

Significance
Salmonella enterica serovar Typhimurium (S. typhimurium) is a bacterial foodborne pathogen that causes significant morbidity and mortality worldwide. Nucleotide-binding oligomerization domain–like receptor family pyrin domain containing 12 NLRP12 is a key innate immune molecule that regulates intestinal inflammation and cancer. However, its physiological function in microbial infection is not fully understood. We found that NLRP12 is a key suppressor of innate immune signaling during salmonellosis. Mice lacking NLRP12 are hyperresistant to S. typhimurium infection, and macrophages deficient in NLRP12 produce high levels of proinflammatory cytokines and other molecules contributing to pathogen clearance. Our work revealed that NLRP12-mediated dampening of host immune defenses is used by S. typhimurium to ensure its persistence and survival in host tissues. Modulation of NLRP12 activity could be useful in the prevention and treatment of salmonellosis.
Abstract
The nucleotide-binding oligomerization domain (NOD)-like receptor family pyrin domain containing 12 (NLRP12) plays a protective role in intestinal inflammation and carcinogenesis, but the physiological function of this NLR during microbial infection is largely unexplored. Salmonella enterica serovar Typhimurium (S. typhimurium) is a leading cause of food poisoning worldwide. Here, we show that NLRP12-deficient mice were highly resistant to S. typhimurium infection. Salmonella-infected macrophages induced NLRP12-dependent inhibition of NF-κB and ERK activation by suppressing phosphorylation of IκBα and ERK. NLRP12-mediated down-regulation of proinflammatory and antimicrobial molecules prevented efficient clearance of bacterial burden, highlighting a role for NLRP12 as a negative regulator of innate immune signaling during salmonellosis. These results underscore a signaling pathway defined by NLRP12-mediated dampening of host immune defenses that could be exploited by S. typhimurium to persist and survive in the host.
The nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family consists of a large number of intracellular pathogen recognition receptors that function as sensors of microbial-derived and danger-associated molecules in the cytoplasm of host cells. A subset of NLR proteins, including NLRP1, NLRP3, and NLRC4, activate caspase-1 via the formation of a cytosolic multiprotein complex termed the inflammasome (1). These inflammasome-forming NLRs mediate processing of the proinflammatory cytokines pro–IL-1β and pro–IL-18, which are then secreted by the cell. The non–inflammasome-forming members of the NLR family contribute to regulation of other key inflammatory pathways. For example, NOD1 and NOD2 activate NF-κB and MAPK pathways (2–5), whereas NLRP6, NLRC3, NLRC5, and NLRX1 have been demonstrated to regulate inflammation negatively (6–9).
NLRP12 (NALP12, MONARCH-1, or PYPAF7) is a poorly characterized member of the NLR family. It has a tripartite domain structure, which consists of an N-terminal PYRIN domain, a central nucleotide binding site domain, and a C-terminal domain composed of at least 12 leucine-rich repeat motifs (10). In humans, NLRP12 is expressed in peripheral blood leukocytes, including granulocytes, eosinophils, monocytes, and dendritic cells (DCs) (10, 11). Similarly, mouse NLRP12 is highly expressed in bone marrow neutrophils and granulocytes, macrophages, and DCs (12, 13). Genetic studies in humans have shown that mutations in the NLRP12 gene are associated with periodic fever syndromes and atopic dermatitis (14–16). More recent studies have demonstrated that NLRP12 has both inflammasome-dependent and inflammasome-independent roles in health and disease. Our laboratory and others have previously reported that NLRP12 mediates protection against colon inflammation and tumorigenesis in vivo by negatively regulating inflammatory responses (12, 17).
Recent studies have revealed a potential role for NLRP12 during infectious diseases. Vladimer et al. (18) reported that Nlrp12−/− mice are hypersusceptible to Yersinia pestis infection, whereby NLRP12 is required to drive caspase-1 activation and IL-1β and IL-18 release. Another study found that WT and Nlrp12−/− mice exhibit similar host innate responses in lung infections induced by Mycobacterium tuberculosis or Klebsiella pneumoniae (13). However, in vitro studies reported that a synthetic analog cord factor, trehalose-6,6-dimycolate (TDP), from M. tuberculosis and LPS from K. pneumoniae induced substantially elevated levels of TNF-α and IL-6 in Nlrp12−/− bone marrow-derived DCs compared with their WT counterpart, although levels of secreted IL-1β were not changed (13). These results suggest that unlike the case in Yersinia infection, NLRP12 does not contribute to inflammasome-mediated protection against M. tuberculosis and K. pneumoniae infections. Overall, the physiological and functional relevance of NLRP12 in the host defense against infectious diseases is not fully understood.
Salmonella enterica serovar Typhimurium (S. typhimurium) is a Gram-negative intracellular pathogen, and one of the most prevalent etiological agents of gastroenteritis worldwide. Salmonella infection accounts for 93.8 million cases of gastroenteritis annually in the world and is a leading cause of death among bacterial foodborne pathogens in the United States (19, 20). Previous studies have found that members of the Toll-like receptor (TLR) family, especially TLR4, are critical for the recognition and clearance of S. typhimurium (21, 22). One consequence of Salmonella-induced TLR activation is the production of inflammatory cytokines and antimicrobial compounds, including pro–IL-1β, pro–IL-18, IFN-γ, TNF-α, and reactive oxygen species, which are critical mediators for the control of bacterial growth in host tissues (23). In addition to TLR-mediated host responses, certain members of the NLR family, including NLRC4 and NLRP3, initiate inflammasome formation to drive processing and release of IL-1β and IL-18 following Salmonella infection (24, 25). Although the precise signals that trigger NLRP3 activation during Salmonella infection are unknown, NLRC4 is activated by NAIPs, a subset of receptors within the NLR family that detect Salmonella flagellin (mouse NAIP5 and NAIP6) or certain rod (mouse NAIP2) or needle (human NAIP and mouse NAIP1) proteins associated with the Salmonella type III secretion system (26–30). Nevertheless, the functional relevance of NLRP12 in response to Salmonella infection is unknown.
Here, we show that NLRP12 negatively regulates antibacterial host defense during Salmonella infection independent of inflammasomes. NLRP12 inhibited TLR-induced NF-κB activation by dampening phosphorylation of IκBα and ERK, consequently enhancing intracellular bacterial survival. Together, our work unveiled an NLRP12-dependent innate immune pathway that may be strategically exploited by S. typhimurium to persist and survive in the host.
Results
NLRP12 Deficiency Reduces Bacterial Burdens in Response to Salmonella Infection.
To characterize the role of NLRP12 during infection with S. typhimurium in vivo, cohorts of WT and Nlrp12−/− mice were infected by oral gavage, and bacterial burdens in the liver and spleen were analyzed at day 7 postinfection. Notably, liver and spleen homogenates from Nlrp12−/− mice infected with S. typhimurium contained significantly lower numbers of Salmonella colony-forming units compared with WT mice (Fig. 1A). Infected Nlrp12−/− mice had reduced spleen size, less microabscess in liver tissue, and a lower frequency of liver necrosis and liver histopathological scores compared with infected WT mice (Fig. 1 B–E). To examine whether the regulatory role of NLRP12 depended on the route of infection, we inoculated mice i.p. with S. typhimurium and found that, similar to our results with orally infected animals, Nlrp12−/− mice were significantly more resistant to S. typhimurium infection compared with WT controls (Fig. 1A). These results suggest that NLRP12 plays a critical role in negatively regulating bacterial burden and pathological damage in response to Salmonella infection.
Fig. 1.

Absence of NLRP12 protects mice from exacerbated Salmonella infection. WT and Nlrp12−/− mice were infected with 1 × 105 (oral route) or 5 × 103 (i.p. route) cfu of S. typhimurium. (A) Bacterial count in liver and spleen of WT and Nlrp12−/− mice was measured by colony-forming unit (CFU) assay after 5 d (i.p.) or 7 d (oral) postinfection. Gross anatomy of Salmonella-infected spleen (B) and liver (C) collected at day 7 after oral infection. The arrow in C indicates microabscesses in the liver. (D) H&E staining of Salmonella-infected liver collected at day 7 after oral infection. The arrows indicate microabscesses and a necrotic area in the liver. (Magnifications: 4× and 10×.) (E) Histological scores of Salmonella-infected liver at day 7. (F) WT and Nlrp12−/− mice were treated with a combination of broad-spectrum antibiotics for 3 wk or left untreated before oral infection with S. typhimurium (1 × 105 cfu per mouse). Survival of the mice was monitored. d, day. Experiments in A–E were performed without the use of antibiotics. Data are representative of at least three independent experiments (n = 8–10). Data represent the mean ± SEM. *P < 0.05.
The NLRP12-related NLR member NLRP6 was shown to regulate the gut microbiota composition to maintain intestinal homeostasis and systemic immune responses (31). We therefore investigated whether the gut microbiota plays a role in NLRP12-mediated susceptibility to Salmonella infection. To determine whether there are specific changes within the gut microbiota of Nlrp12−/− mice compared with WT mice, we used real-time quantitative PCR analysis to measure the levels of 16S rRNA genes of seven different groups of bacteria in the feces and found that Nlrp12−/− mice had significantly less Enterobacteriaceae and Lactobacillus/Enterococcus and a trend toward harboring a higher level of segmented filamentous bacteria (Fig. S1). We then treated WT and Nlrp12−/− mice with broad-spectrum antibiotics for 3 wk to deplete the gut microbiota before an oral challenge with S. typhimurium. Remarkably, we observed that Nlrp12−/− mice treated with antibiotics displayed a substantial delay in Salmonella-induced lethality compared with antibiotic-treated WT mice (Fig. 1F). This delay in mortality was antibiotic-dependent because Nlrp12−/− mice that had not been treated with antibiotics succumbed to Salmonella infection with similar kinetics as the corresponding WT controls (Fig. 1F). These results suggest that the gut microbiota protects against NLRP12-mediated susceptibility to Salmonella infection.
NLRP12-Mediated Inhibition of Proinflammatory and NO Production Enhances Salmonella Survival.
S. typhimurium is a facultative intracellular pathogen that infects and replicates within macrophages. We thus examined the role of NLRP12 in controlling S. typhimurium replication in macrophages. Real-time PCR analyses revealed that the levels of Nlrp12 transcript were relatively similar in bone marrow-derived macrophages (BMDMs) after 30 min or 2 h of infection with S. typhimurium. The transcript level increased rapidly after 8 h postinfection (Fig. S2). Notably, BMDMs from WT and Nlrp12−/− mice contained similar numbers of S. typhimurium after 2 and 8 h of infection; however, following 24 h of infection, Nlrp12−/− BMDMs contained significantly less viable S. typhimurium compared with WT BMDMs (Fig. 2A). The increased bactericidal activity observed in Nlrp12−/− BMDMs was not due to differences in the levels of Salmonella-induced cell death, because WT and Nlrp12−/− BMDMs released comparable levels of lactate dehydrogenase (LDH) into the cell culture medium following Salmonella infection (Fig. 2B).
Fig. 2.

NLRP12 attenuates antimicrobial killing of Salmonella in macrophages. (A and B) BMDMs from WT and Nlrp12−/− mice were infected with S. typhimurium [multiplicity of infection (MOI) of 5]. (A) Numbers of intracellular bacteria in Salmonella-infected cells were enumerated by colony-forming unit (CFU) assay. (B) Culture supernatants collected at 2, 8, and 24 h were analyzed for LDH release. (C) mRNA was isolated from Salmonella-infected BMDMs, and real-time quantitative PCR (qPCR) analysis of Il-6, Kc, and Tnf-α was performed. (D) Culture supernatant collected at 4 h after Salmonella infection was analyzed for IL-6, KC, and TNF-α by ELISA. (E) Real-time qPCR analysis of inducible nitric oxide synthase (iNOS) in BMDMs infected with S. typhimurium. (F) Cell lysates collected from BMDMs infected with S. typhimurium for 4 h were analyzed for caspase-1 (Casp-1) activation by Western blotting. (G) Culture supernatant collected from BMDMs infected with S. typhimurium for 4 h was analyzed for IL-1β by ELISA. Data represent the mean ± SD of triplicate wells. Data are representative of at least three independent experiments. *P < 0.05; **P < 0.01.
Inflammatory cytokines and reactive oxygen and nitrogen species produced by macrophages control Salmonella growth inside the phagosomal compartment of a host cell. To investigate the role of NLRP12 in regulating the production of proinflammatory cytokines, we analyzed the levels of IL-6, keratinocyte-derived chemokine (KC), and TNF-α mRNA transcripts and proteins and found that these were significantly higher in Salmonella-infected Nlrp12−/− BMDMs compared to wildtype BMDMs (Fig. 2 C and D). Consistently, Nlrp12−/− mice infected with S. typhimurium produced a significantly increased level of the chemokine KC and a trend toward higher IL-6 levels in the liver compared with WT mice (Fig. S3). In addition, the level of inducible NO synthase, which is responsible for NO production in macrophages, was significantly higher in Nlrp12−/− BMDMs infected with S. typhimurium compared with WT BMDMs (Fig. 2E). These results suggest that NLRP12 dampens the production of proinflammatory and antimicrobial molecules in macrophages infected with S. typhimurium.
Recently, Vladimer et al. (18) showed that NLRP12 activates caspase-1 and generates IL-18 production by assembling an inflammasome in response to infection by Y. pestis. To investigate whether NLRP12 triggers inflammasome activation in response to Salmonella infection, we infected WT and Nlrp12−/− BMDMs with S. typhimurium and examined caspase-1 proteolysis and IL-1β production. We did not observe differences in caspase-1 activation or IL-1β production in WT and Nlrp12−/− BMDMs infected with S. typhimurium (Fig. 2 F and G), suggesting that NLRP12-mediated regulation of Salmonella infection is independent of classical inflammasome activation. Therefore, NLRP12 may have differential roles in host defense against different bacterial species. Autophagy is another important cellular process responsible for the degradation of cytosolic organelles, proteins, and infectious agents, including S. typhimurium (32). To investigate whether there is NLRP12 dependency in autophagy-mediated killing, we measured the conversion of autophagy protein LC3I into LC3II in WT and Nlrp12−/− BMDMs infected with S. typhimurium by Western blot analysis. We found no evidence for NLRP12-mediated regulation of autophagy induction (Fig. S4). Collectively, our results suggest that NLRP12 inhibits the production of inflammatory cytokines and NO, which are required for efficient control of S. typhimurium infection.
Salmonella-Induced NLRP12-Mediated Inhibition of NF-κB and MAPK Activation by Targeting Phosphorylation of IκBα and ERK.
Production of proinflammatory cytokines and reactive oxygen species is largely regulated by NF-κB and MAPK signaling pathways. To understand whether NLRP12-mediated inhibition of inflammatory and antimicrobial molecules during Salmonella infection is regulated by NF-κB and MAPK signaling pathways in vivo, we infected WT and Nlrp12−/− mice with S. typhimurium and analyzed phosphorylation of IκBα (an inhibitor of NF-κB) and ERK in liver homogenates by Western blotting. Phosphorylation of IκBα and ERK activation was markedly increased in the liver of Nlrp12−/− mice infected with S. typhimurium relative to the levels observed in WT controls (Fig. 3A). Consistently, increased NF-κB and ERK phosphorylation was associated with increased production of KC and IL-6 in the liver of Nlrp12−/− mice (Fig. S3).
Fig. 3.

NLRP12 suppresses NF-κB and ERK activation during Salmonella infection. (A) Liver tissue samples collected from WT and Nlrp12−/− mice at day 7 after i.p. infection with S. typhimurium were homogenized, and lysates were analyzed for phosphorylated (P)-IκBα, IκBα, P-ERK, and ERK using Western blotting. Densitometric analysis of P-IκBα and P-ERK relative to IκBα and ERK, respectively, was performed. (B) BMDMs from WT and Nlrp12−/− mice were infected with S. typhimurium (MOI of 3), and cell lysates were used to analyze for P-IκBα, IκBα, P-ERK, and ERK by Western blotting. Densitometric analysis of P-IκBα relative to IκBα (C) and P-ERK relative to ERK (D) was performed. BMDMs were stimulated with S. typhimurium (E, MOI of 3) or LPS (F, 1 μg/mL), and cell lysates were analyzed for P-p105, P-p100, and P-IKKα/β by Western blotting. β-actin was used as a loading control. Data are representative of at least three independent experiments. Data represent the mean ± SEM. *P < 0.05; **P < 0.01.
We also observed that Nlrp12−/− BMDMs infected with S. typhimurium exhibited significantly increased levels of IκBα and ERK phosphorylation over 8 h of infection compared with WT BMDMs (Fig. 3 B–D). We further investigated whether the canonical or noncanonical NF-κB signaling pathway was inhibited by NLRP12 in response to Salmonella infection. Time-dependent phosphorylation of the canonical NF-κB mediators p105 and IkappaB kinase complex α/β (IKKα/β) was observed in WT BMDMs infected with S. typhimurium, whereas phosphorylation of p105 and IKKα/β was enhanced in Nlrp12−/− BMDMs (Fig. 3E). In contrast, phosphorylation of the noncanonical NF-κB mediator p100 was barely detected and did not appear significantly different between Salmonella-infected WT and Nlrp12−/− BMDMs (Fig. 3E), suggesting that NLRP12 may specifically affect canonical NF-κB signaling during S. typhimurium infection. In agreement, Salmonella LPS-induced phosphorylation of p105 and IKKα/β was increased in Nlrp12−/− BMDMs compared with WT BMDMs, whereas phosphorylation of p100 remained unchanged (Fig. 3F). To explore whether NLRP12 regulates ERK and NF-κB signaling independent of one another, we examined the levels of ERK and IκBα phosphorylation in LPS-stimulated Nlrp12−/− BMDMs that were pretreated with the ERK inhibitor U0126 or the IKK2 inhibitor SC-514. IKK2 inhibition led to decreased ERK activation in LPS-treated Nlrp12−/− BMDMs (Fig. 4A), whereas ERK inhibition failed to interfere with elevated IκBα activation in Nlrp12−/− BMDMs (Fig. 4A). These results suggest that increased ERK activation in LPS-stimulated Nlrp12−/− BMDMs represents a downstream effect of increased NF-κB signaling.
Fig. 4.

NLRP12-mediated inhibition of NF-κB and ERK dampens cytokine production and clearance of Salmonella in macrophages. (A) BMDMs from WT and Nlrp12−/− mice were treated with Salmonella LPS. Nlrp12−/− BMDMs were also treated with either the ERK inhibitor U0126 or the IKK2 inhibitor SC-514 before and during LPS stimulation. Cell lysates were collected and analyzed for P-ERK, ERK, P-IκBα, and IκBα by Western blotting. (B–D) WT and Nlrp12−/− BMDMs were treated with SC-514 during S. typhimurium infection. (B) Cell lysates collected after 2 and 24 h of infection were plated for bacterial colony-forming units (CFU). (C) Culture supernatant from BMDMs collected after 16 h of Salmonella infection was analyzed for IL-6 and KC by ELISA. (D) Nitrite in the culture supernatant collected after 24 h of infection was measured with Griess reagents. Data represent the mean ± SD of triplicate wells. Data are representative of at least three independent experiments. *P < 0.05.
Based on our results, we hypothesized that robust NF-κB activation in the absence of NLRP12 is important in suppressing Salmonella bacterial numbers. To address this, we pretreated WT and Nlrp12−/− BMDMs with SC-514 before and during S. typhimurium infection and quantified the levels of bacterial numbers in infected cells. We found increased bacterial counts in WT and Nlrp12−/− BMDMs compared with untreated controls (Fig. 4B). As expected, the IKK2 inhibitor reduced the production of the NF-κB–dependent proinflammatory cytokines IL-6 and KC (Fig. 4C). NF-κB and ERK signaling has also been implicated in the production of reactive nitrogen species, which have been shown to have bactericidal properties and are important for clearance of infectious agents (23, 33). SC-514–mediated inhibition of IKK2 resulted in significantly reduced nitrite levels in culture supernatants of WT and Nlrp12−/− BMDMs infected with S. typhimurium (Fig. 4D), confirming a role for NF-κB signaling in regulating Salmonella-induced NO production. These findings suggest a critical role for NLRP12 in dampening NF-κB activation during Salmonella infection, which consequently leads to excessive intracellular bacterial numbers in the host.
NLRP12-Mediated Suppression of NF-κB and ERK Activation Occurs in the Presence or Absence of Salmonella Flagellin and the Type III Secretion System.
Flagellin and the Salmonella pathogenicity islands (SPI-1 and SPI-2) of the type III secretion system are Salmonella virulence factors that contribute to host invasion and intracellular survival and have been shown to activate NAIPs and NLRC4 (26–30). We therefore investigated whether these Salmonella virulence factors are required for NLRP12-mediated regulation of NF-κB and ERK signaling. We infected WT and Nlrp12−/− BMDMs with isogenic mutants of S. typhimurium SL1344 lacking flagellin (ΔfliCΔfljB) or SPI-1 (ΔsipB) and SPI-2 (Δspi-2) of the type III secretion system. We found that the S. typhimurium ΔfliCΔfljB, ΔsipB, and Δspi-2 strains induced higher levels of ERK and IκBα phosphorylation in Nlrp12−/− BMDMs than in WT BMDMs (Fig. 5 A–C). These findings indicate that the lack of these Salmonella virulence factors does not reduce NLRP12-mediated suppression of NF-κB and ERK signaling. However, stimulation with Salmonella LPS alone induced elevated phosphorylation of IκBα and ERK in Nlrp12−/− BMDMs compared with WT BMDMs (Fig. 5D). Collectively, these results suggest that NLRP12 mediates the suppression of NF-κB and ERK activation in response to WT S. typhimurium or S. typhimurium lacking flagellin or the SPI-1 and SPI-2 type III secretion systems.
Fig. 5.
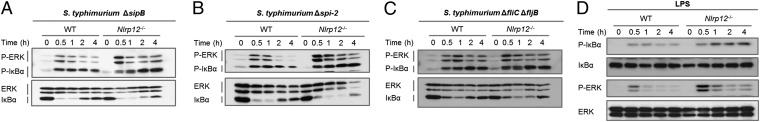
NLRP12 suppresses NF-κB and ERK activation in response to Salmonella LPS but not to flagellin or the SPI-1 or SPI-2 type III secretion system. BMDMs from WT and Nlrp12−/− mice were infected with S. typhimurium ΔsipB (A), Δspi-2 (B), and ΔfliCΔfljB (C) (MOI of 5) or with Salmonella LPS (D). Cell lysates were analyzed for P-ERK, ERK, P-IκBα, and IκBα by Western blotting. Data are representative of at least three independent experiments.
TLR-Independent Stimulation Fails to Trigger NLRP12-Mediated Inhibition of Canonical NF-κB Signals.
In addition to TLRs, the canonical NF-κB pathway can be activated via recognition of proinflammatory cytokine signals by their cognate receptors. To understand whether NLRP12 dampens NF-κB activation triggered by signals induced independent of TLRs, WT and Nlrp12−/− BMDMs were stimulated with IL-1β, IL-6, or TNF-α, followed by analysis of their phosphorylation dynamics of IκBα and ERK. Unlike the case with LPS, phosphorylation of IκBα and ERK was not markedly different in WT and Nlrp12−/− BMDMs in response to these cytokines (Fig. S5 A–C). The cytosolic sensor NOD2 activates NF-κB and MAPK by recognizing bacterial cell component muramyl dipeptide (MDP). When we investigated whether NLRP12 regulates NOD2-dependent activation of NF-κB and MAPK pathways, we observed a similar increase in phosphorylation of IκBα and ERK in WT and Nlrp12−/− BMDMs upon stimulation with MDP (Fig. S5D). In addition, we tested whether NLRP12-dependent inhibition of NF-κB and MAPK pathways is affected when BMDMs were stimulated with lymphotoxin (LT)α1β2 and LT homolog, which exhibits inducible expression, competes with HSV glycoprotein D for HveA and is expressed on T lymphocytes (LIGHT), which activate the LT-β receptor belonging to the TNF family (34). Both LTα1β2 and LIGHT induced similar levels of phosphorylated IκBα and ERK in WT and Nlrp12−/− BMDMs (Fig. S5 E and F). Together, these results indicate that NLRP12 negatively regulates proinflammatory and NO production by specifically targeting TLR-induced NF-κB and ERK signaling during S. typhimurium infection (Fig. 6).
Fig. 6.

Model for NLRP12-mediated inhibition of canonical NF-κB and ERK signaling pathways in response to Salmonella infection. Salmonella infection of host macrophages leads to the activation of TLRs, which induces signal transduction via MyD88 and/or Toll or interleukin-1 receptor domain-containing adaptor inducing interferon-β (Trif) to activate NF-κB and ERK. Phosphorylation of IκBα and subsequent degradation of IκBα (an inhibitor of NF-κB) enables the translocation of NF-κB into the nucleus. ERK, as part of the MAPK pathway, is also phosphorylated. Both NF-κB and ERK activation lead to transcription of genes encoding proinflammatory cytokines and antimicrobial molecules, including iNOS, TNF-α, IL-6, and KC. During S. typhimurium infection, NLRP12 responds to an unknown activator and mediates the inhibition of IκBα and ERK phosphorylation, which results in a reduction in the levels of proinflammatory cytokines and antimicrobial molecules and a decreased capacity to control the replication and spread of S. typhimurium in the host cell. LRRs, leucine-rich repeats; NBD, nucleotide-binding domain; PYD, pyrin domain; SCV, Salmonella-containing vacuole.
Discussion
NLRs regulate innate immune responses against microbial infection via multiple pathways, including formation of caspase-1–activating inflammasome platforms, activation of NF-κB and MAPK, and negative regulation of inflammatory signaling. Studies from our laboratory and others have shown that NLRP12 suppresses colon inflammation through inhibition of NF-κB signaling (12, 17). However, the role of NLRP12 during bacterial infection is less clear. Our study describes a role for NLRP12 in innate immune signaling during salmonellosis. Salmonella is an intracellular pathogen that survives and replicates inside macrophages and epithelial cells. In response to infection, host cells produce an array of proinflammatory cytokines to facilitate pathogen clearance, including IL-1α, IL-1β, TNF-α, IFN-γ, IL-12, IL-18, reactive nitrogen and oxygen species, and antimicrobial peptides (23, 33). Salmonella resists innate host defenses by producing stress resistance factors that prolong survival in host cells, such as production of bacterial antioxidant enzymes that reduce levels of oxygen species in infected cells (23, 33). Here, we showed that NLRP12 plays an anti-inflammatory role during S. typhimurium infection through inhibition of canonical NF-κB activation, which renders the host more susceptible to Salmonella infection (Fig. 6).
The activator of NLRP12 is currently unknown. Our work demonstrated that Salmonella LPS alone induced NLRP12-mediated inhibition of NF-κB activation, which may suggest that an endogenous activator of NLRP12 could potentially be up-regulated by LPS stimulation. Our findings add another layer of complexity to the interplay between host responses and microbial pathogenesis. In the context of Salmonella infection, our study identified NLRP12 as an NLR member that inhibits NF-κB activation. Previously, we showed that NLRP6 inhibited canonical NF-κB signaling following TLR activation and that this NLR promoted increased susceptibility to infection by S. typhimurium, Listeria monocytogenes, and Escherichia coli (8). The fact that multiple members of the NLR family function as negative regulators of NF-κB activation suggests that the excessive and overt inflammation that often results in host tissue damage must be finely controlled. Future studies may investigate whether NLRP6 and NLRP12 play redundant or synergistic roles in dampening immune responses in infectious diseases.
An intriguing observation arising from our study and others, however, is that the type of innate immune response triggered by NLRP12 is multifaceted and pathogen-specific. Indeed, one study showed that NLRP12 drives inflammasome-dependent responses following Yersinia infection (18). Another study found that TDP from M. tuberculosis and LPS from K. pneumoniae induced increased levels of TNF-α and IL-6 in Nlrp12−/− bone marrow-derived DCs more significantly than in WT cells, suggesting that NLRP12 plays a role in suppressing NF-κB signaling in response to these bacterial components (13). The underpinning factor that determines whether NLRP12 suppresses NF-κB activation (e.g., Salmonella, Mycobacterium, and Klebsiella infections) or activates caspase-1 (e.g., Yersinia infection) is an intriguing area for future research.
In conclusion, our results demonstrate that an innate immune signaling pathway defined by NLRP12-mediated inhibition of canonical NF-κB activation can be exploited by S. typhimurium, which leads to reduced production of proinflammatory and antimicrobial molecules. Activation of this regulatory mechanistic pathway allows prolonged persistence and survival of Salmonella in host tissues, and potentially mediates transmission of this organism to other susceptible hosts. Our work therefore suggests that modulation of NLRP12 expression and activation might be beneficial in the prevention and treatment of salmonellosis.
Materials and Methods
Detailed information is presented in SI Materials and Methods. Nlrp12−/− mice were generated as described previously (12). PCR primer sequences are listed in Table S1.
Supplementary Material
Acknowledgments
We thank Anthony Coyle, Ethan Grant, John Bertin (Millennium Pharmaceuticals), Gabriel Nuñez (University of Michigan), and Richard Flavell (Yale University) for the generous supply of mutant mice. M.H.Z. was supported by the Crohn's and Colitis Foundation of America. M.L. is supported by grants from the European Research Council (Grant 281600) and the Fund for Scientific Research-Flanders (Grants G030212N, 1.2.201.10.N.00, and 1.5.122.11.N.00). This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, part of the National Institutes of Health, under Award AR056296; the National Cancer Institute, part of the National Institutes of Health, under Award CA163507; and the National Institute of Allergy and Infectious Diseases, part of the National Institutes of Health, under Award AI101935 (all to T.-D.K.) and by The American Lebanese Syrian Associated Charities (ALSAC).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1317643111/-/DCSupplemental.
References
- 1.Kanneganti TD, Lamkanfi M, Núñez G. Intracellular NOD-like receptors in host defense and disease. Immunity. 2007;27(4):549–559. doi: 10.1016/j.immuni.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Girardin SE, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science. 2003;300(5625):1584–1587. doi: 10.1126/science.1084677. [DOI] [PubMed] [Google Scholar]
- 3.Chamaillard M, et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol. 2003;4(7):702–707. doi: 10.1038/ni945. [DOI] [PubMed] [Google Scholar]
- 4.Inohara N, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem. 2003;278(8):5509–5512. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 5.Girardin SE, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278(11):8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 6.Schneider M, et al. The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-κB. Nat Immunol. 2012;13(9):823–831. doi: 10.1038/ni.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allen IC, et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-κB signaling pathways. Immunity. 2011;34(6):854–865. doi: 10.1016/j.immuni.2011.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anand PK, et al. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature. 2012;488(7411):389–393. doi: 10.1038/nature11250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cui J, et al. NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell. 2010;141(3):483–496. doi: 10.1016/j.cell.2010.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang L, et al. PYPAF7, a novel PYRIN-containing Apaf1-like protein that regulates activation of NF-kappa B and caspase-1-dependent cytokine processing. J Biol Chem. 2002;277(33):29874–29880. doi: 10.1074/jbc.M203915200. [DOI] [PubMed] [Google Scholar]
- 11.Williams KL, Taxman DJ, Linhoff MW, Reed W, Ting JP. Cutting edge: Monarch-1: A pyrin/nucleotide-binding domain/leucine-rich repeat protein that controls classical and nonclassical MHC class I genes. J Immunol. 2003;170(11):5354–5358. doi: 10.4049/jimmunol.170.11.5354. [DOI] [PubMed] [Google Scholar]
- 12.Zaki MH, et al. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell. 2011;20(5):649–660. doi: 10.1016/j.ccr.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allen IC, et al. Characterization of NLRP12 during the in vivo host immune response to Klebsiella pneumoniae and Mycobacterium tuberculosis. PLoS ONE. 2013;8(4):e60842. doi: 10.1371/journal.pone.0060842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jéru I, et al. Identification and functional consequences of a recurrent NLRP12 missense mutation in periodic fever syndromes. Arthritis Rheum. 2011;63(5):1459–1464. doi: 10.1002/art.30241. [DOI] [PubMed] [Google Scholar]
- 15.Jéru I, et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc Natl Acad Sci USA. 2008;105(5):1614–1619. doi: 10.1073/pnas.0708616105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Macaluso F, et al. Polymorphisms in NACHT-LRR (NLR) genes in atopic dermatitis. Exp Dermatol. 2007;16(8):692–698. doi: 10.1111/j.1600-0625.2007.00589.x. [DOI] [PubMed] [Google Scholar]
- 17.Allen IC, et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κB signaling. Immunity. 2012;36(5):742–754. doi: 10.1016/j.immuni.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vladimer GI, et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity. 2012;37(1):96–107. doi: 10.1016/j.immuni.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Majowicz SE, et al. International Collaboration on Enteric Disease ‘Burden of Illness’ Studies The global burden of nontyphoidal Salmonella gastroenteritis. Clin Infect Dis. 2010;50(6):882–889. doi: 10.1086/650733. [DOI] [PubMed] [Google Scholar]
- 20.Barton Behravesh C, et al. FoodNet Working Group Deaths associated with bacterial pathogens transmitted commonly through food: Foodborne diseases active surveillance network (FoodNet), 1996-2005. J Infect Dis. 2011;204(2):263–267. doi: 10.1093/infdis/jir263. [DOI] [PubMed] [Google Scholar]
- 21.Vazquez-Torres A, et al. Toll-like receptor 4 dependence of innate and adaptive immunity to Salmonella: Importance of the Kupffer cell network. J Immunol. 2004;172(10):6202–6208. doi: 10.4049/jimmunol.172.10.6202. [DOI] [PubMed] [Google Scholar]
- 22.Talbot S, et al. Toll-like receptor 4 signalling through MyD88 is essential to control Salmonella enterica serovar typhimurium infection, but not for the initiation of bacterial clearance. Immunology. 2009;128(4):472–483. doi: 10.1111/j.1365-2567.2009.03146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eckmann L, Kagnoff MF. Cytokines in host defense against Salmonella. Microbes Infect. 2001;3(14-15):1191–1200. doi: 10.1016/s1286-4579(01)01479-4. [DOI] [PubMed] [Google Scholar]
- 24.Mariathasan S, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430(6996):213–218. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 25.Broz P, et al. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med. 2010;207(8):1745–1755. doi: 10.1084/jem.20100257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miao EA, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11(12):1136–1142. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Franchi L, et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol. 2006;7(6):576–582. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 28.Zhao Y, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477(7366):596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 29.Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature. 2011;477(7366):592–595. doi: 10.1038/nature10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang J, Zhao Y, Shi J, Shao F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci USA. 2013;110(35):14408–14413. doi: 10.1073/pnas.1306376110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elinav E, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145(5):745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Birmingham CL, Brumell JH. Autophagy recognizes intracellular Salmonella enterica serovar Typhimurium in damaged vacuoles. Autophagy. 2006;2(3):156–158. doi: 10.4161/auto.2825. [DOI] [PubMed] [Google Scholar]
- 33.Shiloh MU, et al. Phenotype of mice and macrophages deficient in both phagocyte oxidase and inducible nitric oxide synthase. Immunity. 1999;10(1):29–38. doi: 10.1016/s1074-7613(00)80004-7. [DOI] [PubMed] [Google Scholar]
- 34.Ware CF. Targeting lymphocyte activation through the lymphotoxin and LIGHT pathways. Immunol Rev. 2008;223:186–201. doi: 10.1111/j.1600-065X.2008.00629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.