

Significance
It is known that G-protein–coupled receptors exhibit several distinct low-energy conformations, each of which might favor binding to different ligands and/or lead to different downstream functions. Understanding the function of such proteins requires knowledge of the ensemble of low-energy configurations that might play a role in this pleiotropic functionality. We present the SuperBiHelix methodology aimed at identifying all the low-energy structures that might play a role in binding, activation, and signaling. SuperBiHelix uses overall template information from all available experimental or theoretical structures and then does very complete (∼13 trillion configurations) sampling of the helix rotations and tilts to predict the ensemble of low-energy structures. We find that different mutations and ligands stabilize different conformations.
Keywords: protein structure prediction, A2A adenosine receptor, β2-adrenergic receptor, constitutive activity, functional selectivity
Abstract
There is overwhelming evidence that G-protein–coupled receptors (GPCRs) exhibit several distinct low-energy conformations, each of which might favor binding to different ligands and/or lead to different downstream functions. Understanding the function of such proteins requires knowledge of the ensemble of low-energy configurations that might play a role in this pleiotropic functionality. We earlier reported the BiHelix method for efficiently sampling the (12)7 = 35 million conformations resulting from 30° rotations about the axis (η) of all seven transmembrane helices (TMHs), showing that the experimental structure is reliably selected as the best conformation from this ensemble. However, various GPCRs differ sufficiently in the tilts of the TMHs that this method need not predict the optimum conformation starting from any other template. In this paper, we introduce the SuperBiHelix method in which the tilt angles (θ, φ) are optimized simultaneously with rotations (η) efficiently enough that it is practical and sufficient to sample (5 × 3 × 5)7 = 13 trillion configurations. This method can correctly identify the optimum structure of a GPCR starting with the template from a different GPCR. We have validated this method by predicting known crystal structure conformations starting from the template of a different protein structure. We find that the SuperBiHelix conformational ensemble includes the higher energy conformations associated with the active protein in addition to those associated with the more stable inactive protein. This methodology was then applied to design and experimentally confirm structures of three mutants of the CB1 cannabinoid receptor associated with different functions.
G-protein–coupled receptors (GPCRs) [also referred to as seven-transmembrane receptors (7TMRs)] are integral membrane proteins that play a central role in transmembrane (TM) signal transduction. This largest superfamily in the human genome with ∼800 receptors identified (1, 2) is activated by a variety of bioactive molecules, including biogenic amines, peptides, and hormones that modulate the activity of 7TMRs to effect regulation of essential physiological processes (e.g., neurotransmission, cellular metabolism, secretion, cell growth, immune defense, and differentiation) through G-protein–coupled and/or β-arrestin–coupled signaling pathways (3) (Fig. 1A). A structural understanding of their pleiotropic function will have a tremendous and broad impact, as the disregulation of these receptors often plays an important role in major disease pathologies (1, 4). Malfunctions in GPCRs play a part in diseases such as ulcers, allergies, migraines, anxiety, psychosis, schizophrenia, hypertension, asthma, congestive heart failure, Parkinson, and glaucoma. GPCRs are of great interest pharmacologically, as the targets of 50% of recently released drugs and 25 of the top 100 best-selling drugs (5).
Fig. 1.

Illustration of the pleiotropic ensemble and its effects. (A) Balanced signaling by GPCRs. (B) Functional/thermodynamic view of GPCR conformations.
The pleiotropic nature of these receptors arises because ∼10–20 conformations of the wild-type (WT) GPCR have sufficiently close energies that one or another can be stabilized by interactions with various ligands, which in turn can lead to ligand-dependent functionality. Moreover, it has been shown (6–8) that even a single mutation can change the relative ordering of these low-lying conformations sufficiently to dramatically change the affinity to various ligands and indeed even the functionality in terms of G-protein coupling and downstream signaling (3). The current structural, thermodynamic, and functional knowledge of GPCRs suggests an emerging conformational-ensemble picture like the one shown in Fig. 1B (9), which provides a schematic of GPCR conformations in different scenarios using WT as a reference. The relative thermodynamic ordering of this pleiotropic ensemble of low-energy GPCR conformations can change depending on the conditions: mutations, presence of ligands, and/or presence of other proteins like G proteins, which in turn modifies the physiological function.
This pleiotropic ensemble also has profound implications for the ability to control receptor pharmacology, especially associated with receptor target-induced side effects, when the targeted receptor activates both beneficial and undesirable signaling pathways. An example is the niacin receptor GPR109A. Niacin is therapeutically beneficial as an antilypolytic agent (via G-protein–mediated pathways), but it also causes cutaneous flushing, which has been directly linked to the activation of β-arrestin 1 pathways (10). An analog of this molecule that does not affect the G-protein pathways but destabilizes the coupling to β-arrestin 1 and blocks that pathway (shown with red arrow in Fig. 1A) will be highly desirable. This requires a structural understanding of how distinct conformations selectively couple to specific pathways under various conditions.
Despite the great interest in GPCRs, progress in obtaining experimental atomic-level structures essential for understanding the nature of activation and for design and optimization of drug candidates has been slow, due to challenges involved in GPCR expression, purification, and crystallization. Breakthroughs in membrane protein structural biology techniques (11) are accelerating GPCR structure determinations and have resulted in structures (crystal/NMR) for ∼19 GPCRs, where a majority have been solved in one inactive form of the receptor. Three of these receptors have also been crystallized in functionally distinct conformations [β2 with Gs (12), meta II rhodopsin (13), and the partially active form of A2A receptor (14)], whose comparison with the respective inactive conformations provides structural insight into their activation. One of these receptors (Neurotensin receptor 1) has been crystallized only in the putatively active conformation (15). Indeed, Kim et al. (16) found for the A3 adenosine receptor that the optimum conformation for binding four selective agonists was the 15th in the hierarchy of conformations for the apo protein, whereas four selective antagonists preferred either the second or the third conformation. Thus, each receptor putatively has multiple active conformations (Fig. 1B), making it essential to obtain a method of predicting GPCR structures that does not require homologous experimental structures and that can determine the ensemble of low-lying structures.
To provide the means for determining this ensemble of low-lying seven-helix bundle conformations, we propose and validate here the SuperBiHelix method, which aims to select from a complete set of seven-helix packings or conformations the ensemble of low-energy GPCR structures that could play a role in ligand binding and/or activation. The SuperBiHelix method builds upon the BiHelix method (17). We showed earlier that, starting with the known X-ray structure, the BiHelix method identifies correctly the optimum packing from the (12)7 = 35 million conformations differing by independent 30° rotations about the axes of the seven transmembrane helices. However, starting with some template (previous experimental or predicted structure) the BiHelix method does not necessarily identify the correct tilts of the helices (7, 9, 18) in the membrane. In this paper, we generalize the BiHelix concept to optimize the helical tilts (θ, φ) simultaneous with the helical rotations (η), which we refer to as SuperBiHelix. We demonstrate here that, starting with the X-ray template, say for A2A, the SuperBiHelix procedure outlined below correctly predicts the structure for β2-adrenergic receptor and vice versa. Also, the method is predictive as exemplified by the design of CB1 receptor mutants with altered G-protein coupling efficacy, which were later confirmed experimentally by GRPγS assays (7).
Methodology
SuperBiHelix and SuperCombiHelix.
The SuperBiHelix procedure starts with a GPCR bundle template, obtained either from X-ray experiments or from previously predicted and validated GPCR structures. This template is defined by the 6 × 7 = 42 degrees of freedom: x, y, h, θ, ϕ, and η values for each of the seven TM helices, as described earlier (17). Because some helices may have kinks and bends, the helical axis is defined as its least moment of inertia. The hydrophobic center (HPC) residue h is the residue that crosses the z = 0 plane, which is defined as the plane that runs through the center of the lipid bilayer. This center is calculated from the hydrophobic profile obtained either by our PredicTM procedure (19) or by homology to the template structure prealigned to an implicit membrane, as in the OPM database (20). The x axis is defined along the vector pointing from the HPC of TM3 (which is near the middle of the bundle), to the HPC of TM2. These definitions of the x axis and z axis implicitly define the y axis. The (x, y) position of the helix HPCs in the z = 0 reference plane is defined by the template and is not currently sampled by SuperBiHelix (this could be included but at substantially increased cost). The helix HPC residue (h) could be optimized by translating the helix along its helical axis, but we do not currently do this for the standard SuperBiHelix method. The coordinates (x, y, h) are expected to be optimized during atomistic membrane bilayer molecular dynamics of the predicted ensemble of conformations. This leaves 3 degrees of freedom to be sampled for each of the seven helices: θ, the tilt angle of the TM helix axis from the z axis (that is perpendicular to the membrane plane); ϕ, the sweep (or azimuthal) angle of the helix axis about the z axis; and η, the rotation of the helix around the helical axis.
The SuperBiHelix procedure starts with an input GPCR bundle determined by 42 template variables defined earlier and would normally be called a homology model. Just as in the BiHelix method (17), we approximate the energy as the sum over pairwise interactions between each of the 12 pairs of interacting helices. For each of these interacting pairs, we sample θ, ϕ, and η, while ignoring the other five helices, leading to the BiHelix energies. SCREAM (21) is used to predict the side-chain placements, which are then minimized for 10 steps with the backbone fixed (to resolve any bad contacts). This procedure is illustrated in Fig. 2A.
Fig. 2.
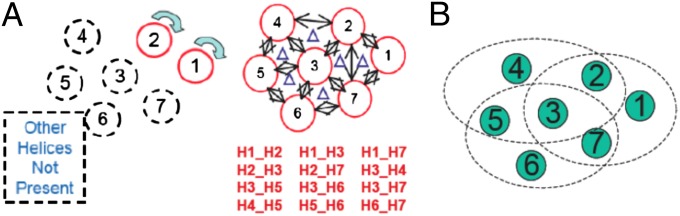
(A) Diagram of the SuperBiHelix method, showing how the seven-helix TM bundle is partitioned into 12 independent helix pairs. The θ, ϕ, and η values for each helix in the pair are sampled with the other helices not present. (B) To efficiently determine a subset of conformations for each helix most likely to lead to the lowest energy bundles, we partition the seven-helix bundle into three QuadHelix bundles: TM1-TM2-TM3-TM7, TM2-TM3-TM4-TM5, and TM3-TM5-TM6-TM7.
Once the 12 pairs of BiHelix energies have been determined for all possible combinations of θ, ϕ, and η, we combine these energies to predict a mean field energy of the entire bundle for effectively (3*5*5)7 ∼ 1013 conformations. These BiHelix energies are partitioned into intrahelical and interhelical components to avoid multiple counting of the intrahelical contributions, and the energy of the entire complex is then calculated, as described in SI Text (17). Although the calculation of the energy of a complex based on its BiHelix energies is very fast, the calculation of all possible configurations is still computationally expensive. In practice, we generally sample three values of θ, five values of ϕ, and five values of η, leading to (3*5*5)7 ∼ 1013 total bundle conformations. To minimize the number of total bundle energies to be calculated, we developed a procedure to determine which conformations for each helix are most favorable.
To determine the conformations for each helix likely to lead to low-energy bundles, we partitioned the seven-helix bundle into three QuadHelix bundles, as shown in Fig. 2B. It is feasible to estimate the total BiHelix energies of the three QuadHelix bundles because it requires only 3*(3*5*5)4 ∼ 108 bundle energies. The 2,000 structures with the lowest energy for each QuadHelix are listed by increasing energy. Then, the best 36 conformations for each helix are selected. This list of 36 conformations for a specific helix depends on how many QuadHelix bundles contain the helix. For helices 1, 4, and 6, which only exist in one QuadHelix bundle, the 36 unique single helix conformations are selected that correspond to the lowest energy QuadHelix conformations. For helices 2, 5, and 7, which exist in two QuadHelix bundles, the top 18 unique single-helix conformations are selected independently from both QuadHelix bundles and then combined. Finally, for helix 3, which exists in all three QuadHelix bundles, the top 12 unique single-helix conformations are selected independently and combined. If a helix conformation is the same in two QuadHelix bundles, then the next configuration in the latter QuadHelix (going in the order TM1-TM2-TM3-TM7, TM2-TM3-TM4-TM5, and TM3-TM5-TM6-TM7) is chosen. At the end, 36 unique configurations for each TM helix are selected. Last, from each individual helical conformation list, the 36 conformations for each helix are used to calculate the energy of 367 ∼ 8 × 1010 full bundles. We then output the 2,000 best energy structures from this procedure.
In the SuperComBiHelix step, these top 2,000 helical bundles (from SuperBiHelix) are built and the side chains are reassigned with SCREAM (21), so they will likely have different conformations than in the BiHelix limit. Then the structure is minimized for 10 steps. This energy ranking for SuperComBiHelix will be different (and more accurate) than that for SuperBiHelix because all seven helices are present instead of just two. This procedure results in the ensemble of low-energy conformations most likely to play a role in binding of ligands and activation of the GPCR.
Extensive testing on these methods, shown below in Validation, led to several improvements in the procedure. During the side-chain prediction steps in SuperBiHelix and SuperComBiHelix, SCREAM must be used with a 0.5-Å resolution library instead of the 1.0-Å resolution library that is the default for SCREAM. Additionally, for the best results we mutate the final two residues of the C and N termini to alanine for each helix during the SuperBiHelix step. Then, before the SuperComBiHelix step, these mutated alanine residues are mutated back to their original residues for the building of the full bundles. This step reduces artificial long-range electrostatic interactions between charged groups that would be located in the polar head group region of the lipid bilayer.
Binding Site Prediction.
For the ligand docking step, we use techniques developed as part of the DarwinDock/GenDock protocols (22), which aims at sampling all possible poses before evaluating energies and then groups the poses into families ordered by the energy of the family head to minimize the number of poses used for energy evaluation. The docking methodology is described in SI Text.
Validation
SuperBiHelix on Crystal Helices in the Correct Template.
The SuperBiHelix and SuperComBiHelix procedures were developed by testing them on helices from one crystal structure in the template of another crystal structure. During these procedures, the extracellular/intracellular loops and the N/C termini were not present, assuming that they do not exert a significant effect on the TM helix bundle conformations. It is also important to determine whether performing SuperBiHelix and SuperComBiHelix on a crystal structure itself returns the original structure. Therefore, SuperBiHelix and SuperComBiHelix were run on the A2A adenosine receptor (23) and the β2-adrenergic crystal structure (24), sampling θ with values of −10°, 0°, and 10°, ϕ with values of −30°, −15°, 0°, 15°, and 30°, and η with values of −30°, −15°, 0°, 15°, and 30°. BiHelix was not rerun beforehand because we had previously analyzed (9) that the η values would be less than 30° from the crystal structure.
We first tested how well the QuadHelix protocol worked. We validated that, in order for the original crystal structure to show up in the best energy SuperBiHelix and SuperComBiHelix structures, the crystal conformation for each helix must be in that helix’s top 36 conformations. The ranking of the crystal conformation for each helix for the A2A adenosine receptor and the β2-adrenergic receptor is shown in the Table S1. The crystal structure conformation for each helix is in the top 36 helical conformations. In fact, for many helices, the crystal structure is the best conformation. Indeed, the worst ranking for a helix is for TM6 in the β2-adrenergic receptor, which ranked number 12. This validates that the QuadHelix protocol works well for the A2A adenosine receptor and the β2-adrenergic receptor crystal structures. It also suggests that it might have been sufficient to have limited the selection to, say, the best 24 rather than the best 36.
We next determined whether the ranking of the top 2,000 SuperBiHelix structures is improved by SuperComBiHelix. The SuperBiHelix and SuperComBiHelix results are shown for the β2-adrenergic receptor crystal structure in Fig. 3. In the SuperBiHelix structures, the crystal structure is rank 78, and after SuperComBiHelix, it is rank 25. Not only does SuperComBiHelix cause significant improvement in the rank of the crystal structure, it also slightly improves the backbone root-mean-squared deviation (rmsd) values of the top 10 structures. Another observation to note is that these backbone rmsd values are very low (<0.6 Å), showing that we are capturing near-native conformations.
Fig. 3.

SuperBiHelix and SuperComBiHelix results for the β2-adrenergic receptor crystal structure. The top 10 structures for both SuperBiHelix and SuperComBiHelix are shown, along the with crystal structure, which is outlined by dashed lines. The crystal shows up as rank 78 for SuperBiHelix and rank 25 for SuperComBiHelix.
The SuperBiHelix and SuperComBiHelix results for the A2A adenosine receptors crystal structure are in Fig. 4. The rank of the crystal structure goes from second in SuperBiHelix to sixth in SuperComBiHelix. Although SuperComBiHelix makes the crystal structure rank slightly worse than in SuperBiHelix, SuperComBiHelix significantly improves the backbone rmsd values of the top 10 structures (≤1.0 Å).
Fig. 4.

SuperBiHelix and SuperComBiHelix results for the A2A adenosine receptor crystal structure. The top 10 structures for both SuperBiHelix and SuperComBiHelix are shown, which include the crystal structure, outlined by dashed lines. The color scale is show in Fig. 3.
The SuperComBiHelix results of both the β2-adrenergic and A2A adenosine crystal structures show that the largest variation is in the sweep angles (φ) of the helices. Neither receptor shows any variation in TM1 and TM2. The sweep angle of TM3 differs from the crystal structure in the A2A adenosine receptor, but not in the β2-adrenergic receptor. The sweep angles of TM4, TM5, TM6, and TM7 vary for both receptors. Finally, TM5 is the only helix whose η value changes from the crystal structure, for both receptors. Thus, it seems that TM5 is most flexible in both receptors. Thus, SuperBiHelix and SuperComBiHelix are successful for both the β2-adrenergic and A2A adenosine crystal structures in returning near-native conformations, when starting from the correct template.
SuperBiHelix on Crystal Helices in an Incorrect Template.
The SuperBiHelix and SuperComBiHelix methods were tested on the β2-adrenergic receptor (24) in the A2A adenosine receptor (23) template, and vice versa. The differences between the x, y, θ, ϕ, and η values for the two templates are given in the Table S2. x and y have only a small amount of variation between templates, supporting the SuperBiHelix procedure, which does not sample x and y. Additionally, ϕ and η vary more among the templates than θ, so more ϕ and η values will need to be sampled than θ values.
To test how well SuperBiHelix predicts structures when helices are in the incorrect template, β2-adrenergic crystal helices were given the x, y, ϕ, θ, and η values of the A2A adenosine template, and SuperBiHelix/SuperComBiHelix steps were run, sampling θ with values of −10°, 0°, and 10°; ϕ with values of −30°, −15°, 0°, 15°, and 30°; and η with values of −30°, −15°, 0°, 15°, and 30°. BiHelix was not run beforehand because η only varies by −18.8° to +4.8° between the two templates. For all of the validation runs, each helix in the original crystal structure was minimized without the other helices present, so that the procedure was not biased toward the crystal structure. The results in Fig. 5 show that the SuperBiHelix and SuperComBiHelix procedures cause the backbone rmsd of predicted conformations (to the β2-adrenergic receptor crystal structure) to go from 2.0 to 1.6 Å, a modest improvement. The 2.0-Å rmsd to the β2-adrenergic crystal structure of the original structure in the incorrect template, represented by all yellow in Fig. 5, is the rmsd achieved by using homology modeling. The lowest possible rmsd, given the angles sampled, is 1.2 Å. The conformation of TM1 is predicted quite poorly, most likely because TM1 has the fewest interactions with other helices: it does not have direct interaction with TM3 like the other TM helices. Additionally, Table S2 shows that, for the x and y values, which are not sampled by SuperBiHelix, TM1 has larger deviations between the β2-adrenergic and A2A adenosine templates than any other helix. Docking of the ligand carazolol (used to crystallize the β2-adrenergic receptor) to structures before and after this conformational sampling procedure shows that this sampling is able to improve the pharmacophore of the protein–ligand interactions (see SI Text for results).
Fig. 5.

SuperComBiHelix results for β2-adrenergic receptor helices in the A2A adenosine template (Upper) and A2A adenosine receptor helices in the β2-adrenergic template (Lower). The rmsd is the backbone rmsd to the target crystal structure. The structure outlined by dashed lines is the structure closest to the crystal structure, given the angles sampled. The structure in all yellow (representing all 0° angles) is the original structure in the incorrect template. The color scale is show in Fig. 3.
For A2A adenosine helices with the x, y, ϕ, θ, and η values of the β2-adrenergic template, SuperBiHelix and SuperComBiHelix causes the backbone rmsd of the predicted conformations (to the A2A adenosine receptor crystal structure) to go from 2.1 to 1.4 Å. This is a good improvement, given that the best rmsd possible, given the angles sampled, is 1.2 Å. The 2.1-Å rmsd to the A2A adenosine crystal structure of the original structure in the incorrect template, represented by all yellow in Fig. 5, is the rmsd achieved by using homology modeling. Docking of the ligand ZM241385 (used to crystallize the A2A adenosine receptor) to structures before and after this conformational sampling procedure shows an improvement in the pharmacophore (see SI Text for results), similar to the case of carazolol docking to predicted β2-adrenergic receptor structure.
We emphasize here that the ligand is not present during the SuperBiHelix and SuperComBiHelix procedures. The crystal structures are typically determined with the ligand bound, so the ligand-free crystal structure need not be the lowest energy structure. The presence of the ligand could change the order of the structures. Additionally, the presence of the loops could change the ordering of the structures. However, even ignoring such factors, the SuperBiHelix procedure provides useful predictions of new GPCR structures.
The Effect of SuperBiHelix on Binding Site Predictions.
Although rmsd is a reasonable metric for testing SuperBiHelix, it does not take ligand binding into account. One of the main purposes of predicting GPCR structures is for drug design, so it is important to measure how well ligand binding can be predicted in structures predicted by SuperBiHelix. Thus, ZM241385 was docked into the A2A adenosine structures predicted from the β2-adrenergic template before and after SuperBiHelix and SuperComBiHelix, and carazolol was docked into the β2-adrenergic structures predicted from the A2A templates before and after SuperBiHelix and SuperComBiHelix. Then these docked results were compared with the ligand-bound crystal structures to see whether SuperBiHelix improved docking. The ligands were also docked into the ligand-free crystal structure for purposes of comparison.
For ZM241385 docked into the ligand-free A2A adenosine crystal structure, the contact rmsd is 2.4 Å. For the A2A adenosine helices in the β2-adrenergic template before SuperBiHelix, the lowest contact rmsd in the final 13 docked structures is 4.6 Å, and after SuperComBiHelix it is 3.4 Å. Thus, SuperBiHelix makes the binding site much more like that of the crystal structure. As seen in Fig. 6, the docked ligand in the best energy SuperComBiHelix structure is very similar to the pose in the crystal structure. They both make strong hydrogen bonds with N253(TM6). The docked ligand in the structure before SuperBiHelix takes a different pose and does not form any hydrogen bonds with N253(TM6).
Fig. 6.
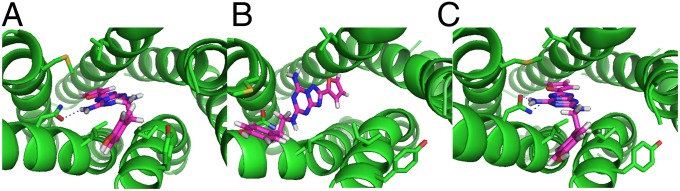
(A) The ZM241385-bound A2A adenosine crystal structure. (B) ZM241385 docked into the A2A adenosine helices in the β2-adrenergic template before SuperBiHelix. (C) ZM241385 docked into the A2A adenosine helices in the β2-adrenergic template after SuperBiHelix.
We discuss the results for the carazolol docking in SI Text, with the docked structures seen in Fig. S1. SuperBiHelix improves the binding-site predictions for both β2-adrenergic helices in the A2A adenosine template and A2A adenosine helices in the β2-adrenergic template, but it has more effect on the A2A adenosine helices in the β2-adrenergic template. This agrees with the rmsd calculations for SuperBiHelix, in which there is a larger effect on the A2A adenosine helices in the β2-adrenergic template than the β2-adrenergic helices in the A2A adenosine template.
Can SuperBiHelix Predict Inactive and Active Conformations of a Receptor?
The GPCR conformation of an activated GPCR is expected to have higher energy than the inactive conformation, making it a challenge to identify these higher energy conformations because, without the agonist and without a nearby G protein, these states might be too high for SuperBiHelix/SuperCombiHelix to identify. We rely on energy ordering the final set of conformations without ligand or G protein and there could be too many nonactive states in between. However, for a case in which a receptor mutant is known to be easily activated, we might expect that our predicted ensemble of low-energy configurations starting with that template might include the active conformation of that mutant receptor (discussed below). We also describe the rhodopsin case in SI Text, in which using helices from an inactive state (rhodopsin) in the active-like ligand-free opsin template (seen in Fig. S2) allowed us to test this approach.
To determine how well SuperBiHelix can recognize active and inactive forms of the same receptor, we looked at the mutant of the cannabinoid receptor CB1 that is constitutively active. Kendall and coworkers identified a single-point mutant T3.46A that was completely inactive and one (T3.46I) that was more active than the WT receptor based on ligand-binding profiles (6) and recently confirmed by the GTPγS assays (7). To determine the origin of these major changes in activity, the SuperBiHelix/SuperCombiHelix methods were applied to the three receptor forms (WT and two single-point mutants T3.46A and T3.46I) to predict the ensemble of low-energy seven-helix bundle conformations. We found substantially different TM helix packings among the WT and mutant receptors that lead to markedly different coupling of the charged residues near each receptor’s cytoplasmic region (Fig. 7 A–C). Both WT and T3.46A exhibited TM3+TM6 coupling, known to be critical to keep GPCRs inactive. The fully inactive T3.46A mutant constrained TM6 further through a TM2+TM6 coupling (R2.37+D6.30) explaining its full inactivity. In contrast, the highly constitutively active T3.46I mutant showed no coupling of TM6 to TM2 or TM3, but rather had a TM5+TM6 coupling, similar to that observed in active GPCR crystal structures. This shows that SuperBiHelix is able to sample and capture conformations with structural differences that explain the binding and activation assays leading to concepts consistent with those extracted from GPCR crystal structures as well.
Fig. 7.

(A–C) Predicted structures of the T3.46A, WT, and T3.46I CB1 GPCRs, showing the salt bridges and hydrogen bonds formed on the cytoplasmic side. (D) Comparison of basal GTPγS binding to HEK293 cell membranes expressing the CB1 receptors including the double mutants (8).
To further validate these findings, we designed double mutants to reverse the activity of the single mutants. Thus, we predicted that the two double mutants (T3.46A/R2.37A and T3.46A/R2.37Q) would regain WT constitutive activity. This prediction was subsequently confirmed experimentally (see SI Text for details) by the GTPγS assays (Fig. 7D), validating the structures predicted with the SuperBiHelix methodology (7). The structures lead to an activation mechanism for CB1 that explains all experiments and that may play a role in other GPCRs. Additional mutants have also been designed and then tested in GTPγS assays (8), lending strong support to this activation mechanism.
The CB1 example shows that the SuperBiHelix methodology can be used to predict and design (based on testable hypothesis) active conformations. SuperBiHelix was able to guide experiments by predicting gain of function experiments (like making the inactive T3.46A mutant constitutively active by adding a well-chosen mutation). Thus, coupled to experiments to provide functional validations, SuperBiHelix methodology can be used to provide very specific tests of specific hypotheses probing the structural basis of GPCR activation.
Discussion
In addition to CB1 (7, 8), SuperBiHelix has been applied successfully to the adenosine A3 receptor (16) and other adenosine receptors (22), serotonin 5-HT2B and 5-HT2C receptor (25), the histamine H3 receptor (26), the CCR5 receptor (27, 28), TAS2R38 bitter taste receptor (29), and the V2 vasopressin receptor (30). The predicted GPCR structures in these studies were validated by predicting the binding sites and energies for known series of ligands and comparing with experimental mutagenesis, binding, and/or functional data. These predicted ligand–protein structures provided molecular-level interpretations for structural or functional observations in the CCR5 and V2 receptors. Indeed, for A3 adenosine receptor (16), it was found that all four selective agonists preferred the 15th WT conformation, whereas the four selective antagonists all preferred the second or the third conformations. Moreover, all of the agonists caused the “trigger” Trp in TM6 (6.48) to switch from vertical before binding to horizontal after binding.
We showed in Validation that SuperBiHelix does better than homology modeling. β2-Adrenergic crystal helices in the A2A adenosine template have a 2.0-Å homology model rmsd, which SuperBiHelix improves to 1.6-Å rmsd. Similarly, A2A adenosine crystal helices in the β2-adrenergic template have a 2.1-Å homology model rmsd, which SuperBiHelix improves to 1.4 Å. In community-wide assessments of structure prediction methods (31, 32) aimed at GPCRs, the SuperBiHelix method has performed well at predicting the receptor structures. Prediction of ligand binding sites (without using prior mutagenesis data on the ligand or similar ligands) has not performed as well because docking of ligands to predicted protein structures depends highly on the accuracy of the protein structure. Homology-based methods have not led to the prediction of multiple receptor conformations like that possible with the SuperBiHelix method. Only a handful of methods (18, 32) have been able to predict multiple conformations based on some level of rigorous conformational sampling.
SuperBiHelix and SuperComBiHelix allow for the efficient sampling of GPCR conformational space. This makes it possible to predict structures of receptors that are dissimilar to any experimental crystal structure. It also predicts an ensemble of low-lying structures, mirroring the flexibility of GPCR structures. When helices from one crystal structure are placed into the template of another structure, SuperBiHelix and SuperComBiHelix successfully move the experimental helices closer to their original template. The procedure also improves binding-site predictions and makes ligand-binding calculations more accurate. The success of SuperBiHelix and SuperComBiHelix on experimental crystal structures can now lead to better predictions of GPCR structures and binding sites, and therefore more successful rational drug design. The computational methodology can also be used to probe GPCR activation as was highlighted by designing a constitutively active mutant based on an inactive CB1 receptor mutant. It shows the strength of the methodology to complement and guide experiments in exploring many structural hypotheses of GPCR activation and function.
Supplementary Material
Acknowledgments
This work was financially supported by funds donated to the Materials and Process Simulation Center. J.K.B. acknowledges the Department of Energy Computational Science Graduate Fellowship. The computers used were funded by grants from Defense University Research Instrumentation Program and by National Science Foundation (equipment part of Materials Research Science and Engineering Center).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1321233111/-/DCSupplemental.
References
- 1.Lagerström MC, Schiöth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov. 2008;7(4):339–357. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- 2.Abrol R, Goddard WA., 3rd . G protein-coupled receptors: Conformational “gatekeepers” of transmembrane signal transduction and diversification. In: Adams JD, Parker KK, editors. Extracellular and Intracellular Signalling. Cambridge, UK: RSC; 2011. pp. 188–229. [Google Scholar]
- 3.Kenakin T, Miller LJ. Seven transmembrane receptors as shapeshifting proteins: The impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev. 2010;62(2):265–304. doi: 10.1124/pr.108.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lundstrom K. Latest development in drug discovery on G protein-coupled receptors. Curr Protein Pept Sci. 2006;7(5):465–470. doi: 10.2174/138920306778559403. [DOI] [PubMed] [Google Scholar]
- 5.Klabunde T, Hessler G. Drug design strategies for targeting G-protein-coupled receptors. ChemBioChem. 2002;3(10):928–944. doi: 10.1002/1439-7633(20021004)3:10<928::AID-CBIC928>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 6.D’Antona AM, Ahn KH, Kendall DA. Mutations of CB1 T210 produce active and inactive receptor forms: Correlations with ligand affinity, receptor stability, and cellular localization. Biochemistry. 2006;45(17):5606–5617. doi: 10.1021/bi060067k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scott CE, Abrol R, Ahn KH, Kendall DA, Goddard WA., 3rd Molecular basis for dramatic changes in cannabinoid CB1 G protein-coupled receptor activation upon single and double point mutations. Protein Sci. 2013;22(1):101–113. doi: 10.1002/pro.2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahn KH, Scott CE, Abrol R, Goddard WA, Kendall DA. Computationally-predicted CB1 cannabinoid receptor mutants show distinct patterns of salt-bridges that correlate with their level of constitutive activity reflected in G protein coupling levels, thermal stability, and ligand binding. Proteins. 2013;81(8):1304–1317. doi: 10.1002/prot.24264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abrol R, Kim SK, Bray JK, Griffith AR, Goddard WA., 3rd Characterizing and predicting the functional and conformational diversity of seven-transmembrane proteins. Methods. 2011;55(4):405–414. doi: 10.1016/j.ymeth.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walters RW, et al. beta-Arrestin1 mediates nicotinic acid-induced flushing, but not its antilipolytic effect, in mice. J Clin Invest. 2009;119(5):1312–1321. doi: 10.1172/JCI36806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blois TM, Bowie JU. G-protein-coupled receptor structures were not built in a day. Protein Sci. 2009;18(7):1335–1342. doi: 10.1002/pro.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rasmussen SG, et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477(7366):549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choe HW, et al. Crystal structure of metarhodopsin II. Nature. 2011;471(7340):651–655. doi: 10.1038/nature09789. [DOI] [PubMed] [Google Scholar]
- 14.Xu F, et al. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332(6027):322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White JF, et al. Structure of the agonist-bound neurotensin receptor. Nature. 2012;490(7421):508–513. doi: 10.1038/nature11558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim S-K, Riley L, Abrol R, Jacobson KA, Goddard WA., 3rd Predicted structures of agonist and antagonist bound complexes of adenosine A3 receptor. Proteins. 2011;79(6):1878–1897. doi: 10.1002/prot.23012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abrol R, Bray JK, Goddard WA., 3rd Bihelix: Towards de novo structure prediction of an ensemble of G-protein coupled receptor conformations. Proteins. 2011;80(2):505–518. doi: 10.1002/prot.23216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhattacharya S, et al. Critical analysis of the successes and failures of homology models of G protein-coupled receptors. Proteins. 2013;81(5):729–739. doi: 10.1002/prot.24195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abrol R, Griffith AR, Bray JK, Goddard WA., 3rd Structure prediction of G protein-coupled receptors and their ensemble of functionally important conformations. Methods Mol Biol. 2012;914:237–254. doi: 10.1007/978-1-62703-023-6_14. [DOI] [PubMed] [Google Scholar]
- 20.Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. OPM: Orientations of proteins in membranes database. Bioinformatics. 2006;22(5):623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- 21.Kam VWT, Goddard WA. Flat-bottom strategy for improved accuracy in protein side-chain placements. J Chem Theory Comput. 2008;4(12):2160–2169. doi: 10.1021/ct800196k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goddard WA, 3rd, et al. Predicted 3D structures for adenosine receptors bound to ligands: Comparison to the crystal structure. J Struct Biol. 2010;170(1):10–20. doi: 10.1016/j.jsb.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 23.Jaakola VP, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322(5905):1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cherezov V, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318(5854):1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim SK, Li Y, Abrol R, Heo J, Goddard WA., 3rd Predicted structures and dynamics for agonists and antagonists bound to serotonin 5-HT2B and 5-HT2C receptors. J Chem Inf Model. 2011;51(2):420–433. doi: 10.1021/ci100375b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim SK, Fristrup P, Abrol R, Goddard WA., 3rd Structure-based prediction of subtype selectivity of histamine H3 receptor selective antagonists in clinical trials. J Chem Inf Model. 2011;51(12):3262–3274. doi: 10.1021/ci200435b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berro R, et al. Use of G-protein-coupled and -uncoupled CCR5 receptors by CCR5 inhibitor-resistant and -sensitive human immunodeficiency virus type 1 variants. J Virol. 2013;87(12):6569–6581. doi: 10.1128/JVI.00099-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grunbeck A, et al. Genetically encoded photo-cross-linkers map the binding site of an allosteric drug on a G protein-coupled receptor. ACS Chem Biol. 2012;7(6):967–972. doi: 10.1021/cb300059z. [DOI] [PubMed] [Google Scholar]
- 29.Tan J, Abrol R, Trzaskowski B, Goddard WA., 3rd 3D structure prediction of TAS2R38 bitter receptors bound to agonists phenylthiocarbamide (PTC) and 6-n-propylthiouracil (PROP) J Chem Inf Model. 2012;52(7):1875–1885. doi: 10.1021/ci300133a. [DOI] [PubMed] [Google Scholar]
- 30.Carpentier E, et al. Identification and characterization of an activating F229V substitution in the V2 vasopressin receptor in an infant with NSIAD. J Am Soc Nephrol. 2012;23(10):1635–1640. doi: 10.1681/ASN.2012010077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Michino M, et al. Community-wide assessment of GPCR structure modelling and ligand docking: GPCR Dock 2008. Nat Rev Drug Discov. 2009;8(6):455–463. doi: 10.1038/nrd2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kufareva I, Rueda M, Katritch V, Stevens RC, Abagyan R. Status of GPCR modeling and docking as reflected by community-wide GPCR Dock 2010 assessment. Structure. 2011;19(8):1108–1126. doi: 10.1016/j.str.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.