

Significance
The colonization of the intestinal track by the trillions of bacteria that live in the gut (i.e., the gut microbiome) is a dynamic process. Through the use of next-generation sequencing and mathematical models, we were able to quantify the interpopulation interactions that occurred after a germfree mouse was inoculated with a murine microbiome. This approach was significant because we were able to quantify these interactions in vivo without the use of cultivation. Although colonization is most commonly associated with the development of a neonate’s microbiome, understanding the ecological mechanisms involved in the successional process is critical to understanding the recovery of the microbiome following antibiotic therapies, changes in diet, and shifts in health.
Keywords: microbiome, microbial ecology, culture independent, 454 sequencing, dynamical systems
Abstract
Understanding the nature of interpopulation interactions in host-associated microbial communities is critical to understanding gut colonization, responses to perturbations, and transitions between health and disease. Characterizing these interactions is complicated by the complexity of these communities and the observation that even if populations can be cultured, their in vitro and in vivo phenotypes differ significantly. Dynamic models are the cornerstone of computational systems biology and a key objective of computational systems biologists is the reconstruction of biological networks (i.e., network inference) from high-throughput data. When such computational models reflect biology, they provide an opportunity to generate testable hypotheses as well as to perform experiments that are impractical or not feasible in vivo or in vitro. We modeled time-series data for murine microbial communities using statistical approaches and systems of ordinary differential equations. To obtain the dense time-series data, we sequenced the 16S ribosomal RNA (rRNA) gene from DNA isolated from the fecal material of germfree mice colonized with cecal contents of conventionally raised animals. The modeling results suggested a lack of mutualistic interactions within the community. Among the members of the Bacteroidetes, there was evidence for closely related pairs of populations to exhibit parasitic interactions. Among the Firmicutes, the interactions were all competitive. These results suggest future animal and in silico experiments. Our modeling approach can be applied to other systems to provide a greater understanding of the dynamics of communities associated with health and disease.
Analysis of microbial communities is complicated by the communities’ large size, diversity, and recalcitrance to culturing (1). Furthermore, even if a population can be cultured and studied under in vitro conditions, there is no guarantee that the observed phenotypes replicate an in vivo phenotype. An underused strategy for describing in vivo phenotypes is the use of quantitative models to describe temporal patterns of biodiversity. Generation of mathematical models can facilitate the inference of relative growth rates, mechanisms of interaction, and responses to perturbations (2–5).
Mathematical models are powerful because they provide a method to explain past experiments and predict the outcomes of future experiments. A commonly used approach in microbial ecology literature is to develop correlation-based networks to describe the co-occurrence and dynamics of populations (6–8). These models are helpful for providing an initial description of the interaction network; however, they ignore the possibility that a relationship can be asymmetrical with one partner benefiting and the other being hindered. Furthermore, application of these methods to time-series data violates assumptions of independence between observations (9). A second approach that has received attention in theoretical literature has been the use of systems of differential equations (3, 8, 10, 11). This approach does take into account the possibility of asymmetrical interactions and allows one to measure relative growth rates. In addition, more sophisticated models can incorporate nonlinear interactions and interactions involving more than two populations. The challenges of this approach are the choice of an adequate mathematical representation, the complexity and scalability, and the need for dense time-series datasets (12).
One viable system for modeling microbial communities is the colonization of germfree mice. Germfree animals provide a tractable model system to study these processes because of their receptivity to diverse community structures. For example, to determine the role of the host in shaping the structure of the microbiome, the gut contents of mice were inoculated into germfree zebrafish and the gut contents of zebrafish were inoculated into germfree mice (13). The stabilized community structure suggested that the host selected for populations that would result in a community resembling the conventional community structure. Others have attempted to humanize germfree mice by colonizing mice with human feces and have shown that the community is stably maintained between generations of mice (14). Neither of these studies described the dynamics of colonization or compared how the successional patterns following colonization of germfree mice with mouse, human, or zebrafish gut contents compared. A recent study investigated the conventionalization of the jejunum and cecum at three time points (1, 7, and 21 d after colonization) in germfree mice and observed that even though a climax community was provided to the germfree mouse, it was necessary for the gut community to undergo successional processes over 21 d and eventually did resemble the inoculating community (15). These studies suggest that there are nonrandom processes driven by the host and the ecological dynamics that shape the structure of the gut microbiome.
The colonization of the gastrointestinal tract at birth and following antibiotic perturbations has many important ramifications for human health. In infants, necrotizing enterocolitis is a critical problem for children born prematurely where the colonization of the gut microbiome is thought to have gone awry (16). In healthy children, whether they are born vaginally or via Caesarian section is thought to impact the populations of bacteria that dominate their fully colonized community (17). These can be thought of as examples of primary succession where a new community forms with no influence from previous community members and is analogous to the long-term successional patterns observed on volcanic lava flows (18). Later in life, antibiotic therapies are known to perturb the structure of the gut microbiome, and for most people the community largely reassembles to its previous state (19); however, in others, they can become colonized by opportunists such as Clostridium difficile. An effective treatment for individuals for whom additional antibiotic therapy is unable to clear the infection is a microbiome transplant where the patient is colonized with fecal material from a donor resulting in a new community structure that C. difficile is unable to colonize (20). Each of these examples can be thought of as examples of secondary succession where a new community forms following a perturbation but the previous populations influence the successional process and are analogous to the recovery of a forest following a fire (18). Numerous recent studies have explored secondary successional patterns in host-associated communities and the forces that can alter the course of these patterns (e.g., refs. 21–23). Processes involved in primary succession during the colonization of the sterile gastrointestinal tract have received limited attention (24, 25). Just as it is important to understand the successional forces for macroecological systems for conservation purposes, it is critical that we understand the successional forces in microbial systems to understand how perturbations can positively and negatively affect human health.
There are a number of interesting ecological questions that could be addressed by colonization experiments. First, do microbial communities assemble following neutral or deterministic processes? Understanding those processes would enable the identification of therapies that would allow one to develop a healthy microbiome where it might not normally. Second, what are the characteristics of a successful population seeking to colonize an established community? Such populations could be either pathogens or probiotics and understanding their mechanism of colonization could permit their exclusion or inclusion. Finally, what types of interactions exist between populations within a microbial community and what are their mechanisms? Traditional microbiology has focused on studying populations in isolation and extrapolating their physiology to a complex community. This approach makes the implicit assumption that results of in vitro studies predict what is occurring in vivo and that bacterial populations have largely neutral interactions with each other. However, even in a microbiome-centric approach it is difficult to determine the types and prevalence of interactions that drive microbial communities. Addressing these and other questions is challenging given the large fraction of bacteria that are still recalcitrant to culturing and the shear diversity of most microbial communities.
In the current study, we were interested in addressing this final question using mathematical models. Specifically, we developed correlation and differential equation models that captured operational taxonomic unit (OTU) dynamics with a differential equation system. Our differential equation framework builds on the rich ecological modeling literature by using the generalized Lotka–Volterra (gLV) equation (26). Both modeling frameworks were developed using dense time series to infer the network structure of a microbial community after its inoculation into germfree mice. To obtain the dense time-series data, we sequenced the 16S rRNA gene from DNA isolated from the fecal material of germfree mice colonized with cecal contents of conventionally raised animals (15). Our modeling approaches indicated that the assembly of the murine gut community involved the complex interactions of its members and resulted in a stable community.
Results
Colonization of Germfree Mice with a Mature Community Results in Reproducible Successional Patterns.
We observed the community dynamics of five germfree C57BL/6 mice that were inoculated with the cecal contents of a single adult C57BL/6 mouse over 21 d. The alpha diversity of the fecal communities from these mice increased during the 4 d following colonization and then plateaued for the remainder of the study (Fig. S1). Ordination of the community structures using nonmetric dimensional scaling of the distances between community structures suggested that although there was considerable intermouse variation immediately after colonization, the gut communities generally tracked each other well (Fig. S2). This was further demonstrated by quantifying the intramouse variation based on the 1-d distance between samples for each mouse (Fig. 1A) and the average intermouse variation for the same day (Fig. 1B). Combined, these results indicated that the communities varied widely over the first 4 to 5 d after colonization and that they then stabilized and replicated one another as the communities developed over the remainder of the study.
Fig. 1.

Average distance between the gut community structure from the present day and the previous day (A) and to the other mice on the same day (B) during the 21 d after colonization. Error bars represent 95% confidence intervals.
Temporal Dynamics of Bacterial Populations During Colonization.
The most abundant OTUs during the 21 d after colonization were related to members of the Bacteroidetes, Firmicutes, Verrucomicrobia, and Proteobacteria (Fig. 2). In general, the community was dominated by OTUs within the Bacteroidetes that affiliated within the Porphyromonadaceae. These OTUs had flat profiles with the exception of OTU 4, which was related to members of the genus Barnesiella and was initially dominant before becoming rare by the fifth day after colonization. Among the Firmicutes, OTUs 9 and 11 were related to members of the genus Lactobacillus and family Erysipelotrichaceae, respectively. These two OTUs appeared to tradeoff as the dominant member of the Firmicutes during the period of this study. OTU 8, a relative of the family Enterobacteriaceae, had its greatest abundance in the days immediately after colonization. Finally, OTU 18, a relative of the genus Akkermansia, climbed and crashed in abundance over the first 5 d after colonization. Although there appeared to be a significant amount of stochastic variation across the duration of this experiment, the overall reproducibility of these patterns between animals suggested that deterministic inter-OTU interactions drove the successional patterns (Fig. 1 and Fig. S2).
Fig. 2.

Average relative abundance for the most abundant OTUs that classified within the phyla Bacteroidetes, Firmicutes, Proteobacteria, and Verrucomicrobia during the 21 d after colonization. Error bars represent 95% confidence interval for the OTU across the five mice.
Modeling by Correlation-Based Analysis.
To obtain a measure of association between OTUs while incorporating their abundance during the colonization process, we inferred Pearson correlation coefficients using a recently described method that is robust for analyzing relative-abundance data (27) (Fig. 3). We identified 61 associations that had a P value less than 10−4; 34 were positive (R > 0.25) and 27 were negative (R < −0.25). When we considered the significant associations between OTUs within the same taxonomic order (n = 10 associations) or phylum (n = 18 associations), there were an equal number of positive and negative interactions. These results suggest that there was not an increased fraction of negative interactions between closely related OTUs as might be expected if closely related organisms shared similar phenotypes.
Fig. 3.

Association network describing the co-occurrence patterns between OTUs that had a correlation value less than −0.25 or larger than 0.25. OTUs 8, 22, 36, and 38 had an average relative abundance of at least 0.5% in individual mice but were not included in the correlation analysis because they did not meet the threshold abundance in all mice.
Quantifying Inter-OTU Interactions Using Ordinary Differential Equation-Based Models.
We then formulated and fit a system of ordinary differential equations (ODEs) to the OTU relative-abundance temporal data. The relative abundances of those OTUs that did not meet the threshold criteria (as described in Materials and Methods, i.e., relative abundance >1% for at least a single time point) were pooled and represented an average of 18.8% of the 16S rRNA gene fragments sequenced from each sample. This screening procedure resulted in the modeling of 16 OTUs and the composite category of rare OTUs (i.e., 17 OTUs total). Considering the size and complexity of the ODE system, as well as the potential instability of the numerical solver during the optimization process, the model fit was excellent (R2 = 0.81, Figs. S3–S8).
The model allowed us to quantify the relative magnitudes of the intrinsic growth rates (i.e., α) and the interactions between OTUs (i.e., β). The predicted relative in vivo growth rates indicated significant variation across the modeled populations (Table 1). Across the 17 OTUs, all had growth rates significantly above zero. In addition, there was wide variation in the growth rates of OTUs that affiliated within the same taxonomic group; however, there was no significant difference in the growth rates of the OTUs affiliated with the Bacteroidetes and Firmicutes (Wilcox test; P = 0.89). The predicted interaction matrix from the model allowed us to quantify the types of relationships between OTUs based on the sign of the values in the β-matrix (Fig. 4). Of the 136 pairs of interactions between the 17 OTUs, 91 were competitive (i.e., both OTUs hindered; −/−), 22 were parasitic (i.e., one OTU benefited and the other was hindered; +/−), 17 were ammensalistic (i.e., one OTU is hindered and the other is unaffected; −/0), 4 were commensal (i.e., one OTU was promoted and the other was unaffected; +/0), 2 were neutral (i.e., neither OTU was affected by the other; 0/0), and none were mutualistic (i.e., both OTUs benefited; +/+). When we required the interaction values to have an absolute value greater than 2.0, there were 90 neutral, 33 ammensalistic, 6 parasitic, 5 commensal, and 2 competitive interactions. These results indicate that during colonization, the strongest interactions involve a cost for one OTU and at best no cost to the other OTU (i.e., competition and ammensalism; n = 35 of 45 nonneutral interactions). Next we sought to characterize whether the types of interactions varied based on phylogeny. We observed a significant difference in the types of interactions between the Firmicutes and Bacteroidetes (Fisher exact test, P = 0.05). Among the 10 interactions between Firmicutes, all of them were competitive, whereas among the 28 interactions between Bacteroidetes, 17 were competitive, 10 were parasitic, 6 were ammensalistic, 2 were commensal, and 1 was neutral. When we guided our analysis by the phylogeny of a representative sequence from each OTU, we observed that among the Bacteroidetes there were 4 pairs of closely related OTUs (Fig. 4); 3 of these were parasitic and 1 was competitive. Together the fits of the α- and β-parameters in our model indicates that although the populations have comparable growth rates, the gut environment is highly competitive and numerous populations keep the others from dominating the community.
Table 1.
The taxonomic description and modeled growth rate of OTUs included in the system of the ODE model
| OTU | Phylum | Class | Order | Family | Genus | Predicted growth rate (SE) |
| 6 | Bacteroidetes | Bacteroidia | Bacteroidales | Incertae sedis | Incertae sedis | 0.59 (0.06) |
| 12 | Bacteroidetes | Bacteroidia | Bacteroidales | Incertae sedis | Incertae sedis | 1.21 (0.03) |
| 1 | Bacteroidetes | Bacteroidia | Bacteroidales | Porphyromonadaceae | Incertae sedis | 1.34 (0.08) |
| 2 | Bacteroidetes | Bacteroidia | Bacteroidales | Porphyromonadaceae | Incertae sedis | 1.16 (0.07) |
| 5 | Bacteroidetes | Bacteroidia | Bacteroidales | Porphyromonadaceae | Incertae sedis | 1.01 (0.06) |
| 7 | Bacteroidetes | Bacteroidia | Bacteroidales | Porphyromonadaceae | Incertae sedis | 1.29 (0.05) |
| 10 | Bacteroidetes | Bacteroidia | Bacteroidales | Porphyromonadaceae | Incertae sedis | 1.15 (0.02) |
| 3 | Bacteroidetes | Bacteroidia | Bacteroidales | Porphyromonadaceae | Barnesiella | 0.52 (0.06) |
| 4 | Bacteroidetes | Bacteroidia | Bacteroidales | Bacteroidaceae | Bacteroides | 0.43 (0.14) |
| 11 | Firmicutes | Erysipelotrichia | Erysipelotrichales | Erysipelotrichaceae | Incertae sedis | 0.77 (0.03) |
| 25 | Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | Incertae sedis | 1.46 (0.03) |
| 9 | Firmicutes | Bacilli | Lactobacillales | Lactobacillaceae | Lactobacillus | 0.83 (0.03) |
| 19 | Firmicutes | Bacilli | Lactobacillales | Lactobacillaceae | Lactobacillus | 0.83 (0.02) |
| 27 | Firmicutes | Bacilli | Lactobacillales | Lactobacillaceae | Lactobacillus | 0.84 (0.06) |
| 8 | Proteobacteria | γ-Proteobacteria | Enterobacteriales | Enterobacteriaceae | Incertae sedis | 0.43 (0.05) |
| 18 | Verrucomicrobia | Verrucomicrobiae | Verrucomicrobiales | Verrucomicrobiaceae | Akkermansia | 0.73 (0.09) |
| Rare | NA | NA | NA | NA | NA | 1.36 (0.07) |
OTUs are grouped by taxonomy. NA, the Rare OTU is a mixture of sequences from various taxa.
Fig. 4.

Heat map of the modeled parameters that describe the interactions between OTUs (βij). Each value represents the effect of the OTU in the row on the OTU represented by the column. Because the relationships are asymmetrical, so is the matrix. The rows and columns are sorted together according to the neighbor joining phylogenetic tree that was calculated using a representative sequence from each OTU.
Discussion
Modeling microbial communities has a long history that had yet to be explored by fitting a dynamic model to a community of bacteria. Early theoretical models provided a plausible simulation of both the mouse gut and continuous-flow mixed cultures (28). More recently, the dynamics of bacteria have been modeled using linear models using dietary perturbations of synthetic communities in germfree mice (29). The advantage of a modeling approach is that it allows scientists to explore mechanisms of interaction using organisms that are difficult or as yet unculturable in their natural environment.
Most of the techniques applied to model microbial communities are extremely useful for uncovering pair-wise interactions but do not adequately describe the intrinsic dynamic nature of them. By construction, memory is embedded in longitudinal data: observations are dependent over time and each data point should not be treated as a static snapshot. This work represents a systematic attempt to use a dynamic model as a network inference tool. We used ODEs in the form of a gLV system to accurately describe the time courses of OTUs across a time span of 21 d for five different mice. Numerical solution and parameter estimation for systems of ODEs are sensitive to model instability (e.g., ill conditioning, stiffness) and fitting issues (e.g., multiple minima, high nonlinearity). One approach to improving parameter estimation is to superimpose constraints on the parameters; however, unless these constraints can be justified both biologically and mathematically, they should not be enforced because the results can be greatly affected. We were able to reconstruct a plausible network of interactions that recapitulated the dynamics and interactions of all of the mice; however, the model formulation captured only first-order interactions between species. Regardless, our model has provided rich information into the mechanisms regulating the dynamics of the murine intestinal microbiota. We are confident that by introducing a model generation and selection scheme, the complexity of the network will be greatly reduced and the parameter estimates and confidence intervals will be more robust.
The use of mathematical and computational models to study complex biological processes is becoming increasingly productive. Technological advances are enabling virtual in silico experiments to explore and answer questions that are problematic to address in the wet-laboratory. An important goal of these virtual in silico experiments is to improve mechanistic insights while reducing uncertainties and prioritizing hypotheses for future testing. Several examples emerge from the current study. First, it was interesting that the intrinsic growth rate of OTU08, a member of the Enterobacteriaceae, was among the two slowest in the community. This is in contrast to the rapid in vitro growth rates for the Enterobacteriaceae, which are typically on the order of two to three doublings per h (30), whereas those of the Firmicutes and Bacteroidetes are considerably slower (31). Further experimental and modeling studies could further develop the relatively simple model of the α-terms to incorporate data describing the metabolome or interaction with the host immune system to better understand the discrepancy between in vivo and in vitro observations. Second, we observed that three of four pairs of closely related populations of Bacteroidetes had parasitic interactions with each other, whereas the interactions between the Firmicutes were all competitive. Gnotobiotic competition experiments could be used to test this hypothesis further by colonizing mice with Bacteroidetes or Firmicutes that vary in their relatedness. Alternatively, cultivation of these populations and sequencing their genomes could reveal overlap in the functional repertoire of the Bacteroidetes but not the Firmicutes. With such results, it would then be possible to further refine models and perform other experiments in silico. For example, instead of focusing on the taxonomic composition of communities, advances have been made in other fields using trait-based models to describe the collection of the phenotypes needed to maintain a stable community (32). Both taxonomy and phenotype-based models could provide complementary insights into the dynamics of microbial communities. Furthermore, they would allow the marrying of multiple omics-based technologies (e.g., 16S rRNA, metatranscriptomics, and metabolomics) to understand the mechanisms behind colonization resistance to pathogens and probiotics.
Modeling microbial communities is dependent on a number of factors. The availability of dense time-series datasets is limited as there are few studies that have more than 10 samples per individual animal with a fixed time increment (14, 33). The advent of next-generation sequencing and sample multiplexing makes obtaining these datasets more practical. The deep sequencing afforded by these technologies poses a tradeoff between the number of reads per sample and the number of samples to sequence. For modeling, additional samples collected every 6 or 12 h instead of every day would be preferred to additional reads per sample because it is impractical to model more populations than we have here, and the model would provide greater granularity of dynamics. Second, experimental validation and model improvement and selection are necessary next steps. Unfortunately, in complex communities such as the murine gut microbiome, it is impossible to selectively remove a population and observe the resulting dynamics. Alternatively, experiments could be developed to test predictions of the model such as the parasitism between the related Bacteroidetes OTUs. If they could be cultured, these pairs of populations could be competed against each other during colonization of germfree animals or in vitro.
The colonization of a germfree mouse is an important model for gaining insights into the development of the infant microbiome, recovery from antibiotic therapy, and other perturbations. Interestingly, even though the mice in this study were cohoused, the gut community initially assembled in a highly variable process and by 5 d after colonization began to follow a reproducible path toward a stable gut community. The successional pattern that we observed was characterized by the transition between different dominant populations that were often from the same taxonomic lineage. Modeling these populations has allowed us to generate additional hypotheses into the nature of in vivo growth rates, niche partitioning, and the degree to which the observed successional pattern was deterministic or neutral. Although the colonization of germfree mice is a necessarily artificial system, it allows us to address a number of interesting ecological questions. This work complements a growing area of research that uses inter- and intraspecies microbiota transplants and that generally find the host models its microbiome to resemble its own conventional community structure and find that the colonized microbiome only partially resembles the inoculating community (e.g., refs. 13–15).
Mathematical modeling of microbial communities provides an entry into the mechanisms that govern the interactions between bacterial populations that are recalcitrant to cultivation in their natural setting. Here we have considered a community assembling within a host; however, these methods could easily be expanded to engineered and aquatic ecosystems. Beyond modeling the interactions within the community, future models could incorporate the interactions between these populations and their environment. Dynamic systems models allow scientists to quantify the processes they observed as well as make predictions about the behaviors of populations. The combination of high-throughput DNA sequencing technologies, tractable model systems, and systems of differential equations provide a powerful combination to better understand the dynamics of microbial communities.
Materials and Methods
Animals and Animal Care.
This study was approved by the University Committee on Use and Care of Animals at the University of Michigan. As described previously (15), the cecal contents of an adult C57BL/6 mouse were homogenized and inoculated into five germfree adult female C57BL/6 mice at the University of Michigan Germ-Free Mouse Facility. The mice were cohoused within a germfree isolator for 21 d after colonization. Fecal samples were obtained daily from each animal and were immediately frozen.
16S rRNA Gene Sequencing and Curation.
Following bead beating, DNA was isolated from each fecal pellet using the HTP PowerSoil DNA extraction kit (MO BIO) according to the manufacturers specifications on an epMotion 5075 (Eppendorf) liquid handling workstation. The V35 region of the 16S rRNA gene was then amplified from each DNA sample and sequenced on a 454 Titanium DNA sequencer (Roche) at the Baylor College of Medicine Human Genome Sequencing Facility (33). Using the mothur software package (34), we used the PyroNoise denoising algorithm, aligned the resulting reads against the SILVA SEED reference alignment, and performed a preclustering step to further reduce sequencing and PCR errors (35–38). We culled any reads that did not have at least 450 flows, had more than one mismatch to the barcode sequence or more than two mismatches to the primer sequence, did not map to the correct region of the 16S rRNA gene, were flagged as chimeric using UCHIME in the de novo detection mode (39), or were classified as being eukaryotic, chloroplasts, or mitochondria when using the RDP 16S rRNA training set (40). In parallel to the mouse sample, we sequenced and processed a mock community consisting of 21 reference genomic DNAs where we knew the actual 16S rRNA gene sequences (i.e., a mock community). Analysis of the error rates for sequencing the mock community revealed an observed sequencing error rate of 0.01% (36). The curated sequences were clustered into OTUs based on a 3% dissimilarity cutoff using the average neighbor algorithm (41). The final OTU counts and taxonomy tables are listed in Dataset S1.
Community Analysis.
Alpha diversity was measured using the inverse Simpson index and the number of observed OTUs (42). Beta diversity was measured using the 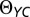 community distance metric because it applies an even weighting across the community distribution (43). Correlation-based analyses were performed using the SparCC algorithm, which limits the number of false correlations identified due to OTU data being based on compositional data (27). Furthermore, to avoid identifying spurious interactions as being significant due to the lack of independence between time points, we used a stringent maximum P value of 10−4. SparCC correlation and P value matrices are listed in Dataset S2. To limit the effects of uneven sampling we randomly sampled 2,900 sequences from each sample. This number of sequences was selected to maintain the largest number of samples with as many reads possible. Measurements of alpha- and beta-diversity were performed by rarefying the dataset to 2,900 sequences per sample with 1,000 iterations.
community distance metric because it applies an even weighting across the community distribution (43). Correlation-based analyses were performed using the SparCC algorithm, which limits the number of false correlations identified due to OTU data being based on compositional data (27). Furthermore, to avoid identifying spurious interactions as being significant due to the lack of independence between time points, we used a stringent maximum P value of 10−4. SparCC correlation and P value matrices are listed in Dataset S2. To limit the effects of uneven sampling we randomly sampled 2,900 sequences from each sample. This number of sequences was selected to maintain the largest number of samples with as many reads possible. Measurements of alpha- and beta-diversity were performed by rarefying the dataset to 2,900 sequences per sample with 1,000 iterations.
Dynamic Modeling of the Gut Community.
Network reconstruction of the germfree time-course data were formulated as a constrained nonlinear least squares problem. In detail, the mathematical model formulation is represented by the following gLV system of ODEs:
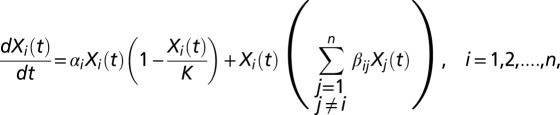 |
where the parameters αi can be interpreted as the intrinsic growth rate of species i, and βij as the interaction strength between species i and j (positive, neutral or negative). Here the βij are analogous to elements of a co-occurrence and association matrix. The parameter K represents the carrying capacity term in the logistic growth component for the entire community (i.e., at any time 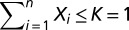 ). Given the complexity of the gLV model and obvious concerns of scalability, we chose to model only OTUs with relative abundances greater than 1% at any time point for each mouse. To make the comparison across mice meaningful, we modeled the same OTUs across all five mice, even if not all of them fulfilled the 1% relative-abundance threshold for all of the mice. We then specified a fully connected gLV model where all of the species interact with each other. The resulting system of ODEs contained 17 equations (i.e., n = 17) and 289 parameters (i.e., 17 parameters for each equation, 1 αi and 16 βij). The constrained least squares problem was implemented and solved in MATLAB [Version 7.13.0.564 (R2011b); MathWorks] using the function find minimum of constrained nonlinear multivariable function (FMINCON) in the Optimization Toolbox and several numerical solvers (both explicit and implicit methods, as available in MATLAB). To speed up computation, we ran the fittings within the Parallel Computing Toolbox. See ref. 44 for details on the fitting algorithm implementation. Briefly, we superimposed nonnegative constraints on the intrinsic growth rates (αi ≥ 0, i = 1, 2, …, 17) without any constraints on the interaction terms (βij) and set K = 1. In addition, we used the observed relative abundances from the first day following colonization as the initial conditions. For the initial conditions of the parameters, we varied them between
). Given the complexity of the gLV model and obvious concerns of scalability, we chose to model only OTUs with relative abundances greater than 1% at any time point for each mouse. To make the comparison across mice meaningful, we modeled the same OTUs across all five mice, even if not all of them fulfilled the 1% relative-abundance threshold for all of the mice. We then specified a fully connected gLV model where all of the species interact with each other. The resulting system of ODEs contained 17 equations (i.e., n = 17) and 289 parameters (i.e., 17 parameters for each equation, 1 αi and 16 βij). The constrained least squares problem was implemented and solved in MATLAB [Version 7.13.0.564 (R2011b); MathWorks] using the function find minimum of constrained nonlinear multivariable function (FMINCON) in the Optimization Toolbox and several numerical solvers (both explicit and implicit methods, as available in MATLAB). To speed up computation, we ran the fittings within the Parallel Computing Toolbox. See ref. 44 for details on the fitting algorithm implementation. Briefly, we superimposed nonnegative constraints on the intrinsic growth rates (αi ≥ 0, i = 1, 2, …, 17) without any constraints on the interaction terms (βij) and set K = 1. In addition, we used the observed relative abundances from the first day following colonization as the initial conditions. For the initial conditions of the parameters, we varied them between 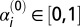 and
and 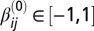 . We applied a standard cross-validation approach to account for model variability and assess the robustness of our estimates (45). To perform this approach we performed 20 fittings per mouse where we randomly held 10% of the time points and performed the fitting. We then calculated the mean and SE for each parameter distribution and built 95% confidence intervals for the estimates. We also tried to use the Hessian calculation returned by the MATLAB function FMINCON to estimate the confidence intervals for αi and βij (46); however, all of the methods available for numerically approximating the Hessian matrix (i.e., dense quasi-Newton approximation, limited memory large-scale quasi-Newton approximation, finite differences of the gradient) returned sloppy confidence intervals despite the fact that the constraints of the nonlinear least squares implementation are all linear. The α-values and β-matrices are listed in Dataset S3.
. We applied a standard cross-validation approach to account for model variability and assess the robustness of our estimates (45). To perform this approach we performed 20 fittings per mouse where we randomly held 10% of the time points and performed the fitting. We then calculated the mean and SE for each parameter distribution and built 95% confidence intervals for the estimates. We also tried to use the Hessian calculation returned by the MATLAB function FMINCON to estimate the confidence intervals for αi and βij (46); however, all of the methods available for numerically approximating the Hessian matrix (i.e., dense quasi-Newton approximation, limited memory large-scale quasi-Newton approximation, finite differences of the gradient) returned sloppy confidence intervals despite the fact that the constraints of the nonlinear least squares implementation are all linear. The α-values and β-matrices are listed in Dataset S3.
Supplementary Material
Acknowledgments
The authors thank Nicole Falkowski and Merritt G. Gillilland for the technical assistance provided. This work was supported by National Institutes for Health Grants R01GM095356, R01HG005975, U19AI090871, and P30DK034933 (to P.D.S.); R01GM095356 (to S.M.); U19AI090871 and P30DK034933 (to G.B.H.); and R01GM095356 (to J.F.P.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The standard flowgram files (SFF) files and minimum information about a marker gene sequence (MIMARKS) table reported in this paper are available at www.mothur.org/germ_free_model.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1311322111/-/DCSupplemental.
References
- 1.Riesenfeld CS, Schloss PD, Handelsman J. Metagenomics: Genomic analysis of microbial communities. Annu Rev Genet. 2004;38:525–552. doi: 10.1146/annurev.genet.38.072902.091216. [DOI] [PubMed] [Google Scholar]
- 2.Larsen P, Hamada Y, Gilbert J. Modeling microbial communities: Current, developing, and future technologies for predicting microbial community interaction. J Biotechnol. 2012;160(1-2):17–24. doi: 10.1016/j.jbiotec.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 3.O’Dwyer JP, Lake JK, Ostling A, Savage VM, Green JL. An integrative framework for stochastic, size-structured community assembly. Proc Natl Acad Sci USA. 2009;106(15):6170–6175. doi: 10.1073/pnas.0813041106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gerber GK, Onderdonk AB, Bry L. Inferring dynamic signatures of microbes in complex host ecosystems. PLOS Comput Biol. 2012;8(8):e1002624. doi: 10.1371/journal.pcbi.1002624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freter R, Brickner H, Fekete J, Vickerman MM, Carey KE. Survival and implantation of Escherichia coli in the intestinal tract. Infect Immun. 1983;39(2):686–703. doi: 10.1128/iai.39.2.686-703.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilbert JA, et al. Defining seasonal marine microbial community dynamics. ISME J. 2012;6(2):298–308. doi: 10.1038/ismej.2011.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barberán A, Bates ST, Casamayor EO, Fierer N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012;6(2):343–351. doi: 10.1038/ismej.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faust K, et al. Microbial co-occurrence relationships in the human microbiome. PLOS Comput Biol. 2012;8(7):e1002606. doi: 10.1371/journal.pcbi.1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sokal RR, Rohlf FJ. 1995. Biometry: The Principles and Practice of Statistics in Biological Research (Freeman, New York), 3rd Ed, p 887.
- 10.Kirschner DE, Blaser MJ. The dynamics of Helicobacter pylori infection of the human stomach. J Theor Biol. 1995;176(2):281–290. doi: 10.1006/jtbi.1995.0198. [DOI] [PubMed] [Google Scholar]
- 11.Schluter J, Foster KR. The evolution of mutualism in gut microbiota via host epithelial selection. PLoS Biol. 2012;10(11):e1001424. doi: 10.1371/journal.pbio.1001424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faust K, Raes J. Microbial interactions: From networks to models. Nat Rev Microbiol. 2012;10(8):538–550. doi: 10.1038/nrmicro2832. [DOI] [PubMed] [Google Scholar]
- 13.Rawls JF, Mahowald MA, Ley RE, Gordon JI. Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell. 2006;127(2):423–433. doi: 10.1016/j.cell.2006.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turnbaugh PJ, et al. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med. 2009;1(6):6ra14. doi: 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gillilland MG, 3rd, et al. Ecological succession of bacterial communities during conventionalization of germ-free mice. Appl Environ Microbiol. 2012;78(7):2359–2366. doi: 10.1128/AEM.05239-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stewart CJ, et al. The preterm gut microbiota: Changes associated with necrotizing enterocolitis and infection. Acta Paediatr. 2012;101(11):1121–1127. doi: 10.1111/j.1651-2227.2012.02801.x. [DOI] [PubMed] [Google Scholar]
- 17.Dominguez-Bello MG, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci USA. 2010;107(26):11971–11975. doi: 10.1073/pnas.1002601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Begon M, Harper JL, Townsend CR. Ecology: Individuals, Populations, and Communities. 3rd Ed. Malden, MA: Blackwell Science; 1996. [Google Scholar]
- 19.Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6(11):e280. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kassam Z, Hundal R, Marshall JK, Lee CH. Fecal transplant via retention enema for refractory or recurrent Clostridium difficile infection. Arch Intern Med. 2012;172(2):191–193. doi: 10.1001/archinte.172.2.191. [DOI] [PubMed] [Google Scholar]
- 21.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: Human gut microbes associated with obesity. Nature. 2006;444(7122):1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 22.Wu GD, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334(6052):105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koren O, et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell. 2012;150(3):470–480. doi: 10.1016/j.cell.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. Development of the human infant intestinal microbiota. PLoS Biol. 2007;5(7):e177. doi: 10.1371/journal.pbio.0050177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koenig JE, et al. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4578–4585. doi: 10.1073/pnas.1000081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Law R, Blackford JC. Self-assembling food webs: A global viewpoint of coexistence of species in Lotka-Volterra communities. Ecology. 1992;73(2):567–578. [Google Scholar]
- 27.Friedman J, Alm EJ. Inferring correlation networks from genomic survey data. PLOS Comput Biol. 2012;8(9):e1002687. doi: 10.1371/journal.pcbi.1002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kirschner D, Freter R. Mathematical models of colonization and persistence in bacterial infections. In: Nataro J, Blaser M, Cunningham-Rundles S, editors. Persistent Bacterial Infections. Washington, DC: ASM Press; 2000. pp. 79–100. [Google Scholar]
- 29.Faith JJ, McNulty NP, Rey FE, Gordon JI. Predicting a human gut microbiota’s response to diet in gnotobiotic mice. Science. 2011;333(6038):101–104. doi: 10.1126/science.1206025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bremer H, Dennis P. Modulation of chemcial composition and other parameters of the cell growth rate. In: Neidhardt F, editor. Escherichia coli and Salmonella. Vol 2. Washington, DC: ASM Press; 1996. pp. 1553–1569. [Google Scholar]
- 31.Martens EC, et al. Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol. 2011;9(12):e1001221. doi: 10.1371/journal.pbio.1001221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allison SD. A trait-based approach for modelling microbial litter decomposition. Ecol Lett. 2012;15(9):1058–1070. doi: 10.1111/j.1461-0248.2012.01807.x. [DOI] [PubMed] [Google Scholar]
- 33.Schloss PD, et al. Stabilization of the murine gut microbiome following weaning. Gut Microbes. 2012;3(4):383–393. doi: 10.4161/gmic.21008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schloss PD, et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schloss PD. A high-throughput DNA sequence aligner for microbial ecology studies. PLoS ONE. 2009;4(12):e8230. doi: 10.1371/journal.pone.0008230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schloss PD, Gevers D, Westcott SL. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE. 2011;6(12):e27310. doi: 10.1371/journal.pone.0027310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quince C, Lanzen A, Davenport RJ, Turnbaugh PJ. Removing noise from pyrosequenced amplicons. BMC Bioinformatics. 2011;12:38. doi: 10.1186/1471-2105-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pruesse E, et al. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35(21):7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27(16):2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73(16):5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schloss PD, Westcott SL. Assessing and improving methods used in operational taxonomic unit-based approaches for 16S rRNA gene sequence analysis. Appl Environ Microbiol. 2011;77(10):3219–3226. doi: 10.1128/AEM.02810-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Magurran AE. 2004. Measuring Biological Diversity (Blackwell Publishing, Malden, MA)
- 43.Yue JC, Clayton MK. A similarity measure based on species proportions. Comm Statist Theory Methods. 2005;34(11):2123–2131. [Google Scholar]
- 44.Marino S, Voit EO. An automated procedure for the extraction of metabolic network information from time series data. J Bioinform Comput Biol. 2006;4(3):665–691. doi: 10.1142/s0219720006002259. [DOI] [PubMed] [Google Scholar]
- 45.Arlot S, Celisse A. A survey of cross-validation procedures for model selection. Statistics Surveys. 2010;4:40–79. [Google Scholar]
- 46.Seber GAF, Wild CJ. Nonlinear Regression. Hoboken, NJ: Wiley Interscience; 2003. p 768. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.