

Significance
Many persistent and chronic bacterial infections are associated with the formation of large cell aggregates and biofilms that are difficult to treat. This includes respiratory and urinary tract infections, infections on medical devices, and infections of the ear, gums, and heart. One mechanism used by bacteria to aggregate and form biofilms involves the expression of self-associating surface-located autotransporter proteins such as Antigen 43 (Ag43). Here we present the crystal structure of the functional passenger domain of Ag43 and demonstrate that its unique L-shaped structure drives the formation of cell aggregates via a molecular Velcro-like handshake mechanism. This work provides insight into the structure–function mechanisms that facilitate bacterial interactions during infection.
Keywords: Ag43, virulence factor, structural biology, urinary tract infection
Abstract
Aggregation and biofilm formation are critical mechanisms for bacterial resistance to host immune factors and antibiotics. Autotransporter (AT) proteins, which represent the largest group of outer-membrane and secreted proteins in Gram-negative bacteria, contribute significantly to these phenotypes. Despite their abundance and role in bacterial pathogenesis, most AT proteins have not been structurally characterized, and there is a paucity of detailed information with regard to their mode of action. Here we report the structure–function relationships of Antigen 43 (Ag43a), a prototypic self-associating AT protein from uropathogenic Escherichia coli. The functional domain of Ag43a displays a twisted L-shaped β-helical structure firmly stabilized by a 3D hydrogen-bonded scaffold. Notably, the distinctive Ag43a L shape facilitates self-association and cell aggregation. Combining all our data, we define a molecular “Velcro-like” mechanism of AT-mediated bacterial clumping, which can be tailored to fit different bacterial lifestyles such as the formation of biofilms.
Autotransporter (AT) proteins represent a large family of diverse secreted and outer-membrane proteins present in numerous Gram-negative bacteria, including all classes of Proteobacteria as well as numerous species of Chlamydiales and Fusobacteria (1–4). They are a core component of a molecular mechanism for the delivery of cargo protein domains across the outer membrane, and are often associated with important biological functions such as adhesion, aggregation, toxicity, and biofilm formation. All AT proteins are defined by a specific domain architecture that comprises an N-terminal signal sequence that directs secretion of the protein across the inner membrane via the general secretory pathway, a passenger (α) domain that is either anchored to the cell surface or released into the external milieu and determines the functional characteristics of the protein, and a translocation (β) domain that inserts into the outer membrane (5). Originally, AT proteins were thought to possess structural properties that facilitate their independent transport across the bacterial membrane and final routing to the cell surface (3). However, it is now known that accessory factors such as components of the Bam complex, periplasmic chaperones (SurA, Skp, DegP, and FkpA), and the translocation and assembly module contribute to the secretion of some AT proteins (6, 7).
The AIDA-I–type AT proteins represent the largest and most diverse group of the AT superfamily (8). In Escherichia coli, 16 AIDA-I–type subgroups have been identified, which differ in the length and sequence of their passenger domain and their functional properties (8). Although these AT proteins show substantial sequence and functional variability, they are predicted to adopt common domain architectures; the transporter β-domain folds into the canonical β-barrel, and the functional passenger domain generally incorporates a right-handed β-helix (9).
Antigen 43 (Ag43) is one of the best-studied AIDA-I–type AT proteins. This phase-variable AT protein is produced as a 1,039-amino acid preprotein incorporating an N-terminal signal peptide that directs translocation across the cytoplasmic membrane into the periplasm and a classical passenger (α43)–translocation (β43) domain structure (Fig. 1A). Processing of Ag43 occurs between the functional α43 and the translocating β43 domains; however, the two subunits remain in contact via noncovalent interactions (3), with the α43 domain protruding ∼10 nm from the cell surface, where it is free to interact with Ag43 passenger domains presented on the surface of neighboring cells.
Fig. 1.

Domain organization and structure of the Ag43a passenger domain (α43a). (A) Schematic showing the arrangement of the protein domains: signal peptide (SP), passenger domain (α-domain), autochaperone domain (AC), and translocator domain (β-domain). (B) Ribbon representation of the secondary-structure elements of the α43a β-helix with β-strands and loops colored in blue and green, respectively. Loop 1 and loop 2 protruding from the β-spine are colored in red, and the four β-hairpins are shown in black. (C) Top view of the β-helix showing the internal side-chain stacking. Residues are shown in stick representation. (D) Stereoview of the 2Fo − Fc electron density map contoured at 1σ of the cross-section of the α43a β-helix (residues 90L–108H).
Ag43 is found in most E. coli pathotypes, including uropathogenic E. coli (UPEC) (10). Many UPEC strains also possess multiple copies of the Ag43-encoding agn43 (or flu) gene (10, 11). Well-characterized functions of Ag43 include cell aggregation and biofilm formation (12–14). Sequence variation within the α43 domain of Ag43 paralogues is associated with different functional properties. For example, the reference UPEC strain CFT073 contains two Ag43 variants (Ag43a and Ag43b) that share 85% sequence identity in the functional α-domain; only Ag43a mediates strong aggregation and biofilm formation and long-term colonization of the mouse urinary tract (10). Ag43 from UPEC strain UTI89 is also associated with the formation of intracellular bacterial communities (IBCs) that resemble biofilms and contribute to chronic urinary tract infection (UTI) (15).
The huge diversity of AT proteins in Gram-negative bacteria supports their role in multiple facets of pathogenesis. Although important virulence functions have been ascribed to several different AT proteins, their mechanism of action remains to be elucidated. This is particularly evident in the case of AIDA-I AT proteins, for which our current knowledge of structure–function relationships is either nonexistent or very limited and insufficient to enable a defined mechanistic model for their role in pathogenesis (3, 16). Here we investigate the structure–function relationships of the prototypic AIDA-I–type AT Ag43a from UPEC and show that its functional α43a domain contains a β-helical structure. Remarkably, unlike other structurally characterized β-helices, α43a has an L shape, and our work shows a critical interplay between the bent β-helical scaffold and the virulence activity of this protein. Our data also provide unique insight into the mechanism of self-association of Ag43, which may represent a universal mechanism of AT-mediated bacterial aggregation.
Results
Architecture of the Ag43a Passenger Domain.
We determined the crystal structure of the functional passenger domain of Ag43a (α43a) from UPEC CFT073. The structure was determined by single anomalous diffraction (SAD) and refined to an Rfree of 16.76% (R factor 21.56%) at 2.5-Å resolution (SI Appendix, Table S1). The α43 domain of Ag43a folds into a three-stranded β-helix structure comprising 23 complete turns (Fig. 1 B and C). Each rung of the β-helix consists of three β-strands linked by loop regions; on average, there are 20 residues per turn (Fig. 1D). Unlike other β-helical AT proteins, which possess a rather straight overall architecture (9), the crystal structure of α43a revealed an L-shaped protein consisting of a 13-rung parallel β-helix domain (the stem of the letter L), followed by three rungs flanked by four β-hairpin motifs that bend the protein by about 110° and a C-terminal seven-rung parallel β-helix domain (the bottom of the letter L) (Fig. 1B). Consequently, superposition of the L-shaped β-spine in α43a with previously characterized passenger domains is rather poor (rmsd values range from 3.5 to 9.3 Å) (SI Appendix, Table S2). Small-angle X-ray scattering data were collected on α43a to establish whether the bent β-helix observed in the crystal is representative of the structure of the protein in solution. Structural parameters such as the radius of gyration (Rg) and the maximum linear dimension (Dmax) of the molecule are in excellent agreement with the crystal structure (SI Appendix, Table S3). Further, the predicted scattering profile of α43 agrees with the experimental data, and the low-resolution structure obtained by ab initio modeling also agrees with the crystal structure (SI Appendix, Fig. S1), providing convincing evidence that the protein has an l shape in solution.
The top section of the β-helix in α43a (the stem of the letter L) is twisted as though the two ends of this section were rotated against each other, resulting in a distorted prism-like morphology where the triangular cross-sections are rotated by about 45° (Fig. 1C), generating long grooves that run diagonally along the surface of the protein (SI Appendix, Fig. S2 A and B). The interior of the α43a β-helix is populated predominantly by aliphatic residues forming long stacks (Fig. 1C); however, the core of the N-terminal part of the β-helix (the stem of the letter L) also revealed a stacking of polar residues in consecutive turns, with the side chains oriented to create an extensive network of interstrand hydrogen bonds (Fig. 1C). Additionally, although the interior of the helix is almost completely packed, water molecules traverse through the stem of the L-shaped α43a in single file (SI Appendix, Fig. S2C).
Self-Association of Ag43a.
Ag43 is a self-recognizing AT protein that promotes bacterial cell aggregation through self-interaction (17). Although at low protein concentration we did not observe any oligomerization of recombinant α43a (SI Appendix, Fig. S1D), size-exclusion chromatography, blue native PAGE, and sedimentation velocity analytical ultracentrifugation analyses indicated that α43a can oligomerize in vitro upon concentration (SI Appendix, Fig. S3).
To gain insight into the molecular mechanism of self-association used by Ag43a, we analyzed the oligomerization of α43a molecules in the crystal lattice. The α43a oligomers were obtained by generating the crystallographically related subunits, which revealed a tightly packed α43a dimer where the two twisted β-helical molecules coil around each other in a trans configuration (each monomer relates to the other by a twofold screw axis perpendicular to the axis of the β-helix) (Fig. 2A). This α43a crystallographic dimer resembles the trans oligomerization mechanism described for the Haemophilus influenzae Hap AT adhesin (18). However, whereas Hap–Hap assembly is only stabilized by weak van der Waals interactions, investigation of the α43a dimer interfaces (interfaces 1 and 1′) using the web-based software PISA (19) identified hydrogen bonds and salt bridges stabilizing α43a–α43a assembly. Each α43a molecule contains ladders of polar and charged residues in the N-terminal part of the edge joining faces 2 (F2) and 3 (F3) and the middle section of the F3 face (Fig. 2B), forming 18 hydrogen bonds and 2 electrostatic interactions (interfaces 1 and 1′ contain 9 hydrogen bonds and 1 salt bridge each).
Fig. 2.

α43a–α43a interactions. (A) Self-association of α43a molecules in trans configuration showing interfaces 1 and 1′. A close-up view of the packing interface is shown; each interface contains nine hydrogen bonds [N29–T256 (two hydrogen bonds), N60–T256, N60–T237, D79–T237, N96–R200, T97–R200, T98–R200, G115–R200] and a salt bridge between the R59 and E216 side chains. (B) Close-up view of the packing interface of modeled α43b molecules indicating the predicted hydrogen-bond interactions (D29–T255, R59–N215, N60–T236, N60–T255, D79–T236, S78–N215, T97–R199, G114–R199). The α43b interface residues that are not conserved in α43a are colored in magenta.
The crystal packing also revealed more complex α43a oligomers, such as assemblies of two pairs of dimers interacting via the N-terminal section of the F1–F2 edge (rungs 3–7). This secondary interaction (interface 2) buries the side chains of small hydrophobic residues populating the loops linking F1 and F2, forming a tetramer stabilized by van der Waals forces (SI Appendix, Fig. S4 A and B). Additionally, each α43a interacts with a third α43a subunit via hydrogen bonds and hydrophobic interactions between the N-terminal section of the F3–F1 edge (rungs 3–8, molecule 1) and the F2 face adjacent to the structure-bending β-hairpin (interface 3). These interactions include nine hydrogen-bond interactions (SI Appendix, Fig. S4 A and B).
Structural Comparison of α43a and α43b.
UPEC strain CFT073 encodes two nonidentical copies of Ag43, Ag43a (c3655) and Ag43b (c1273) (10). These proteins share 90% sequence identity overall, with their predicted α-domains sharing 85% sequence identity (SI Appendix, Fig. S5). Despite their high sequence similarity, these proteins mediate different degrees of cell aggregation; whereas Ag43a promotes strong aggregation, the aggregation mediated by Ag43b is less pronounced (10). Given the similarity between these two proteins, they are likely to present a very similar overall structure. To shed light on the observed functional differences between the two Ag43 proteins from CFT073, we modeled the passenger domain of Ag43b by substituting the appropriate residues in the α43a scaffold and selecting the most stable conformations with no steric clashes. Analysis of the α43b–α43b symmetry at interface 1 using PISA showed that Ag43b–Ag43b self-association is possible despite a reduced number of hydrogen bonds to stabilize this dimer, primarily due to modification of two residues involved in the α43a dimer (α43a N96 and T98 are G and V, respectively, in α43b). Additionally, substitution of α43a E216 into α43b N results in an α43b dimer interface lacking the two electrostatic interactions present in α43a–α43a (R59–E216) (Fig. 2B). The Ag43b model also indicated that variation in its primary sequence (compared with Ag43a) would impede its oligomerization through interfaces 2 and 3 (SI Appendix, Fig. S4C). Conversely, our model showed that Ag43a and Ag43b could interact through interfaces 1 and 1′ with 14 hydrogen bonds and 1 electrostatic interaction (Fig. 3A).
Fig. 3.

Heterologous association of Ag43a–Ag43b adhesins. (A) Modeled heterologous head-to-tail association of α43a and α43b, showing close-up views of interfaces 1 and 1′ and indicating the predicted interactions. Interface 1: eight hydrogen bonds [N29–T255, R59–N215, N60–T236, N60–T255, D79–T236, N96–R199, G115–R199 (two hydrogen bonds)]; interface 1′: six hydrogen bonds (D29–T256, D60–T237, N60–T256, D79–T237, G96–R200, G114–R200); and a salt bridge between the R59 and E216 side chains. (B–E) Ag43a–Ag43b interaction drives E. coli cell aggregation. Fluorescence micrographs of E. coli expressing (B) Ag43a (green) or Ag43b (red), (C) pBAD/Myc-His A (green) or pBAD/Myc-His A (red), (D) Ag43a (green) or pBAD/Myc-His A (red), and (E) pBAD/Myc-His A (green) or Ag43b (red). Panels are representative images from two independent experiments.
Ag43a–Ag43b Interaction Mediates Cell Aggregation.
Our structural model shows that the head-to-tail association through interface 1 can accommodate heterologous association between Ag43a and Ag43b. To investigate this interaction in vivo, we used two previously described isogenic E. coli strains that are chromosomally tagged with genes encoding different fluorescent proteins (20), transformed with plasmids containing the agn43a or agn43b gene under the control of the arabinose-inducible araBAD promoter (10). E. coli Ag43a+ cells (colored green) were observed to interact with Ag43b+ cells (colored red) in cell aggregates forming 1 h after mixing the two strains (Fig. 3B). No cell aggregates were observed for differentially tagged strains each containing the empty vector pBAD/Myc-His A (Fig. 3C), confirming that cell-aggregate formation was mediated by the presence of Ag43a and Ag43b on the bacterial cell surface. To eliminate the possibility that the presence of Ag43b+ cells in the mixed cell aggregates was nonspecific (i.e., due to “trapping” of Ag43b+ cells into aggregates formed by Ag43a+ cells via Ag43a–Ag43a interactions), Ag43a+ (green) and pBAD/Myc-His A (red) cells or Ag43b+ (red) and pBAD/Myc-His A (green) cells were mixed equally and examined by confocal microscopy. In both cases, the resulting cell aggregates only contained cells of the tagged strain expressing Ag43, whereas cells containing the empty vector remained primarily in suspension (Fig. 3 D and E). Taken together, our results provide further insight into the self-recognizing properties of Ag43 by demonstrating that the passenger domains of two nonidentical homologs from the same UPEC strain can interact with each other to drive E. coli aggregation.
Ag43-Mediated Aggregation Occurs by α43a trans Association.
Our findings from the analysis of α43a dimers in the crystal lattice suggested that interfaces 1 and 1′ are important for passenger-domain association. To test this, we constructed a mutant with seven amino acid substitutions located in the α43a interface 1 loops (7 loop: N29G, R59G, N60G, S78G, D79G, N96G, T98G) (SI Appendix, Fig. S5), designed to eliminate the ladder of nine hydrogen bonds identified in the packing interface (Fig. 2). Plasmid constructs harboring the wild-type and 7 loop mutant agn43 gene were transformed into the previously characterized E. coli agn43 null strain MS427 (21), which is unable to mediate cell aggregation (22). These strains were then examined in cell-aggregation assays to determine whether the mutations introduced into Ag43a altered its function. Disrupting interface 1 interactions by removing all existing H bonds completely abolished Ag43a-mediated cell aggregation, and the aggregation profile of the E. coli strain expressing the Ag43a 7 loop mutant was indistinguishable from that of a strain completely lacking Ag43a (Fig. 4A). This dramatic loss in cell aggregation was not due to lack of expression of the mutant Ag43a protein on the cell surface, as demonstrated by Western blot analysis of heat-released proteins (Fig. 4A, Inset) and immunofluorescence labeling using polyclonal Ag43a antisera (Fig. 4C). We also generated two independent mutants with substitution of Ag43a E216 (E216A and E216N), designed to eliminate the two electrostatic interactions present in the α43a–α43a dimer (R59–E216) (SI Appendix, Fig. S5). Neither of the two substitution mutants was affected in mediating cell–cell aggregation compared with WT Ag43a (SI Appendix, Fig. S6A).
Fig. 4.

Functional characterization of Ag43a mutants. E. coli aggregation profiles of strains expressing (A) ag43a 7 loop mutant (N29G, R59G, N60G, S78G, D79G, N96G, T98G) or (B) ag43a hairpin deletion mutants ΔH1, ΔH2, and ΔH1,H2. Mutants were compared with isogenic control strains containing ag43a WT or empty vector. Ag43a protein production at the bacterial cell surface was examined in all Ag43a-overexpressing cultures using an anti-Ag43a polyclonal serum by (A and B, Insets) heat release of α43a from the cell surface and Western blot analysis or (C) immunofluorescence labeling and confocal laser microscopy. α43a was detected in strains containing ag43a-encoding plasmids (WT and mutants) but not the empty vector control (−).
The contribution of interfaces 2 and 3 in Ag43a-mediated cell–cell aggregation was evaluated by substituting G110 and N66, respectively, with a tryptophan (G110W and N66W) (SI Appendix, Fig. S5). The introduction of a bulky residue in these positions was predicted to compromise the α43a–α43a interaction via interfaces 2 and 3, respectively (similar to the effect observed in the modeled α43b). In cell-aggregation assays, no effect was observed on the ability of the Ag43a G110W and N66W substitution mutants to mediate bacterial cell aggregation compared with WT Ag43a (SI Appendix, Fig. S6A). Taken together, our results show that only interface 1 is necessary for Ag43a self-association. This interface is stabilized by multiple hydrogen bonds and two electrostatic interactions, and our data show that the latter are not essential for association.
Modification of α43a L Shape Abolishes Cell Aggregation.
Two sets of Ag43a deletion mutants were generated to examine the importance of the unique structural features of Ag43a in its function as a self-recognizing AT adhesin. In the first set of mutants, we disrupted the two long loops identified in the β-helix of α43a as protruding from the β-spine (SI Appendix, Fig. S5) by deleting residues in loop 1 (L1, 37LELGPDSDENT47) and loop 2 (L2, 141AEGGPESENVS151) and leaving G36/G48/G49 and G140/S152/G153 to link the contiguous β-strands, respectively. Two Ag43a single-loop deletion mutants (ΔL1 and ΔL2) and a double-loop deletion mutant (ΔL1,L2) were constructed, expressed in E. coli MS427, and tested in cell-aggregation assays. No difference was observed in the ability of the three Ag43a loop-deletion mutants to mediate cell–cell aggregation compared with WT (SI Appendix, Fig. S6 B and C). In the second set of mutants, we introduced changes aimed at removing the L-shaped bending observed in the overall architecture of Ag43a. This was achieved by deleting the residues corresponding to two of the β-hairpins present in α43a (H1, 268AATVTGTNRLGAFSVVA284; H2, 341GAAVSGTRSDGKAFSIG357) (SI Appendix, Fig. S5), which, based on our structural data, would result in a straight β-helix. Indeed, sedimentation velocity analyses of native α43a and the double–β-hairpin deletion mutant (α43a ΔH1,H2) showed that the frictional ratio (i.e., shape) of the latter is larger than the native protein (f/f03 values are 2.4 and 1.5, respectively), indicating that α43aΔH1,H2 is significantly more extended (SI Appendix, Fig. S7). Disruption of either or both of the two β-hairpins completely abolished Ag43a-mediated cell aggregation (Fig. 4B), whereas mutant protein production and transport to the cell surface was confirmed by Western blot analysis (Fig. 4B, Inset) and immunofluorescence labeling (Fig. 4C). In conclusion, our data demonstrate that the unique L shape of Ag43a is required for Ag43a–Ag43a association and bacterial clumping.
Discussion
AT proteins comprise a superfamily of extremely common but diverse secreted and outer-membrane proteins involved in the pathogenesis of many Gram-negative bacteria (1, 2). These extracellular proteins are highly immunogenic, and AT proteins such as pertactin (from Bordetella pertussis) and NadA (from Neisseria meningitidis serotype B) are integral components of human vaccines (23, 24). Despite their abundance and the increasing amount of data linking AT proteins to bacterial virulence, very little structural information is available at a molecular level for these proteins. In fact, AT proteins are significantly underrepresented in the Protein Data Bank (PDB), with only 1 full-length AT structure, 7 AT passenger domains, 5 AT β-domains, and 12 small domains of trimeric autotransporters deposited among the ∼87,500 structures currently available in the PDB (9, 18, 25–36). None of these entries belong to the large family of AIDA-I–type AT proteins from Gamma-Proteobacteria (3, 16).
In this study, we investigated the structure–function relationships of Ag43, a prototypical AIDA-I AT. We selected UPEC CFT073 as our study model, which is a reference pyelonephritis isolate with two agn43 copies encoded in its genome: the aggregation- and biofilm-dominant Ag43a and the nonidentical Ag43b. We structurally characterized the passenger domain of Ag43a (α43a), an AT protein that contributes to UPEC disease pathogenesis by promoting strong aggregation, biofilm growth, and persistence in the urinary tract (10).
UPEC is responsible for 75–95% of all uncomplicated UTI cases (37). These infections cause significant morbidity and are of increasing concern due to the frequent occurrence of multidrug-resistant strains (38). In mouse infection models, the expression of Ag43 by UPEC contributes to bladder colonization and IBC formation (10, 15). Our structural data suggest that the scaffold in α43a appears to be evolutionarily optimized for its aggregation function and provide unique insight into its role in biofilm formation. α43a folds into a right-handed three-stranded β-helix, but unlike previously structurally characterized AT proteins, this adhesin is an L-shaped protein, where the long arm of the L is twisted over its axis, resulting in a distorted solenoid-like morphology (Fig. 1B). The distinctive irregular twisted structure in α43a generates long grooves running diagonally along its surface, which may be well-suited to resist shear forces that could break apart cell aggregates, such as those encountered by UPEC in the urinary tract. α43a contains intricate networks of hydrogen bonds that stabilize the structure along the axis of the β-stalk. Consecutive turns of the β-helix are tightly linked by hydrogen bonds between parallel strands as well as stacks of hydrogen-bonded polar residues decorating the outside of the β-helix, which cross-link and fasten different sections of the protein along the vertical length. The core of α43a also has ladders of polar residues and a single-file water chain running down the interior of the solenoid, which are interconnected via hydrogen bonds to form a 3D framework (SI Appendix, Fig. S2C). The hydrogen-bonded chain of water molecules may provide a dual effect by enhancing the strength of the protein and providing some flexibility to its frame. Finally, the L shape of α43a may strengthen its “foundations” by increasing the contact surface and the number of interactions between the α43a and β43a domains, thus enhancing the attachment of the protein to the cell surface.
Our structural and functional data suggest that α43a molecules from adjacent cells interact in a head-to-tail conformation (Figs. 2 and 4) resembling Hap–Hap self-oligomerization (18). This allows us to propose a model for a universal mechanism of AT-driven bacterial aggregation, which resembles a modified molecular Velcro, where interacting bacterial surfaces are covered with hook-like AT structures that in close proximity can self-assemble via a “handshake” mechanism to form fastened aggregated communities (Fig. 5A). Our AT protein architecture indicates that this general AT aggregation mechanism can be tailored to fit the lifestyle demands of the bacterium producing each AT. Although Hap associations are stabilized by weak van der Waals interactions, our results suggest that Ag43 α43–α43 dimer formation is dependent on multiple interactions, with hydrogen bonds, electrostatic interactions, and van der Waals interactions participating in dimer stabilization (Fig. 2A). The stronger α43a–α43a interface might enhance cell aggregation in high shear-force environments. Furthermore, this general mechanism supports the observed ability of self-associating ATs to form heterologous associations with other β-helical AT proteins (39). In this study, we benefited from the naturally occurring Ag43a variant Ag43b, and showed that in silico and in vivo these two proteins can associate in a head-to-tail manner to form heterologous aggregates (Fig. 3).
Fig. 5.
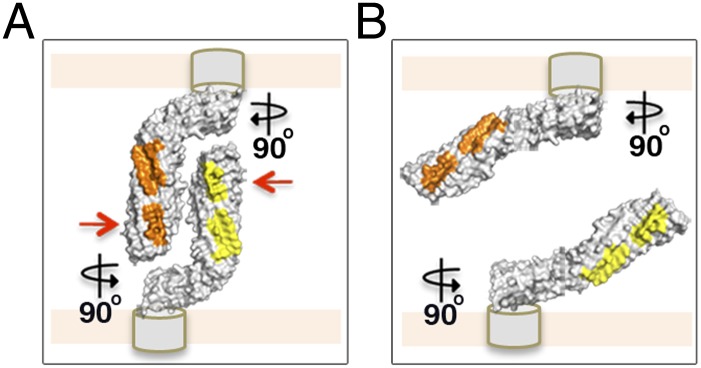
Model of a molecular “Velcro-like” mechanism of bacterial cell aggregation mediated by Ag43a self-assembly in a head-to-tail conformation. (A) Close-up view of the self-associating α43a molecules rotated 90°. The α43a functional domains are predicted to interact with the β-translocating domain (depicted as gray cylinders) using the foot of the L, which will position the long stem of the protein almost perpendicular to the cell surface, making the interacting surfaces (shown in yellow and orange) easily accessible to neighboring molecules. Mutation of the interacting residues in interface 1 (red arrows) completely abolishes Ag43-mediated cell aggregation. (B) Loss of the overall L shape in α43a likely changes the positioning of the long stem of the protein relative to the cell surface, from an easily accessible perpendicular arrangement to a nearly parallel layout, compromising the ability of α43a to interact with Ag43 molecules on adjacent bacterial cells.
Finally, the functional characterization of a series of Ag43a mutants revealed that hydrogen-bond interactions at interface 1 and the unique L-shaped architecture of Ag43a are critical for its self-recognizing properties. The lack of aggregation mediated by the 7 loop mutant is consistent with previous domain-swap experiments that localized its binding region to the N-terminal one-third of the protein (14). In the Ag43 β-hairpin deletion mutants, loss of the L shape may alter the position of the long stem of the protein relative to the cell surface, from an easily accessible perpendicular arrangement to a nearly parallel layout, which would compromise the ability of α43a to interact with Ag43 molecules present on the surface of neighboring bacterial cells (Fig. 5 A and B). This elongated parallel positioning could also decrease the number of Ag43a molecules coating the surface of the cell due to steric hindrance. Indeed, we observed a reduction in the level of passenger domain present on the surface of cells expressing the double-hairpin mutant in our in vivo experiments by Western blot analysis (Fig. 4B, Inset), which is not likely, however, to fully account for the dramatic aggregation deficiency observed for this mutant, as other mutations with similarly reduced passenger-domain production (i.e., E216N and ΔL1,L2) retained a WT-like aggregation profile (SI Appendix, Fig. S6). Last, removal of the L shape could also affect the attachment of the Ag43 α-domain to the cell surface by weakening the interaction with its cognate membrane β-domain.
Taken together, our results provide an attractive molecular Velcro-like mechanism of AT-mediated interactions between different cells, which drives the formation of bacterial aggregates and biofilms that impart enhanced resistance to host immune factors and antibiotics. Notably, most self-associating ATs are predicted to contain β-helical structures in their functional passenger domain. Future work will be required to test how the universal mechanism we propose here can accommodate important differences between AT proteins, including length, amino acid sequence, and domain composition. The requirement of an L-shaped α43a conformation for Ag43-mediated cell aggregation also suggests this may be a common feature shared by other self-associating AIDA-I–type AT proteins. Finally, the molecular Velcro-like mechanism of Ag43 binding suggests that strategies to block α43–α43 interaction, possibly based on the use of synthetic peptides or specific single-domain antibody fragments, could represent a novel approach to prevent aggregation and biofilm formation by UPEC.
Materials and Methods
Production of Native and Selenomethionine-Labeled α43a.
The coding sequence for the α-domain of Ag43a (residues 53–553; locus tag c3655) was amplified from E. coli CFT073 using Agn43 LIC F and Agn43 LIC R primers containing ligation-independent cloning (LIC) overhangs (SI Appendix, Table S4). The amplified gene was inserted into a vector encoding an N-terminal His6 tag followed by thioredoxin (TRX) and the tobacco etch virus (TEV) protease cleavage site. The protein was expressed in E. coli BL21 (DE3) pLysS using autoinduction (24 h at 29 °C). TRX-histidine–tagged α43a was purified by Ni-affinity chromatography and cleaved using His-tagged TEV protease. After removing the His-tagged TEV protease and TRX-His6, α43a was purified to homogeneity by gel-filtration chromatography (ÄKTA, GE Healthcare) using a Superdex S-75 (GE Healthcare) column pre-equilibrated in buffer A (25 mM Hepes–NaOH, pH 7, 150 mM NaCl) followed by ion-exchange chromatography (Mono Q 5/50 GL; GE Healthcare) using buffer B (25 mM Hepes–NaOH, pH 8, 50 mM NaCl) and buffer C (25 mM Hepes–NaOH, pH 8, 500 mM NaCl). The selenomethionine (SeMet)-labeled α43a was expressed in minimal medium containing 50 μg ml−1 SeMet as previously described (40).
Crystallization and Diffraction Data Measurement.
Purified α43a was equilibrated in buffer B and concentrated to 18 mg ml−1. Small stacked plate-like crystals were obtained from solutions consisting of 2.8–2.9 M sodium malonate (pH 4). After optimization, which included using additive screens (Hampton Research), larger stacked plate-shaped crystals (0.5 × 0.25 × 0.05 mm) were obtained in 2.8 M sodium malonate (pH 4) supplemented with 10 mM ATP. Diffraction data for native and SeMet α43a were collected at the 3BM1 protein crystallography beamline at Australian Synchrotron. One-degree oscillation images were collected for a total of 180° in the case of native crystals and 360° for SeMet α43a. Native diffraction was indexed and integrated with XDS (41) and scaled using SCALA (42); SeMet data were integrated and scaled with HKL2000 (43).
Structure Determination and Refinement.
The structure of α43a was solved by SAD phasing of the α43a-SeMet derivative. Phase calculation, density modification, and preliminary model building were carried out using PHENIX AutoSol and AutoBuild (44). PHENIX AutoSol found four Se atoms in the asymmetric unit. The resulting phases were used in AutoBuild for automated building of the protein structure. The model was completed by manual building using Coot (45) and refined against the native 2.5-Å resolution dataset using phenix.refine (44) and TLS (translation/libration/screw) refinement (46). The quality of the α43a model was assessed by MolProbity (http://molprobity.biochem.duke.edu). Molecular figures were generated using PyMOL (47). The PDB ID code is 4KH3.
Construction of Ag43a Mutants.
Cloning of the ag43a gene from UPEC CFT073 into pBAD/Myc-His A (48) has been previously described (10). The resulting vector (pCO4) was used as the parent vector for the construction of all Ag43a mutants used in this study. Deletion mutants were generated by a three-step PCR procedure where two complementary primers flanking the region of interest were first used (SI Appendix, Table S4). PCR products were then double-digested using SacI-KpnI or SacI-StuI and ligated to SacI-KpnI– or SacI-StuI–digested pCO4 plasmid. The Ag43 7 loop mutant was synthesized by Epoch Life Science. All constructs were confirmed by sequencing.
Bacterial Cell-Aggregation Assays.
Plasmids encoding ag43a wild-type and mutant variants were transformed into the previously described E. coli agn43 null strain MS427 (49), and protein expression was induced by aerobic growth in LB broth containing 0.2% l-arabinose for 4.5 h at 37 °C. Bacterial suspensions were adjusted to an optical density at 600 nm (OD600) of 3 and left to stand at room temperature. Cell aggregation was followed by sampling three 100-μL aliquots from the upper part of each culture at 30-min intervals and measuring the OD600. Assays were performed in triplicate. For mixed cell-aggregation assays, two isogenic E. coli strains chromosomally tagged with genes encoding cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP) (20) were transformed with pBAD/Myc-His A and agn43a- or agn43b-encoding plasmids pCO4 and pCO3, respectively (10). Assays were performed as described above, except that at time 0 the strains were mixed at a 1:1 ratio in various combinations. Samples were collected for cell-aggregate examination using a scanning confocal laser microscope (LSM 510 META; Zeiss) equipped for multitrack excitation and detection of CFP and YFP. CFP-tagged cells were colored green and YFP-tagged cells were colored red, and image processing was performed using ZEN software (Zeiss).
Ag43a Immunodetection.
Production and translocation of Ag43 at the bacterial cell surface was evaluated by two immunodetection methods using a rabbit polyclonal serum against α43a from CFT073 Ag43a (c3655). Western blot analysis was used to detect α43a released in the supernatant of OD600-matched E. coli cultures following mild heat treatment (60 °C, 2 min). Culture conditions used were identical to those used for bacterial cell-aggregation assays. Non–heat-treated cells from the same cultures were fixed with 3% (wt/vol) paraformaldehyde on glass slides and incubated with rabbit anti-α43a serum followed by goat anti-rabbit IgG-Alexa 546 (Life Technologies). Slides mounted with ProLong Gold antifade reagent (Life Technologies) were examined under a scanning confocal laser microscope.
Supplementary Material
Acknowledgments
We thank Prof. Jenny Martin for support. We acknowledge the use of the Australian Synchrotron and the University of Queensland Remote Operation Crystallization and X-Ray Diffraction Facility. This work was supported by Australian National Health and Medical Research Council (NHMRC) (APP1042651) and Australian Research Council (ARC) (DP1097032) Grants. B.H., M.T., A.E.W., and M.A.S. are supported by a La Trobe Institute for Molecular Science Fellowship, an ARC Discovery Early Career Researcher Award (DE120101143), an NHMRC Peter Doherty Fellowship (9569864), and an ARC Future Fellowship (FT100100662), respectively.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The crystallography, atomic coordinates, and structure factors reported in this paper have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 4KH3).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1311592111/-/DCSupplemental.
References
- 1.Benz I, Schmidt MA. Structures and functions of autotransporter proteins in microbial pathogens. Int J Med Microbiol. 2011;301(6):461–468. doi: 10.1016/j.ijmm.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 2.Kajava AV, Steven AC. The turn of the screw: Variations of the abundant beta-solenoid motif in passenger domains of type V secretory proteins. J Struct Biol. 2006;155(2):306–315. doi: 10.1016/j.jsb.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 3.Henderson IR, Navarro-Garcia F, Desvaux M, Fernandez RC, Ala’Aldeen D. Type V protein secretion pathway: The autotransporter story. Microbiol Mol Biol Rev. 2004;68(4):692–744. doi: 10.1128/MMBR.68.4.692-744.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Celik N, et al. A bioinformatic strategy for the detection, classification and analysis of bacterial autotransporters. PLoS ONE. 2012;7(8):e43245. doi: 10.1371/journal.pone.0043245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henderson IR, Cappello R, Nataro JP. Autotransporter proteins, evolution and redefining protein secretion. Trends Microbiol. 2000;8(12):529–532. doi: 10.1016/s0966-842x(00)01853-9. [DOI] [PubMed] [Google Scholar]
- 6.Ieva R, Bernstein HD. Interaction of an autotransporter passenger domain with BamA during its translocation across the bacterial outer membrane. Proc Natl Acad Sci USA. 2009;106(45):19120–19125. doi: 10.1073/pnas.0907912106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Selkrig J, et al. Discovery of an archetypal protein transport system in bacterial outer membranes. Nat Struct Mol Biol. 2012;19(5):506–510. doi: 10.1038/nsmb.2261. [DOI] [PubMed] [Google Scholar]
- 8.Wells TJ, Totsika M, Schembri MA. Autotransporters of Escherichia coli: A sequence-based characterization. Microbiology. 2010;156(Pt 8):2459–2469. doi: 10.1099/mic.0.039024-0. [DOI] [PubMed] [Google Scholar]
- 9.Leyton DL, Rossiter AE, Henderson IR. From self sufficiency to dependence: Mechanisms and factors important for autotransporter biogenesis. Nat Rev Microbiol. 2012;10(3):213–225. doi: 10.1038/nrmicro2733. [DOI] [PubMed] [Google Scholar]
- 10.Ulett GC, et al. Functional analysis of antigen 43 in uropathogenic Escherichia coli reveals a role in long-term persistence in the urinary tract. Infect Immun. 2007;75(7):3233–3244. doi: 10.1128/IAI.01952-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roche A, McFadden J, Owen P. Antigen 43, the major phase-variable protein of the Escherichia coli outer membrane, can exist as a family of proteins encoded by multiple alleles. Microbiology. 2001;147(Pt 1):161–169. doi: 10.1099/00221287-147-1-161. [DOI] [PubMed] [Google Scholar]
- 12.Henderson IR, Meehan M, Owen P. Antigen 43, a phase-variable bipartite outer membrane protein, determines colony morphology and autoaggregation in Escherichia coli K-12. FEMS Microbiol Lett. 1997;149(1):115–120. doi: 10.1111/j.1574-6968.1997.tb10317.x. [DOI] [PubMed] [Google Scholar]
- 13.Danese PN, Pratt LA, Dove SL, Kolter R. The outer membrane protein, antigen 43, mediates cell-to-cell interactions within Escherichia coli biofilms. Mol Microbiol. 2000;37(2):424–432. doi: 10.1046/j.1365-2958.2000.02008.x. [DOI] [PubMed] [Google Scholar]
- 14.Klemm P, Hjerrild L, Gjermansen M, Schembri MA. Structure-function analysis of the self-recognizing Antigen 43 autotransporter protein from Escherichia coli. Mol Microbiol. 2004;51(1):283–296. doi: 10.1046/j.1365-2958.2003.03833.x. [DOI] [PubMed] [Google Scholar]
- 15.Anderson GG, et al. Intracellular bacterial biofilm-like pods in urinary tract infections. Science. 2003;301(5629):105–107. doi: 10.1126/science.1084550. [DOI] [PubMed] [Google Scholar]
- 16.Webb CT, Heinz E, Lithgow T. Evolution of the β-barrel assembly machinery. Trends Microbiol. 2012;20(12):612–620. doi: 10.1016/j.tim.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 17.Schembri MA, Hjerrild L, Gjermansen M, Klemm P. Differential expression of the Escherichia coli autoaggregation factor antigen 43. J Bacteriol. 2003;185(7):2236–2242. doi: 10.1128/JB.185.7.2236-2242.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meng G, Spahich N, Kenjale R, Waksman G, St Geme JW., III Crystal structure of the Haemophilus influenzae Hap adhesin reveals an intercellular oligomerization mechanism for bacterial aggregation. EMBO J. 2011;30(18):3864–3874. doi: 10.1038/emboj.2011.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu Q, et al. Statistical analysis of interface similarity in crystals of homologous proteins. J Mol Biol. 2008;381(2):487–507. doi: 10.1016/j.jmb.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reisner A, Molin S, Zechner EL. Recombinogenic engineering of conjugative plasmids with fluorescent marker cassettes. FEMS Microbiol Ecol. 2002;42(2):251–259. doi: 10.1111/j.1574-6941.2002.tb01015.x. [DOI] [PubMed] [Google Scholar]
- 21.Kjaergaard K, Schembri MA, Ramos C, Molin S, Klemm P. Antigen 43 facilitates formation of multispecies biofilms. Environ Microbiol. 2000;2(6):695–702. doi: 10.1046/j.1462-2920.2000.00152.x. [DOI] [PubMed] [Google Scholar]
- 22.Reisner A, Haagensen JA, Schembri MA, Zechner EL, Molin S. Development and maturation of Escherichia coli K-12 biofilms. Mol Microbiol. 2003;48(4):933–946. doi: 10.1046/j.1365-2958.2003.03490.x. [DOI] [PubMed] [Google Scholar]
- 23.Serruto D, Bottomley MJ, Ram S, Giuliani MM, Rappuoli R. The new multicomponent vaccine against meningococcal serogroup B, 4CMenB: Immunological, functional and structural characterization of the antigens. Vaccine. 2012;30(Suppl 2):B87–B97. doi: 10.1016/j.vaccine.2012.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poolman JT, Hallander HO. Acellular pertussis vaccines and the role of pertactin and fimbriae. Expert Rev Vaccines. 2007;6(1):47–56. doi: 10.1586/14760584.6.1.47. [DOI] [PubMed] [Google Scholar]
- 25.Emsley P, Charles IG, Fairweather NF, Isaacs NW. Structure of Bordetella pertussis virulence factor P.69 pertactin. Nature. 1996;381(6577):90–92. doi: 10.1038/381090a0. [DOI] [PubMed] [Google Scholar]
- 26.van den Berg B. Crystal structure of a full-length autotransporter. J Mol Biol. 2010;396(3):627–633. doi: 10.1016/j.jmb.2009.12.061. [DOI] [PubMed] [Google Scholar]
- 27.Gangwer KA, et al. Crystal structure of the Helicobacter pylori vacuolating toxin p55 domain. Proc Natl Acad Sci USA. 2007;104(41):16293–16298. doi: 10.1073/pnas.0707447104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson TA, Qiu J, Plaut AG, Holyoak T. Active-site gating regulates substrate selectivity in a chymotrypsin-like serine protease: The structure of Haemophilus influenzae immunoglobulin A1 protease. J Mol Biol. 2009;389(3):559–574. doi: 10.1016/j.jmb.2009.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khan S, Mian HS, Sandercock LE, Chirgadze NY, Pai EF. Crystal structure of the passenger domain of the Escherichia coli autotransporter EspP. J Mol Biol. 2011;413(5):985–1000. doi: 10.1016/j.jmb.2011.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Otto BR, et al. Crystal structure of hemoglobin protease, a heme binding autotransporter protein from pathogenic Escherichia coli. J Biol Chem. 2005;280(17):17339–17345. doi: 10.1074/jbc.M412885200. [DOI] [PubMed] [Google Scholar]
- 31.Barnard TJ, Dautin N, Lukacik P, Bernstein HD, Buchanan SK. Autotransporter structure reveals intra-barrel cleavage followed by conformational changes. Nat Struct Mol Biol. 2007;14(12):1214–1220. doi: 10.1038/nsmb1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhai Y, et al. Autotransporter passenger domain secretion requires a hydrophobic cavity at the extracellular entrance of the β-domain pore. Biochem J. 2011;435(3):577–587. doi: 10.1042/BJ20101548. [DOI] [PubMed] [Google Scholar]
- 33.Oomen CJ, et al. Structure of the translocator domain of a bacterial autotransporter. EMBO J. 2004;23(6):1257–1266. doi: 10.1038/sj.emboj.7600148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tajima N, Kawai F, Park SY, Tame JR. A novel intein-like autoproteolytic mechanism in autotransporter proteins. J Mol Biol. 2010;402(4):645–656. doi: 10.1016/j.jmb.2010.06.068. [DOI] [PubMed] [Google Scholar]
- 35.Hartmann MD, et al. Complete fiber structures of complex trimeric autotransporter adhesins conserved in enterobacteria. Proc Natl Acad Sci USA. 2012;109(51):20907–20912. doi: 10.1073/pnas.1211872110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grijpstra J, Arenas J, Rutten L, Tommassen J. Autotransporter secretion: Varying on a theme. Res Microbiol. 2013;164(6):562–582. doi: 10.1016/j.resmic.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 37.Hooton TM. Clinical practice. Uncomplicated urinary tract infection. N Engl J Med. 2012;366(11):1028–1037. doi: 10.1056/NEJMcp1104429. [DOI] [PubMed] [Google Scholar]
- 38.Totsika M, et al. Uropathogenic Escherichia coli mediated urinary tract infection. Curr Drug Targets. 2012;13(11):1386–1399. doi: 10.2174/138945012803530206. [DOI] [PubMed] [Google Scholar]
- 39.Sherlock O, Schembri MA, Reisner A, Klemm P. Novel roles for the AIDA adhesin from diarrheagenic Escherichia coli: Cell aggregation and biofilm formation. J Bacteriol. 2004;186(23):8058–8065. doi: 10.1128/JB.186.23.8058-8065.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heras B, et al. Dehydration converts DsbG crystal diffraction from low to high resolution. Structure. 2003;11(2):139–145. doi: 10.1016/s0969-2126(03)00005-4. [DOI] [PubMed] [Google Scholar]
- 41.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 1):72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 43.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 44.Adams PD, et al. PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58(Pt 11):1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 45.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 46.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 4):439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 47. DeLano WL (2002) The PyMOL Molecular Graphics System (DeLano Scientific, San Carlos, CA)
- 48.Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177(14):4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kjaergaard K, Schembri MA, Hasman H, Klemm P. Antigen 43 from Escherichia coli induces inter- and intraspecies cell aggregation and changes in colony morphology of Pseudomonas fluorescens. J Bacteriol. 2000;182(17):4789–4796. doi: 10.1128/jb.182.17.4789-4796.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.