, and Keegan Hines for comments on the manuscript. The work was supported by National Institutes of Health Grants NS077821 (to R.W.A.), AA020992 (to R.W.A.), and NS078152 (to J.Y.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1321697111/-/DCSupplemental.
References
- 1.Marty A, Neher E. Potassium channels in cultured bovine adrenal chromaffin cells. J Physiol. 1985;367:117–141. doi: 10.1113/jphysiol.1985.sp015817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fettiplace R, Fuchs PA. Mechanisms of hair cell tuning. Annu Rev Physiol. 1999;61:809–834. doi: 10.1146/annurev.physiol.61.1.809. [DOI] [PubMed] [Google Scholar]
- 3.Kang J, Huguenard JR, Prince DA. Voltage-gated potassium channels activated during action potentials in layer V neocortical pyramidal neurons. J Neurophysiol. 2000;83(1):70–80. doi: 10.1152/jn.2000.83.1.70. [DOI] [PubMed] [Google Scholar]
- 4.Smith MR, Nelson AB, Du Lac S. Regulation of firing response gain by calcium-dependent mechanisms in vestibular nucleus neurons. J Neurophysiol. 2002;87(4):2031–2042. doi: 10.1152/jn.00821.2001. [DOI] [PubMed] [Google Scholar]
- 5.Edgerton JR, Reinhart PH. Distinct contributions of small and large conductance Ca2+-activated K+ channels to rat Purkinje neuron function. J Physiol. 2003;548(Pt 1):53–69. doi: 10.1113/jphysiol.2002.027854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meredith AL, Thorneloe KS, Werner ME, Nelson MT, Aldrich RW. Overactive bladder and incontinence in the absence of the BK large conductance Ca2+-activated K+ channel. J Biol Chem. 2004;279(35):36746–36752. doi: 10.1074/jbc.M405621200. [DOI] [PubMed] [Google Scholar]
- 7.Pyott SJ, Glowatzki E, Trimmer JS, Aldrich RW. Extrasynaptic localization of inactivating calcium-activated potassium channels in mouse inner hair cells. J Neurosci. 2004;24(43):9469–9474. doi: 10.1523/JNEUROSCI.3162-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sausbier M, et al. Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc Natl Acad Sci USA. 2004;101(25):9474–9478. doi: 10.1073/pnas.0401702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burdyga T, Wray S. Action potential refractory period in ureter smooth muscle is set by Ca sparks and BK channels. Nature. 2005;436(7050):559–562. doi: 10.1038/nature03834. [DOI] [PubMed] [Google Scholar]
- 10.Rancz EA, Häusser M. Dendritic calcium spikes are tunable triggers of cannabinoid release and short-term synaptic plasticity in cerebellar Purkinje neurons. J Neurosci. 2006;26(20):5428–5437. doi: 10.1523/JNEUROSCI.5284-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Q, Chen B, Ge Q, Wang ZW. Presynaptic Ca2+/calmodulin-dependent protein kinase II modulates neurotransmitter release by activating BK channels at Caenorhabditis elegans neuromuscular junction. J Neurosci. 2007;27(39):10404–10413. doi: 10.1523/JNEUROSCI.5634-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen B, Liu P, Zhan H, Wang ZW. Dystrobrevin controls neurotransmitter release and muscle Ca2+ transients by localizing BK channels in Caenorhabditis elegans. J Neurosci. 2011;31(48):17338–17347. doi: 10.1523/JNEUROSCI.3638-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knaus HG, Garcia-Calvo M, Kaczorowski GJ, Garcia ML. Subunit composition of the high conductance calcium-activated potassium channel from smooth muscle, a representative of the mSlo and slowpoke family of potassium channels. J Biol Chem. 1994;269(6):3921–3924. [PubMed] [Google Scholar]
- 14.Shen KZ, et al. Tetraethylammonium block of Slowpoke calcium-activated potassium channels expressed in Xenopus oocytes: Evidence for tetrameric channel formation. Pflugers Arch. 1994;426(5):440–445. doi: 10.1007/BF00388308. [DOI] [PubMed] [Google Scholar]
- 15.Xia XM, Ding JP, Lingle CJ. Inactivation of BK channels by the NH2 terminus of the beta2 auxiliary subunit: An essential role of a terminal peptide segment of three hydrophobic residues. J Gen Physiol. 2003;121(2):125–148. doi: 10.1085/jgp.20028667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orio P, Latorre R. Differential effects of beta 1 and beta 2 subunits on BK channel activity. J Gen Physiol. 2005;125(4):395–411. doi: 10.1085/jgp.200409236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brenner R, et al. BK channel beta4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nat Neurosci. 2005;8(12):1752–1759. doi: 10.1038/nn1573. [DOI] [PubMed] [Google Scholar]
- 18.Semenov I, Wang B, Herlihy JT, Brenner R. BK channel beta1-subunit regulation of calcium handling and constriction in tracheal smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2006;291(4):L802–L810. doi: 10.1152/ajplung.00104.2006. [DOI] [PubMed] [Google Scholar]
- 19.Pyott SJ, et al. Cochlear function in mice lacking the BK channel alpha, beta1, or beta4 subunits. J Biol Chem. 2007;282(5):3312–3324. doi: 10.1074/jbc.M608726200. [DOI] [PubMed] [Google Scholar]
- 20.Zeng XH, Benzinger GR, Xia XM, Lingle CJ. BK channels with beta3a subunits generate use-dependent slow afterhyperpolarizing currents by an inactivation-coupled mechanism. J Neurosci. 2007;27(17):4707–4715. doi: 10.1523/JNEUROSCI.0758-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yan J, Aldrich RW. LRRC26 auxiliary protein allows BK channel activation at resting voltage without calcium. Nature. 2010;466(7305):513–516. doi: 10.1038/nature09162. [DOI] [PubMed] [Google Scholar]
- 22.Yan J, Aldrich RW. BK potassium channel modulation by leucine-rich repeat-containing proteins. Proc Natl Acad Sci USA. 2012;109(20):7917–7922. doi: 10.1073/pnas.1205435109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elkins T, Ganetzky B, Wu CF. A Drosophila mutation that eliminates a calcium-dependent potassium current. Proc Natl Acad Sci USA. 1986;83(21):8415–8419. doi: 10.1073/pnas.83.21.8415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Atkinson NS, Robertson GA, Ganetzky B. A component of calcium-activated potassium channels encoded by the Drosophila slo locus. Science. 1991;253(5019):551–555. doi: 10.1126/science.1857984. [DOI] [PubMed] [Google Scholar]
- 25.Pallanck L, Ganetzky B. Cloning and characterization of human and mouse homologs of the Drosophila calcium-activated potassium channel gene, slowpoke. Hum Mol Genet. 1994;3(8):1239–1243. doi: 10.1093/hmg/3.8.1239. [DOI] [PubMed] [Google Scholar]
- 26.Tseng-Crank J, et al. Cloning, expression, and distribution of functionally distinct Ca2+-activated K+ channel isoforms from human brain. Neuron. 1994;13(6):1315–1330. doi: 10.1016/0896-6273(94)90418-9. [DOI] [PubMed] [Google Scholar]
- 27.Cui J, Cox DH, Aldrich RW. Intrinsic voltage dependence and Ca2+ regulation of mslo large conductance Ca-activated K+ channels. J Gen Physiol. 1997;109(5):647–673. doi: 10.1085/jgp.109.5.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cox DH, Cui J, Aldrich RW. Allosteric gating of a large conductance Ca-activated K+ channel. J Gen Physiol. 1997;110(3):257–281. doi: 10.1085/jgp.110.3.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horrigan FT, Cui J, Aldrich RW. Allosteric voltage gating of potassium channels I. Mslo ionic currents in the absence of Ca2+ J Gen Physiol. 1999;114(2):277–304. doi: 10.1085/jgp.114.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horrigan FT, Aldrich RW. Allosteric voltage gating of potassium channels II. Mslo channel gating charge movement in the absence of Ca2+ J Gen Physiol. 1999;114(2):305–336. doi: 10.1085/jgp.114.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Horrigan FT, Aldrich RW. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J Gen Physiol. 2002;120(3):267–305. doi: 10.1085/jgp.20028605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cui J, Yang H, Lee US. Molecular mechanisms of BK channel activation. Cell Mol Life Sci. 2009;66(5):852–875. doi: 10.1007/s00018-008-8609-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Latorre R, Morera FJ, Zaelzer C. Allosteric interactions and the modular nature of the voltage- and Ca2+-activated (BK) channel. J Physiol. 2010;588(Pt 17):3141–3148. doi: 10.1113/jphysiol.2010.191999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holmgren M, Smith PL, Yellen G. Trapping of organic blockers by closing of voltage-dependent K+ channels: Evidence for a trap door mechanism of activation gating. J Gen Physiol. 1997;109(5):527–535. doi: 10.1085/jgp.109.5.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Y, Holmgren M, Jurman ME, Yellen G. Gated access to the pore of a voltage-dependent K+ channel. Neuron. 1997;19(1):175–184. doi: 10.1016/s0896-6273(00)80357-8. [DOI] [PubMed] [Google Scholar]
- 36.del Camino D, Holmgren M, Liu Y, Yellen G. Blocker protection in the pore of a voltage-gated K+ channel and its structural implications. Nature. 2000;403(6767):321–325. doi: 10.1038/35002099. [DOI] [PubMed] [Google Scholar]
- 37.del Camino D, Yellen G. Tight steric closure at the intracellular activation gate of a voltage-gated K+ channel. Neuron. 2001;32(4):649–656. doi: 10.1016/s0896-6273(01)00487-1. [DOI] [PubMed] [Google Scholar]
- 38.Flynn GE, Zagotta WN. Conformational changes in S6 coupled to the opening of cyclic nucleotide-gated channels. Neuron. 2001;30(3):689–698. doi: 10.1016/s0896-6273(01)00324-5. [DOI] [PubMed] [Google Scholar]
- 39.Li W, Aldrich RW. Unique inner pore properties of BK channels revealed by quaternary ammonium block. J Gen Physiol. 2004;124(1):43–57. doi: 10.1085/jgp.200409067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Contreras JE, Holmgren M. Access of quaternary ammonium blockers to the internal pore of cyclic nucleotide-gated channels: Implications for the location of the gate. J Gen Physiol. 2006;127(5):481–494. doi: 10.1085/jgp.200509440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Piskorowski RA, Aldrich RW. Relationship between pore occupancy and gating in BK potassium channels. J Gen Physiol. 2006;127(5):557–576. doi: 10.1085/jgp.200509482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilkens CM, Aldrich RW. State-independent block of BK channels by an intracellular quaternary ammonium. J Gen Physiol. 2006;128(3):347–364. doi: 10.1085/jgp.200609579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li W, Aldrich RW. State-dependent block of BK channels by synthesized shaker ball peptides. J Gen Physiol. 2006;128(4):423–441. doi: 10.1085/jgp.200609521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Contreras JE, Srikumar D, Holmgren M. Gating at the selectivity filter in cyclic nucleotide-gated channels. Proc Natl Acad Sci USA. 2008;105(9):3310–3314. doi: 10.1073/pnas.0709809105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang QY, Zeng XH, Lingle CJ. Closed-channel block of BK potassium channels by bbTBA requires partial activation. J Gen Physiol. 2009;134(5):409–436. doi: 10.1085/jgp.200910251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou Y, Xia XM, Lingle CJ. Cysteine scanning and modification reveal major differences between BK channels and Kv channels in the inner pore region. Proc Natl Acad Sci USA. 2011;108(29):12161–12166. doi: 10.1073/pnas.1104150108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen X, Aldrich RW. Charge substitution for a deep-pore residue reveals structural dynamics during BK channel gating. J Gen Physiol. 2011;138(2):137–154. doi: 10.1085/jgp.201110632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Doyle DA, et al. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science. 1998;280(5360):69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 49.Armstrong CM. Voltage-gated K channels. Sci STKE. 2003;2003(188):re10. doi: 10.1126/stke.2003.188.re10. [DOI] [PubMed] [Google Scholar]
- 50.Cuello LG, Jogini V, Cortes DM, Perozo E. Structural mechanism of C-type inactivation in K+ channels. Nature. 2010;466(7303):203–208. doi: 10.1038/nature09153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cuello LG, et al. Structural basis for the coupling between activation and inactivation gates in K+ channels. Nature. 2010;466(7303):272–275. doi: 10.1038/nature09136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hackos DH, Chang TH, Swartz KJ. Scanning the intracellular S6 activation gate in the shaker K+ channel. J Gen Physiol. 2002;119(6):521–532. doi: 10.1085/jgp.20028569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sukhareva M, Hackos DH, Swartz KJ. Constitutive activation of the Shaker Kv channel. J Gen Physiol. 2003;122(5):541–556. doi: 10.1085/jgp.200308905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu Y, et al. Intersubunit coupling in the pore of BK channels. J Biol Chem. 2009;284(35):23353–23363. doi: 10.1074/jbc.M109.027789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shi N, Zeng W, Ye S, Li Y, Jiang Y. Crucial points within the pore as determinants of K+ channel conductance and gating. J Mol Biol. 2011;411(1):27–35. doi: 10.1016/j.jmb.2011.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fersht A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. New York: Freeman; 1999. [Google Scholar]
- 57.Johnson JP, Jr, Zagotta WN. Rotational movement during cyclic nucleotide-gated channel opening. Nature. 2001;412(6850):917–921. doi: 10.1038/35091089. [DOI] [PubMed] [Google Scholar]
- 58.Webster SM, Del Camino D, Dekker JP, Yellen G. Intracellular gate opening in Shaker K+ channels defined by high-affinity metal bridges. Nature. 2004;428(6985):864–868. doi: 10.1038/nature02468. [DOI] [PubMed] [Google Scholar]
- 59.Grosman C, Zhou M, Auerbach A. Mapping the conformational wave of acetylcholine receptor channel gating. Nature. 2000;403(6771):773–776. doi: 10.1038/35001586. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.