

Significance
Pathological deposition of mutated Cu/Zn superoxide dismutase (SOD1) accounts for ∼20% of the familial ALS (fALS) cases. Insoluble protein aggregates, immunoreactive for SOD1, have been found in both fALS and sporadic ALS (sALS) patients. To study the molecular origin of SOD1 aggregation, we used a computational approach to discover four segments from SOD1 that form fibril-like aggregates. We found that two of these, 101DSVISLS107 and 147GVIGIAQ153, are likely to trigger the aggregation of full-length SOD1, suggesting common molecular determinants of fALS and sALS.
Keywords: protein aggregation, peptide structure, amyotrophic lateral sclerosis
Abstract
ALS is a terminal disease of motor neurons that is characterized by accumulation of proteinaceous deposits in affected cells. Pathological deposition of mutated Cu/Zn superoxide dismutase (SOD1) accounts for ∼20% of the familial ALS (fALS) cases. However, understanding the molecular link between mutation and disease has been difficult, given that more than 140 different SOD1 mutants have been observed in fALS patients. In addition, the molecular origin of sporadic ALS (sALS) is unclear. By dissecting the amino acid sequence of SOD1, we identified four short segments with a high propensity for amyloid fibril formation. We find that fALS mutations in these segments do not reduce their propensity to form fibrils. The atomic structures of two fibril-forming segments from the C terminus, 101DSVISLS107 and 147GVIGIAQ153, reveal tightly packed β-sheets with steric zipper interfaces characteristic of the amyloid state. Based on these structures, we conclude that both C-terminal segments are likely to form aggregates if available for interaction. Proline substitutions in 101DSVISLS107 and 147GVIGIAQ153 impaired nucleation and fibril growth of full-length protein, confirming that these segments participate in aggregate formation. Our hypothesis is that improper protein maturation and incompletely folded states that render these aggregation-prone segments available for interaction offer a common molecular pathway for sALS and fALS.
ALS is a progressive neurodegenerative disease that affects motor neurons, often causing death within 2 to 5 years. Ninety percent of ALS cases are sporadic (sALS), and their cause is unknown (1). However, the remaining 10% of ALS cases are inherited familial ALS (fALS), ∼20% of which are linked to mutations in the Cu/Zn superoxide dismutase (SOD1) gene.
Mature SOD1 is a 32-kDa homodimeric metalloenzyme, in which each monomer contains a copper ion, zinc ion, and one intrasubunit disulfide bond (2) (Fig. 1A). SOD1 is one of the most abundant proteins in cells, serving to protect organisms against oxidative damage. The loss of protein function does not necessarily lead to disease because SOD1-deficient mice develop mild impairments that are not observed in ALS (3). Instead, the mutated SOD1 seems to have a toxic gain of function that leads to the pathologies of disease. To date, more than 140 dominant disease-related mutations that span nearly the whole protein sequence have been described (http://alsod.iop.kcl.ac.uk/Als/).
Fig. 1.

(A) Ribbon diagram of SOD1 dimer (PDB code: 2C9U) which shows zinc (green) and copper (blue) atoms coordinated in the metal-binding loop. The intrasubunit disulfide bond between Cys57 and Cys146 is shown in gold. The four SOD1 segments predicted to form fibrils are shown in red. (B) Diagram of the 3D-profile Rosetta energies (y axis) calculated for each six-residue segment from the SOD1 sequence (x axis). Segments predicted to form fibrils are highlighted in red.
Several studies suggest sALS and fALS have common mechanisms of pathogenesis associated with accumulation of misfolded SOD1 (4). Evidence to support this has shown that insoluble protein aggregates found in both fALS (5) and sALS (6) patients were SOD1 immunoreactive. Scientists have created transgenic mice that express human SOD1 mutations found in fALS. The mice exhibit behavioral and cellular symptoms similar to human ALS (7), including accumulation of insoluble aggregates (8). In addition, expression in mice of heterozygous wild-type/mutant SOD1 augments disease symptoms relative to homozygous mutant animals (7), implying that the wild-type protein produced by the allele carrying the normal gene enhances the toxicity of the mutant protein in fALS. It was also shown that transgenic mice overexpressing wild-type SOD1 show ALS-like symptoms and large amounts of aggregated SOD1 in the spinal cord and brain (9). These findings suggest that investigating the mechanism of aggregation of wild-type and mutant SOD1 may further our understanding of the molecular origins of both fALS and sALS.
SOD1-containing pathological inclusions in ALS may have amyloid-like properties. First, neuronal tissue from a mouse model that expresses the H46R/H48Q mutations can be stained with Thioflavin S, a fluorescent dye whose properties change in the presence of amyloid-like aggregates (10). Second, in vitro, reduction of the disulfide bond and removal of metals from SOD1 and its mutants lead to the formation of amyloid-like aggregates (11). Third, SOD1 fibrils formed in vitro share common toxic properties with ALS inclusions, such as the ability to induce inflammation (12) and activate microglial cells (13). Fourth, amyloid fibril formation of recombinant SOD1 can be seeded with tissue extracts of SOD1-containing inclusions from ALS transgenic mice (14). All these data suggest the pathologic inclusions share similarities with in vitro formed amyloid fibrils.
It is well established that amyloid fibril formation is driven by the exposure of short aggregation-prone segments. Here, we pinpoint the aggregation-prone regions of SOD1 to clarify the molecular origins of ALS. Using a computational algorithm (15), we identified four segments in SOD1 with high propensities to form fibrils. We experimentally observed that proline substitutions in two of these segments block fibril formation of full-length protein, emphasizing the importance of these segments in amyloid aggregation. Our studies therefore suggest specific segments within the SOD1 sequence that drive the aggregation and perhaps initiate pathology of ALS.
Results
Aggregation-Prone Segments in SOD1.
To find which segments induce aggregation of SOD1, we used the 3D-profile method (15) (Fig. 1B). In SOD1, we find four segments of seven to nine residues with energies favorable for fibril formation: 14VQGIINFE21, 30KVWGSIKGL38, 101DSVISLS107, and 147GVIGIAQ153. We confirmed our predictions by determining that all four segments form fibril-like or needle-like aggregates (Fig. 2).
Fig. 2.

The four predicted segments formed fibrils. Mutations associated with fALS preserve the fibril-forming propensities of 13VQGIINFE21, 101DSVISLS107, and 147GVIGIAQ153. In addition, 13 of the 14 mutated segments that we tested formed aggregates with fibril morphology, confirming the robustness of our predictions. (Scale bar, 100 nm.)
Most in-Segment Disease Mutations Are Compatible with Aggregate Formation.
Twenty-four mutations associated with fALS (http://alsod.iop.kcl.ac.uk/Als/) are located in the four aggregation-prone segments. We found that only 2 of the 24 mutations changed fibril formation propensity of the segments from favorable to unfavorable (Supporting Information, Fig. S1 and Table S1). To validate the computational predictions we synthesized 14 peptides with disease-related mutations. We found that 13 of the 14 mutant peptides formed aggregates with fibril morphology (Fig. 2). The only peptide, G37R, which did not form fibrils was also predicted to have low fibril-formation propensity.
101DSVISLS107 and 147GVIGIAQ153 Form Steric Zipper Structures.
The crystal structures of the two fibril-forming segments 101DSVISLS107 and 147GVIGIAQ153 were solved to 1.4-Å and 1.9-Å resolution, respectively. Each segment forms pairs of β-sheets that mate together through complementary surfaces (Fig. 3 A and B). Such structures are typical for amyloid-forming segments and are known as steric zippers (16) (Fig. 3 A and B).
Fig. 3.

Steric zipper structures of (A) 101DSVISLS107, (B) 147GVIGIAQ153, and (C) 147GVTGIAQ153 (I149T) segments. Off-axis (Right) and down the fibril axis (Left) views of the dry steric zipper interface of 101DSVISLS107, 147GVIGIAQ153, and147GVIGIAQ153. The dry steric zipper of the 147GVTGIAQ153 (I149T) (C) compared with the wild type (B) is stabilized by two hydrogen bonds (dashed lines) formed between Gln153 and the mutated Thr149, showing that this mutation is compatible with the steric zipper. The side-chain carbon atoms and Cα atoms of odd-numbered residues are colored burgundy to distinguish the two faces of the sheets. This was done to illustrate the face-to-back steric zipper packing of 101DSVISLS107 (A) and the face-to-face packing of 147GVIGIAQ153 (B) and 147GVTGIAQ153 (I149T) (C). Blue and green spheres represent water and Zn, respectively.
The structures revealed that the steric zippers formed by 101DSVISLS107 and 147GVIGIAQ153 differ in arrangement. β-Sheets in the 101DSVISLS107 structure interact through their distinct surfaces in the face-to-back packing of class-2 steric zippers (Fig. 3A). In contrast, the 147GVIGIAQ153 steric zipper is formed by identical β-sheet surfaces in face-to-face packing of class-1 steric zippers (Fig. 3B). In the 147GVIGIAQ153 structure, we observed that β-sheets also interact tightly with their opposite sides in back-to-back packing (Supporting Information, Fig. S2A). The area buried and shape complementarity of the two steric zipper interfaces are comparable (Supporting Information, Table S2).
Segments with fALS Mutations also Form Steric Zipper Structures.
To determine how mutations found in fALS change the fibril-forming properties of the wild-type segment, we studied the structure of 147GVTGIAQ153 (I149T) and 147GIIGIAQ153 (V148I). X-ray fiber diffraction of 147GIIGIAQ153 (V148I) showed a cross-β diffraction pattern common to amyloid fibrils (Supporting Information, Fig. S3), but we could not obtain a high-resolution structure. The structure of 147GVTGIAQ153 (I149T) was solved to 1.3 Å (Fig. 3C and Supporting Information, Fig. S2B). The dry interface of 147GVTGIAQ153 (I149T) compared with the wild-type segment is additionally stabilized by two hydrogen bonds formed between Thr and Gln (Fig. 3 B and C). In general, the 147GVTGIAQ153 (I149T) structure reveals that mutations in the steric zipper can change the spatial arrangement of the β-sheet surfaces forming the steric zipper without diminishing its apparent strength.
The 147GVIGIAQ153 Segment Accelerates Fibril Formation of Metal-Free SOD1WT.
We tested whether any of the isolated peptide segments accelerate nucleation or growth of full-length protein fibrils. We hypothesized that if a segment is critical for fibril formation, the presence of that short segment should alter the aggregation kinetics of the full-length protein, possibly by providing a template for fibril nucleation (17). Of the four segments, only 147GVIGIAQ153 from the C terminus accelerated fibril formation of wild-type Cu/Zn superoxide dismutase [metal-free (apo)SOD1WT] in a dose-dependent manner (Fig. 4A and Supporting Information, Fig. S6A). This acceleration was consistently observed, although the required ratio of segment to full-length protein varied with the different preparations of SOD1. These results suggest that 147GVIGIAQ153 might initiate fibril formation of full-length protein.
Fig. 4.

The C-terminal segment 147GVIGIAQ153 nucleates the fibril formation of full-length SOD1. (A) Using a ThT fluorescence assay, we observed that full-length SOD1 coincubated with 147GVIGIAQ153 forms fibrils more rapidly than the full-length protein alone. Adding 15 molar excess of 147GVIGIAQ153 shortened the lag time of aggregate formation nearly 50%. (Right) TEM images of the samples taken after the ThT fluorescence signal reached a plateau. (Upper Right) Micrograph of apoSOD1WT fibrils. (Lower Right) Micrograph of apoSOD1WT coincubated with the same peptide showing that wild-type apoSOD1 fibrils (white arrows) colocalize with 147GVIGIAQ153 needle-like aggregates (black arrows). (B) The 147GVIGIAQ153 segment seeded the aggregation of mutant apoSOD1G93A. Twenty times molar excess of 147GVIGIAQ153 shortened the lag phase of apoSOD1WT aggregation by about one third. apoSOD1G93A coincubated with a 20 molar excess of 147GVIGIAQ153 (Lower Right) formed a mixture of full-length protein fibrils (white arrow) and 147GVIGIAQ153 needle-like aggregates (black arrows). Each point in the ThT fluorescence assays is an average of four replicates.
The 147GVIGIAQ153 Segment Accelerates the Fibril Formation of apoSOD1G93A.
To test whether the 147GVIGIAQ153 segment can nucleate the fibril formation of SOD1 mutants associated with fALS, we followed the aggregation of G93A mutant SOD1 (apoSOD1G93A), one of the best characterized mutants of SOD1 (18). Indeed, similar to apoSOD1WT, fibril formation of apoSOD1G93A was accelerated in the presence of 147GVIGIAQ153 (Fig. 4B). Likewise, we observed variability in the aggregation kinetics and in the quantity of 147GVIGIAQ153 needed to nucleate the fibril formation of the apoSOD1G93A mutant from different protein preparations. In electron micrographs of samples collected after the Thioflavin T (ThT) fluorescence signal reached a plateau, we observed thick rigid fibril-like aggregates of 147GVIGIAQ153 and flexible thin fibrils of apoSOD1G93A (Fig. 4B). In general, we observed that the fibrils of full-length protein tend to cluster with needle-like aggregates of 147GVIGIAQ153, further hinting that the full-length aggregation is facilitated by the presence of this C-terminal segment.
Discussion
SOD1 Contains Four Fibril-Forming Segments.
Aided by a computational algorithm (15), we identified four segments likely involved in SOD1 fibril formation. In isolation from the rest of the protein, all these segments formed aggregates with fibril morphology (Fig. 2, Left). The existence of several aggregation-prone segments in SOD1 unifies conflicting studies that find different regions from the protein participate in aggregate formation (19, 20).
fALS-Linked Mutations Modulate but Do Not Disrupt Aggregation.
There are roughly 140 mutations in SOD1 that are associated with fALS. Of the 14 we tested, 13 formed aggregates (Fig. 2, Left). The ability of the segments to form fibrils was unaffected by the mutations that changed their charge, size, or hydrophobicity. The structure of the mutant segment I149T revealed molecular details of the changes in the steric zipper organization caused by the mutation. Although the steric zippers formed by wild-type and mutant segments have different spatial arrangements, their interaction energies are comparable as judged by the area buried and shape complementarity of the interfaces (Supporting Information, Table S2). Although, many fALS mutations promote aggregation by destabilizing the native protein structure, our results suggest that disease-related mutations generally do not inhibit fibril formation of the aggregation-prone segment and may even enhance it.
Two C-Terminal Segments Dominate Fibril Formation.
Full-length protein with I18P and I35P substitutions formed fibrils similar in morphology to the wild-type protein (Supporting Information, Fig. S5), leading us to conclude that the N-terminal segments 14VQGIINFE21 and 30KVWGSIKGL38 do not form the β-sheet spine of the SOD1 fibrils. Pro substitution at position I104 suppressed fibril formation of wild-type protein, implying that 101DSVISLS107 is involved in fibril nucleation and/or growth (Fig. 5). We also observed change in fibril morphology of the I149P mutant that blocks the aggregation of 147GVIGIAQ153 compared with wild-type fibrils. The I149P mutant formed rod-shaped aggregates that were less than 100 nm in length (Fig. 5), suggesting that the C terminus of SOD1 takes part in fibril elongation and/or forms the β-sheet spine of the SOD1 fibrils. In the samples of both I104P and T149P mutants we observed wild-type fibrils, suggesting that blocking the aggregation of the C-terminal segments may in turn lead to fibril formation through other segments. The two peptide segments 14VQGIINFE21 and 30KVWGSIKGL38 formed aggregates with fibril morphology (Fig. 2), suggesting they can also participate in aggregation of the full-length protein. Thus, for fALS cases in which the fibril-forming segment 147GVIGIAQ153 is missing due to early translation termination of SOD1 (21), the remaining three aggregation-prone segments can participate in aggregate formation.
Fig. 5.
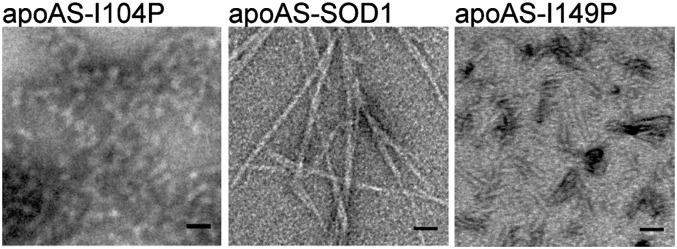
Pro substitutions in 101DSVISLS107 and 147GVIGIAQ153 inhibited aggregation of full-length protein. Wild-type like apoSOD1 with mutated surface C6A and C111S (apoAS-SOD1) in which Ile104 was substituted with Pro, formed amorphous aggregates (Left). There were also occasional fibers like those of the wild-type protein (Center). Short rods were observed in the apoAS-SOD1I149P sample (Right) together with some wild-type-looking fibrils.
C-Terminal Segment 147GVIGIAQ153 Seeds Aggregation.
Using fibril formation assays, we find that addition of the C-terminal segment 147GVIGIAQ153 accelerates the aggregation of wild-type protein (Fig. 4A). In the native state, the 147GVIGIAQ153 segment is buried in the dimer interface and protected from exposure (Fig. 1A). Reduction of the disulfide bond results in enhanced mobility of the disulfide loop and of the adjacent 147GVIGIAQ153 segment. This in turn weakens the interactions across the dimer interface and destabilizes the β-barrel fold. Indeed, the conditions used in this study favor the monomeric state (22), in which the 147GVIGIAQ153 segment is exposed for interaction.
The 147GVIGIAQ153 segment also accelerated the aggregation of the fALS mutant apoSOD1G93A further suggesting the importance of this segment in SOD1 aggregation. In addition, that the wild-type and mutant protein can template each other’s aggregation implies structural similarities between the aggregates. Preformed nuclei of apoSOD1G93A seed the aggregation of wild-type protein (Supporting Information, Fig. S8A) and vice versa (Supporting Information, Fig. S8B). Based on this, we can infer that the aggregation of the wild-type protein influences the aggregation of mutant SOD1. Our experiments show that the 147GVIGIAQ153 segment accelerates the aggregation of both wild-type and mutant protein suggesting that this segment is a common molecular determinant of aggregate formation in sALS and fALS.
Our experiments suggest that fiber formation of SOD1 is initiated by the monomer where the segment 147GVIGIAQ153 is unprotected. Although mature SOD1 is a stable dimer, newly translated SOD1 is a copper-free monomer with reduced Cys, in which the aggregation-prone C terminus is exposed and flexible. Dimer formation is a part of the SOD1 maturation process and facilitated by the copper chaperone for SOD1 (CCS). CCS modifies the nascent SOD1 by inserting copper and oxidizing its intrasubunit disulfide bond; this leads to spontaneous dimer formation (23). Thus, any delay in the posttranslational processing of SOD1 by CCS, such as slow protein folding, sequence mutations, or changes in the cellular environment, lead to increased accumulation of unfolded monomeric intermediates (24). Indeed, pathogenic SOD1 proteins derived from either cultured cells or the spinal cords of transgenic mice tend to be metal-deficient (25) and/or lack the disulfide bond (26).
It is possible that various processes, such as abnormally high expression of SOD1, can also result in an increased pool of aggregation-susceptible monomers. Normally, the cellular concentration of SOD1 is 30-fold more abundant than that of CCS (at both the RNA and protein levels) (27), so CCS must cycle through the pool of newly translated molecules to activate them. Overexpression of wild-type SOD1 caused paralysis in mice (9). Thus, it is likely that the excess SOD1 overwhelms the endogenous CCS system, and the pool of immature wild-type SOD1 molecules aggregates, leading to paralysis which is similar to symptoms in ALS. In short, exposure of the aggregation-prone segments in immature SOD1 could be at the root of both sporadic and familial ALS.
A Common Pathway for fALS and sALS.
Although SOD1-containing aggregates are seen in only a subset of sALS cases, the similarities between SOD1-linked fALS and sALS disease suggest that clarification of the molecular origins of SOD1 aggregation will be informative about ALS cases as a whole. Our hypothesis is that the exposure of one or more fibril-forming segments of SOD1 can lead to aggregation of the full-length protein. Here we identified two segments, 101DSVISLS107 (located at the Greek Key loop) and 147GVIGIAQ153 (located at the C terminus), that are likely to participate in fibril nucleation and growth of SOD1. fALS mutations located within these segments do not prevent fibril formation but do alter the fibril morphology and the steric zipper packing. We speculate that in vivo exposure of these segments could occur before chaperone-assisted maturation of the protein. Overall our data suggest a common molecular origin of fALS and sALS: the exposure and the interaction of aggregation-prone segments at the C terminus of SOD1.
Materials and Methods
Peptides were either purchased (CS Bio, Celtek Bioscience Peptides, and GenScript) or synthesized in the laboratory. 14VQGIINFE21, 101DSVISLS107, and their corresponding mutants were dissolved in water to a final concentration of 2 mM. 147GVIGIAQ153 and its corresponding mutants except for 147RVIGIAQ153 (G147R) and 147GVTGIAQ153 (I149T) were dissolved to 1 mM with water. 147GVTGIAQ153 (I149T) formed fibril-like aggregates when dissolved to 6 mM with water. 147RVIGIAQ153 (G147R) formed fibrils when dissolved to 1 mM in 1 mM EDTA, 25 mM K phosphate, pH 7.0. 30KVWGSIKGL38 and its mutant were dissolved to 60 mM with 0.1 M Tris base (pH was not adjusted). Peptide solutions were incubated at 37 °C with shaking, followed by transmission electron microscopy (TEM) examination after 7–10 d.
GVIGIAQ was crystallized using the hanging drop vapor diffusion method: 1 μL of 0.3 mg/mL 147GVIGIAQ153 was mixed with 1 μL of reservoir solution. The reservoir solution comprised 1 mL of 1 M sodium acetate, pH 4.5, and 1.75 M ammonium sulfate. 147GVTGIAQ153 (I149T) was also crystallized by the hanging drop vapor diffusion method: 1 μL of 6 mg/mL peptide dissolved in water was mixed with 2 μL of reservoir solution. The drop was equilibrated over 1 mL of reservoir solution containing 0.1 M sodium acetate, pH 4.5, and 0.7 M hexanediol. Similarly, 101DSVISLS107 was crystallized using the hanging drop method: 1 µL of 3 mg/mL peptide (filtered through a 0.2-µm filter) was mixed with 1 µL of reservoir solution [0.1 M Mes, pH 6.0, 20% (wt/vol) PEG 6000, and 5 mM ZnCl2]. X-ray diffraction data were collected at beamline 24-ID-E of the Advanced Photon Source and at beamline ID13 of the European Synchrotron Radiation Facility. Data were collected at 100 K with 5° oscillations.
Human SOD1WT and SOD1G93A were expressed in the EGy118 strain of Saccharomyces cerevisiae which lacks the endogenous yeast sod1 gene. The proteins were purified and metals were removed as previously described (22), except that in the last step, 25 mM potassium phosphate, pH 7.0, 1 mM EDTA were used as dialysis buffer. After metal removal, apoSOD1WT and apoSOD1G93A were immediately frozen and stored in −80 °C. The protein was used within 2 wk of freezing. Concentrated protein was thawed on ice and filtered through a 0.2-μm filter before the fibril formation assay. Fibril formation assays were performed with 17 μM apoSOD1WT and apoSOD1G93A in 25 mM potassium phosphate buffer, pH 7.0, 1 mM EDTA, 35 mM Tris(2-carboxyethyl)phosphine (TCEP), and 10 μM ThioflavinT. Fibril formation of full-length apoSOD1 was assayed by monitoring ThioflavinT. Detailed description of the experimental methods can be found in Supporting Information, SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Dr. L. Goldschmidt, Dr. M. Chattopadhyay, Dr. R. Nelson, Dr. B. Chan, and Prof. J. S. Valentine for discussions; Dr. I. Kourinov, Dr. J. Schuermann, Dr. K. Rajashankar, Dr. N. Sukumar, and Dr. S. Banerjee at Advanced Photon Source beamline 24-ID-E and European Synchrotron Radiation Facility beamline ID13 for help with X-ray data collection; Howard Hughes Medical Institute, P01 NS049134, National Institutes of Health AG029430, NIH P01, and Department of Energy for support to the D.E. laboratory; and Department of Veterans Affairs 1I01BX000506, the Judith and Jean Pape Adams Charitable Foundation, and NIH R01 NS39112 for support to the P.J.H. laboratory.
Footnotes
The authors declare no conflict of interest.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org [PDB ID codes 4NIP (GVIGIAQ), 4NIO (GVTGIAQ), and 4NIN (DSVISLS)].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1320786110/-/DCSupplemental.
References
- 1.Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat Rev Neurosci. 2006;7(9):710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- 2.Tainer JA, Getzoff ED, Beem KM, Richardson JS, Richardson DC. Determination and analysis of the 2 A-structure of copper, zinc superoxide dismutase. J Mol Biol. 1982;160(2):181–217. doi: 10.1016/0022-2836(82)90174-7. [DOI] [PubMed] [Google Scholar]
- 3.Reaume AG, et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13(1):43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- 4.Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344(22):1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- 5.Ohi T, Nabeshima K, Kato S, Yazawa S, Takechi S. Familial amyotrophic lateral sclerosis with His46Arg mutation in Cu/Zn superoxide dismutase presenting characteristic clinical features and Lewy body-like hyaline inclusions. J Neurol Sci. 2004;225(1-2):19–25. doi: 10.1016/j.jns.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 6.Forsberg K, et al. Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS ONE. 2010;5(7):e11552. doi: 10.1371/journal.pone.0011552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prudencio M, Durazo A, Whitelegge JP, Borchelt DR. An examination of wild-type SOD1 in modulating the toxicity and aggregation of ALS-associated mutant SOD1. Hum Mol Genet. 2010;19(24):4774–4789. doi: 10.1093/hmg/ddq408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang J, et al. Copper-binding-site-null SOD1 causes ALS in transgenic mice: Aggregates of non-native SOD1 delineate a common feature. Hum Mol Genet. 2003;12(21):2753–2764. doi: 10.1093/hmg/ddg312. [DOI] [PubMed] [Google Scholar]
- 9.Graffmo KS, et al. Expression of wild-type human superoxide dismutase-1 in mice causes amyotrophic lateral sclerosis. Hum Mol Genet. 2013;22(1):51–60. doi: 10.1093/hmg/dds399. [DOI] [PubMed] [Google Scholar]
- 10.Wang J, et al. Fibrillar inclusions and motor neuron degeneration in transgenic mice expressing superoxide dismutase 1 with a disrupted copper-binding site. Neurobiol Dis. 2002;10(2):128–138. doi: 10.1006/nbdi.2002.0498. [DOI] [PubMed] [Google Scholar]
- 11.Chattopadhyay M, et al. Initiation and elongation in fibrillation of ALS-linked superoxide dismutase. Proc Natl Acad Sci USA. 2008;105(48):18663–18668. doi: 10.1073/pnas.0807058105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fiala M, et al. IL-17A is increased in the serum and in spinal cord CD8 and mast cells of ALS patients. J Neuroinflammation. 2010;7:76. doi: 10.1186/1742-2094-7-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roberts K, et al. Extracellular aggregated Cu/Zn superoxide dismutase activates microglia to give a cytotoxic phenotype. Glia. 2013;61(3):409–419. doi: 10.1002/glia.22444. [DOI] [PubMed] [Google Scholar]
- 14.Chia R, et al. Superoxide dismutase 1 and tgSOD1 mouse spinal cord seed fibrils, suggesting a propagative cell death mechanism in amyotrophic lateral sclerosis. PLoS ONE. 2010;5(5):e10627. doi: 10.1371/journal.pone.0010627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldschmidt L, Teng PK, Riek R, Eisenberg D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc Natl Acad Sci USA. 2010;107(8):3487–3492. doi: 10.1073/pnas.0915166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sawaya MR, et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447(7143):453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 17.Ivanova MI, Sievers SA, Sawaya MR, Wall JS, Eisenberg D. Molecular basis for insulin fibril assembly. Proc Natl Acad Sci USA. 2009;106(45):18990–18995. doi: 10.1073/pnas.0910080106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kayatekin C, Zitzewitz JA, Matthews CR. Disulfide-reduced ALS variants of Cu, Zn superoxide dismutase exhibit increased populations of unfolded species. J Mol Biol. 2010;398(2):320–331. doi: 10.1016/j.jmb.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furukawa Y, Kaneko K, Yamanaka K, Nukina N. Mutation-dependent polymorphism of Cu,Zn-superoxide dismutase aggregates in the familial form of amyotrophic lateral sclerosis. J Biol Chem. 2010;285(29):22221–22231. doi: 10.1074/jbc.M110.113597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan PK, et al. Structural similarity of wild-type and ALS-mutant superoxide dismutase-1 fibrils using limited proteolysis and atomic force microscopy. Proc Natl Acad Sci USA. 2013;110(27):10934–10939. doi: 10.1073/pnas.1309613110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orrell RW, et al. Clinical and functional investigation of 10 missense mutations and a novel frameshift insertion mutation of the gene for copper-zinc superoxide dismutase in UK families with amyotrophic lateral sclerosis. Neurology. 2012;48(3):746–751. doi: 10.1212/wnl.48.3.746. [DOI] [PubMed] [Google Scholar]
- 22.Doucette PA, et al. Dissociation of human copper-zinc superoxide dismutase dimers using chaotrope and reductant. Insights into the molecular basis for dimer stability. J Biol Chem. 2004;279(52):54558–54566. doi: 10.1074/jbc.M409744200. [DOI] [PubMed] [Google Scholar]
- 23.Seetharaman SV, et al. Immature copper-zinc superoxide dismutase and familial amyotrophic lateral sclerosis. Exp Biol Med (Maywood) 2009;234(10):1140–1154. doi: 10.3181/0903-MR-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seetharaman SV, et al. Disrupted zinc-binding sites in structures of pathogenic SOD1 variants D124V and H80R. Biochemistry. 2010;49(27):5714–5725. doi: 10.1021/bi100314n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lelie HL, et al. Copper and zinc metallation status of copper-zinc superoxide dismutase from amyotrophic lateral sclerosis transgenic mice. J Biol Chem. 2011;286(4):2795–2806. doi: 10.1074/jbc.M110.186999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sheng Y, Chattopadhyay M, Whitelegge J, Valentine JS. SOD1 aggregation and ALS: Role of metallation states and disulfide status. Curr Top Med Chem. 2012;12(22):2560–2572. doi: 10.2174/1568026611212220010. [DOI] [PubMed] [Google Scholar]
- 27.Kawamata H, Manfredi G. Different regulation of wild-type and mutant Cu,Zn superoxide dismutase localization in mammalian mitochondria. Hum Mol Genet. 2008;17(21):3303–3317. doi: 10.1093/hmg/ddn226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.