

Significance
Plant cell walls contain significant amounts of the polysaccharides cellulose and hemicellulose, which can be depolymerized by enzymes to sugars and upgraded to renewable fuels and chemicals. Traditionally, enzymes for biomass depolymerization were based on naturally occurring hydrolytic enzymes, until the recent discovery of another natural enzymatic paradigm for carbohydrate deconstruction. Namely, lytic polysaccharide monooxygenases (LPMOs), long thought to be hydrolases or carbohydrate-binding modules, were revealed to be oxidative, copper-containing enzymes. These enzymes are receiving significant attention as they could revolutionize biomass deconstruction to upgradeable intermediates for renewable energy applications. Here, we apply quantum mechanical calculations to elucidate the oxidative reaction mechanism to offer predictions into how LPMOs function.
Keywords: C–H activation, copper monooxygenase, GH61, CBM33, biofuels
Abstract
Lytic polysaccharide monooxygenases (LPMOs) exhibit a mononuclear copper-containing active site and use dioxygen and a reducing agent to oxidatively cleave glycosidic linkages in polysaccharides. LPMOs represent a unique paradigm in carbohydrate turnover and exhibit synergy with hydrolytic enzymes in biomass depolymerization. To date, several features of copper binding to LPMOs have been elucidated, but the identity of the reactive oxygen species and the key steps in the oxidative mechanism have not been elucidated. Here, density functional theory calculations are used with an enzyme active site model to identify the reactive oxygen species and compare two hypothesized reaction pathways in LPMOs for hydrogen abstraction and polysaccharide hydroxylation; namely, a mechanism that employs a η1-superoxo intermediate, which abstracts a substrate hydrogen and a hydroperoxo species is responsible for substrate hydroxylation, and a mechanism wherein a copper-oxyl radical abstracts a hydrogen and subsequently hydroxylates the substrate via an oxygen-rebound mechanism. The results predict that oxygen binds end-on (η1) to copper, and that a copper-oxyl–mediated, oxygen-rebound mechanism is energetically preferred. The N-terminal histidine methylation is also examined, which is thought to modify the structure and reactivity of the enzyme. Density functional theory calculations suggest that this posttranslational modification has only a minor effect on the LPMO active site structure or reactivity for the examined steps. Overall, this study suggests the steps in the LPMO mechanism for oxidative cleavage of glycosidic bonds.
Carbohydrates are the most diverse set of biomolecules, and thus, many enzyme classes have evolved to assemble, modify, and depolymerize carbohydrates, including glycosyltransferases, glycoside hydrolases, carbohydrate esterases, and polysaccharide lyases (1). Recently, a new enzymatic paradigm was discovered that employs copper-dependent oxidation to cleave glycosidic bonds in polysaccharides (2–13). These newly classified enzymes, termed lytic polysaccharide monooxygenases (LPMOs), broadly resemble other copper monooxygenases and some hydroxylation catalysts (14–21).
The discovery that LPMOs use an oxidative mechanism has attracted interest both because it is a unique paradigm for carbohydrate modification that employs a powerful C–H activation mechanism, and also because LPMOs synergize with hydrolytic enzymes in biomass conversion to sugars because they act directly on the crystalline polysaccharide surface without the requirement for depolymerization (4, 22, 23), making them of interest in biofuels production. LPMOs were originally characterized as Family 61 glycoside hydrolases (GH61s, reclassified as auxiliary activity 9, AA9) or Family 33 carbohydrate-binding modules (CBM33s, reclassified as AA10), which are structurally similar enzymes found in fungi and nonfungal organisms (22), respectively. In 2005, Vaaje-Kolstad et al. described the synergism (24) of a chitin-active CBM33 (chitin-binding protein, CBP21) with hydrolases, but the mechanism was not apparent. Harris et al. demonstrated that a GH61 boosts hydrolytic enzyme activity on lignocellulosic biomass (2). Vaaje-Kolstad et al. subsequently showed that CBP21 employs an oxidative mechanism to cleave glycosidic linkages in chitin (4).
Following these initial discoveries, multiple features of LPMOs have been elucidated. LPMOs use copper (5–7) and produce either aldonic acids or 4-keto sugars at oxidized chain ends, believed to result from hydroxylation at the C1 or C4 carbon, respectively. Hydroxylation at the C1 carbon is proposed to spontaneously undergo elimination to a lactone followed by hydrolytic ring opening to an aldonic acid, whereas hydroxylation and elimination at C4 yields a 4-keto sugar at the nonreducing end (5–12). The active site is a mononuclear type(II) copper center ligated by a “histidine brace” (5, 12), comprising a bidentate N-terminal histidine ligand via the amino terminus and an imidazole ring nitrogen atom and another histidine residue also via a ring nitrogen atom. Hemsworth et al. reported a bacterial LPMO structure wherein the active site copper ion was photoreduced to Cu(I) (12), and Aachmann et al. demonstrated that Cu(I) binds with higher affinity than Cu(II) in CBP21 (13). A structural study of a fungal LPMO revealed an N-terminal methylation on a nitrogen atom in the imidazole ring of unknown function (5), but some LPMOs are active without this modification (6, 11). LPMOs require reducing agents for activity such as ascorbate (2–8, 10–12), and cellobiose dehydrogenase (CDH), a common fungal secretome component, can potentiate LPMO activity in lieu of a small-molecule reducing agent (7, 8).
Overall, many structural and mechanistic insights have been reported since the discoveries that LPMOs are oxidative enzymes (4–10). However, many questions remain regarding LPMO function (22, 25). Here, we examine the LPMO catalytic mechanism with density functional theory (DFT) calculations on an active site model (ASM) of a fungal LPMO. We seek to (i) understand the identity of the reactive oxygen species (ROS), (ii) compare two hypothesized catalytic mechanisms, and (iii) examine the role of N-terminal methylation in catalysis.
Results
Development of the ASM.
We used the structure from a Thermoascus aurantiacus LPMO (Protein Data Bank, PDB ID code 2YET) to build the ASM (5). Fig. 1A shows a model of the T. aurantiacus LPMO on the cellulose surface. We developed the ASM based on fungal LPMO sequence alignments (3, 9, 11) and DFT geometry optimizations. The initial ASM (before O2 or cellulose are considered) includes His1, His86, His164, Gln173, Tyr175, three crystallographic water molecules, and the copper ion (Fig. 1B). The aforementioned residues best maintain the shape and electronic properties of the metal binding site while providing a computationally tractable system (SI Appendix, Fig. S1 and Table S1). His1 and His86 directly coordinate copper in the equatorial plane to form the histidine brace (5, 12). His164 is conserved, hydrogen bonds to two ordered water molecules, and forms a stabilizing π–π interaction with His86. Gln173 is conserved in fungal LPMOs (5, 12), which hydrogen bonds to the Tyr175 hydroxyl group, the latter of which is axially coordinated to copper. All further calculations use this ASM with the addition of O2 and substrate, and water molecules where needed. As described in Methods, all geometry optimizations in the main text were conducted with the B3LYP functional and the 6–31G(d) basis set. Additional optimizations with the M06-L functional and the 6–31G(d) basis set are presented in SI Appendix. Single point energy calculations were conducted on the final overall barriers for the two mechanisms with the 6–311++G(d,p) basis set. As the LPMO mechanism likely requires spin-crossing events, open shell singlet and triplet species along the catalytic reaction coordinate were fully optimized at the spin-unrestricted level.
Fig. 1.

ASM of the T. aurantiacus LPMO and the initial ROS structure. (A) Illustration of the T. aurantiacus LPMO (gray) with cellulose (green) with the LPMO active site and a cellulose chain in stick. (B) LPMO active site. Residues in stick format are ASM components including three water molecules (red spheres). (C) ASM residues from the structure (2YET, pink stick format with waters in red) and the DFT geometry-optimized structure of initial state [Cu(II)] (blue stick format with water molecules in stick). (D) DFT geometry-optimized structure of the reduced [Cu(I)] state. (E) Stable η1-Cu(II)-superoxo species, [Cu(II)]-O-O⋅. Hydrogen atoms are omitted for clarity (SI Appendix, Fig. S3). DFT optimizations were performed with UB3LYP/6–31G(d) with CPCM diethyl ether.
Enzyme Activation and Initial O2 Binding to the LPMO Active Site.
From the enzyme ASM, we optimized the system geometry with a Cu(II) state (5, 12) (denoted [Cu(II)]) and a reduced Cu(I) state (denoted [Cu(I)]), the latter of which is the hypothesized oxidation state for O2 activation (7, 10, 13). Table 1 lists several bond distances, copper coordination geometry, partial atomic charges (20), and the rmsd from the structure for these optimized states (atom labels in Fig. 1B). The initial state with Cu(II) is shown in Fig. 1C (blue) threaded onto the structure (pink), which closely agrees (rmsd = 0.37 Å for heavy atoms, which is quite good agreement for “theozyme” ASMs) (26). The coordinating nitrogen atoms in the histidine brace are within 1.97–2.08 Å to copper, with one equatorial water molecule. The Tyr175 hydroxyl group (3.08 Å) and a water molecule (2.33 Å) occupy the axial positions for a distorted octahedral geometry with Jahn–Teller distortion, as expected. The optimized geometry for the reduced state is shown in Fig. 1D. Copper is coordinated by the histidine brace (1.91–2.14 Å) and water (2.19 Å), whereas the hydroxyl group of the Tyr175 is located 4.37 Å from the metal, imparting the expected tetrahedral geometry of a Cu(I) center. The atomic charges of copper are 1.48 and 0.92 for the formal Cu(II) and Cu(I) oxidation states, respectively.
Table 1.
Structural and electronic parameters for the ASM as a function of copper oxidation and O2 binding
| Property | 2YET | [Cu(II)] | [Cu(I)] | [Cu(II)-O-O⋅] |
| Cu-Nδ (His1), Å | 2.10 | 1.97 | 1.91 | 1.977 |
| Cu-N (His1), Å | 2.43 | 2.08 | 2.14 | 2.177 |
| Cu-Nε (His86), Å | 2.32 | 1.99 | 1.93 | 1.982 |
| Cu charge | N/A | 1.48 | 0.92 | 1.38 |
| Rmsd, Å | N/A | 0.37 | 0.77 | 0.53 |
| Coordination geom./num. | DO/6 | DO/6 | T/4 | TB/5 |
[Cu(II)] and [Cu(I)] represent the initial resting state and the reducing state, respectively. Cu charge represents Natural Population Analysis charges. Rmsd was calculated only with heavy atoms based on 2YET. DO, distorted octahedral; T, tetrahedral; TB, trigonal bipyramidal.
It is hypothesized that O2 binds to [Cu(I)] to form a Cu(II)-superoxo ([Cu(II)]-O-O⋅) complex (7, 9, 10), but the geometry and coordination of the copper–oxygen complex is unknown. LPMO geometries were DFT optimized with O2 bound in configurations constructed by varying the O1W-Cu-O1-O2 dihedral angle in 30° increments for both end-on (η1) and side-on (η2) configurations in both singlet and triplet states (SI Appendix, Fig. S2). The calculations show that the most stable optimized structure of the [Cu(II)]-O-O⋅ complex is an end-on (η1) configuration (Fig. 1E, Cu-O1-O2 angle = 107.5°). The triplet spin state is the ground state, and is 4.6 kcal/mol more stable than the singlet structure, in agreement with similar end-on copper-O2 adducts (27). The computed free O2 singlet–triplet (S–T) energy gap of 20.9 kcal/mol also agrees well with the experimental value of 20.5 kcal/mol (28). Upon complexation, overlap with the filled Cu 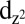 orbital raises the energy of the in-plane O-Oπ* orbital and reduces the S–T gap, although evidently there is not significant separation between frontier orbitals because (as expected for η1 complexes) the triplet is still favored by 4.6 kcal/mol (29). In the complex, the charge transfer of −0.5e from Cu to O2 results in a lengthening of the O-O bond length to 1.31 Å, close to the ideal bond length for Cu(II) superoxide of 1.33 Å (30). There is also significant biradicaloid character as shown by the B3LYP spin densities of 0.59 (O1), 0.67 (O2), and 0.60 (Cu) (SI Appendix, Table S2). The η1-superoxide species, the three nitrogen atoms from the histidine brace, and the equatorially bound water molecule (O1W) form a trigonal bipyramidal geometry around copper. From this species, DFT calculations are used to examine two mechanisms.
orbital raises the energy of the in-plane O-Oπ* orbital and reduces the S–T gap, although evidently there is not significant separation between frontier orbitals because (as expected for η1 complexes) the triplet is still favored by 4.6 kcal/mol (29). In the complex, the charge transfer of −0.5e from Cu to O2 results in a lengthening of the O-O bond length to 1.31 Å, close to the ideal bond length for Cu(II) superoxide of 1.33 Å (30). There is also significant biradicaloid character as shown by the B3LYP spin densities of 0.59 (O1), 0.67 (O2), and 0.60 (Cu) (SI Appendix, Table S2). The η1-superoxide species, the three nitrogen atoms from the histidine brace, and the equatorially bound water molecule (O1W) form a trigonal bipyramidal geometry around copper. From this species, DFT calculations are used to examine two mechanisms.
Oxidative Mechanism with a Cu(II)-Superoxo ROS.
We first examine a “superoxo mechanism” wherein a Cu(II)-superoxo (Cu(II)-O-O⋅↔Cu(III)-O-O−) is the ROS for hydrogen abstraction to form a hydroperoxo species, which then hydroxylates the C1 (or C4) carbon of a glucosyl ring (Fig. 2A). The + superscript denotes the overall system charge and ‡ denotes a transition state (TS). Fig. 2 shows the results describing oxidative attack at C1, and the C4 oxidation results are shown in SI Appendix, Fig. S4. The mechanism starts with [Cu(I)] as the reactant; the energy diagram is reported with respect to [Cu(I)] and O2. The superoxo mechanism (Fig. 2A) begins with the formation of the superoxo intermediate ([Cu(II)]-O-O⋅). Cellobiose, which was chosen to represent the substrate, coordinates to the enzyme [Cu(II)]-O-O⋅ to form 1 (the order of substrate and O2 complexation is discussed below). Restraints are placed on selected carbons (C2 and C5 for both glucosyl units and C4 of nonreducing end and C1 of reducing end units, respectively) to maintain the cellobiose geometry as it would be in the cellulose surface. As the enzyme–substrate geometry is unknown, three enzyme–substrate complexes were tested for C1 and C4 oxidation each by translating the [Cu(II)]-O-O⋅ system along the cellulose chain at distances wherein the oxygen could attack at C1 or C4. Full energy landscapes were computed (SI Appendix, Table S3) for these geometries for both C1 and C4 hydroxylation, and all are within 1–2 kcal/mol for the activation barriers.
Fig. 2.

Superoxo mechanism for polysaccharide hydroxylation by a fungal LPMO. (A) Proposed oxidative mechanism with a Cu(II)-superoxo ([Cu(II)]-O-O⋅) ROS for C1 hydroxylation. C4 hydroxylation results are shown in SI Appendix, Fig. S4. Intermediates and TSs (‡) are shown for each geometry considered. Three enzyme–substrate configurations were considered for both C1 and C4 hydroxylation (SI Appendix, Table S3). (B) Potential energy surface for C1 hydroxylation. Energies in kcal/mol. (C) B3LYP/6–31G(d) optimized structures of intermediates and TSs for the Cu(II)-superoxo ROS mechanism.
Fig. 2B shows the energy landscape for the superoxo mechanism for C1 hydroxylation. As shown in Fig. 2C, hydrogen atom abstraction from the C1 carbon by [Cu(II)]-O-O⋅ proceeds via TS-1 to form intermediate 2 with an activation energy of 34.9 kcal/mol. Hydroxylation of the C1 radical center formed in this C–H abstraction step is performed by 2. Homolytic cleavage of the O-O bond occurs in TS-2 to form 3 with an activation energy of 15.0 kcal/mol relative to 2. Intermediate 3 is a [Cu(II)]-O⋅ species and is 48.7 kcal/mol lower in energy than 2. Cleavage of the glycosidic bond from the hydroxylated cellobiose will occur via elimination (SI Appendix, Fig. S5) (7, 10). The reduction of the [Cu(II)]-O⋅ group in 3 by ascorbic acid (converted to dehydroascorbic acid) is favorable by 17.2 kcal/mol, which returns the enzyme to the initial [Cu(I)] species to complete the catalytic cycle. Ascorbic acid was chosen as a model reducing agent with experimental precedent (4), and we note that we lump the proton transfer (PT) and electron transfer (ET) events into a single step wherein we only consider thermodynamics, not the barriers for these steps, as the ET and PT pathways are unknown. The [Cu(II)]-O-O⋅ ROS is a triplet in its ground state, whereas all singlet and triplet states are essentially isoenergetic (except 3) for subsequent intermediates and TSs along the reaction coordinate. Product 4 is a closed shell singlet and energies for 3 in the triplet and open shell singlet states are calculated to be −39.6 and −46.1 kcal/mol, respectively, so spin crossover from the triplet to open shell singlet state occurs after TS-2. The overall activation barrier for this mechanism is 43.0 kcal/mol (B3LYP/6–31G(d)) or 39.9 kcal/mol with B3LYP/6–311++G(d,p)//B3LYP/6–31G(d). M06-L calculations for this mechanism also exhibit the same overall barrier (43.0 kcal/mol, SI Appendix, Fig. S6). Also, oxidation at the C1 carbon is computed to be marginally more reactive than the C4 carbon by 4.3 kcal/mol in the overall activation barrier (SI Appendix, Fig. S5).
Oxidative Mechanism with a Cu(II)-Oxyl ROS.
Considering the high energy barrier for the Cu(II)-superoxo mechanism, a more oxidatively powerful ROS will likely be required for polysaccharide hydroxylation. Thus, we consider C–H oxidation by a Cu(II)-oxyl ROS, dubbed the “oxyl mechanism” (Fig. 3) as this species has been previously predicted to exhibit a more strongly oxidative character than Cu(II)-superoxo (31, 32). We hypothesize that a Cu(II)-oxyl ROS, [Cu(II)]-O⋅, could be formed by direct reduction of a superoxo species, [Cu(II)]-O-O⋅, by ascorbic acid to form water. Cu(II)-oxyl (Cu(II)-O⋅↔Cu(III)-O−) species have been implicated in several bioinorganic mechanisms to date (20, 33, 34). From this system, we test two different geometries for oxidative attack at the C1 and C4 carbons to evaluate different enzyme–substrate poses (SI Appendix, Fig. S7). The computed energy difference between [Cu(II)]-O-O⋅ and [Cu(II)]-O⋅ is 3.1 kcal/mol. As with the previous mechanism, we lump the ET and PT steps into a single step. SI Appendix, Fig. S8 shows the thermodynamics of the single ET and PT steps (assuming these are coupled), but again we do not explicitly consider barriers, as the ET and PT pathways and order of transfer are unknown. C–H abstraction from the C1 carbon of cellobiose by [Cu(II)]-O⋅ occurs via TS-5 (Fig. 3C), producing a [Cu(II)]-OH complex and a carbon-centered radical on cellobiose (Fig. 3C, 6). TS-5 lies 15.4 kcal/mol higher in energy than 5 (Fig. 3C), which, compared with the 34.9 kcal/mol computed for hydrogen abstraction by a [Cu(II)]-O-O⋅ species, shows the [Cu(II)]-O⋅ ROS to be significantly more reactive. The [Cu(II)]-OH complex transfers the OH group via an oxygen-rebound mechanism to the cellobiose via TS-6 (Fig. 3C) to form 4′ (Fig. 3C), hydroxylating cellobiose, and reforming the [Cu(I)] complex to complete the cycle. Cleavage of the glycosidic bond after hydroxylation occurs via elimination, which occurs spontaneously or is facilitated by an acid (7). We calculated the relative energy of the intermediates along the elimination and hydrolysis pathway (SI Appendix, Fig. S5) and the overall reaction is downhill to the products after substrate hydroxylation (ΔE = −6.3 kcal/mol).
Fig. 3.

Oxyl mechanism for polysaccharide hydroxylation by a fungal LPMO. (A) Proposed oxidative mechanism with a [Cu(II)]-O⋅ ROS for C1 hydroxylation. C4 hydroxylation results are shown in SI Appendix, Fig. S4. Intermediates and TSs (‡) are shown for each geometry, with an overall system charge of +1. Two enzyme–substrate configurations were considered for both C1 and C4 hydroxylation (SI Appendix, Fig. S7). (B) Potential energy surface for C1 hydroxylation. Energies are in kcal/mol. (C) B3LYP/6–31G(d) optimized structures of intermediates and TSs for the [Cu(II)]-O⋅ ROS mechanism.
The [Cu(II)]-O⋅ ROS is a triplet in its ground state (the open shell singlet is computed with B3LYP to be 4.0 kcal/mol higher), whereas for all subsequent radical pairs along the reaction coordinate, open shell singlet and triplet states are essentially isoenergetic. The 4′ is a closed shell singlet, so spin crossover from the triplet to singlet ground state occurs after TS-5. The 4′ is 39.5 kcal/mol lower in energy than the [Cu(II)]-O⋅ ROS. Comparing both hypothesized mechanisms involving different ROSs, for the Cu(II)-superoxo species C–H abstraction followed by OH transfer is rate limiting and lies 43.0 kcal/mol above the ROS intermediate, whereas for the Cu(II)-oxyl, C–H abstraction is rate limiting and lies 18.8 kcal/mol or 24.0 kcal/mol with B3LYP/6–311++G(d,p)//B3LYP/6–31G(d) above the relevant ROS. Our calculations thus suggest that the active catalyst is Cu(II)-oxyl, because this ROS yields a reasonable energy barrier at biologically relevant temperatures. This perspective is unaltered whether a different level of theory is used, as the M06-L functional results in an 18.1 kcal/mol overall activation barrier (SI Appendix, Fig. S6), and different enzyme–substrate complex geometries (SI Appendix, Fig. S7) lie within 4.7 and 3.3 kcal/mol at the C1 and C4 carbons, respectively. Selected structural and electronic parameters are provided for both mechanisms in SI Appendix, Tables S4 and S5.
Role of the N-Terminal Methylation in the LPMO Reaction Mechanism.
We also examine the impact of methylation on the N-terminal histidine. This posttranslational modification (PTM) has been hypothesized to modify LPMO reactivity (5, 7, 12). We reexamined both mechanisms without N-methylation on the N-terminal histidine. The rate-limiting C–H abstraction step of [Cu(II)]-O⋅ ROS has a marginally higher activation barrier in the absence of N-methylation by 1.1 kcal/mol (B3LYP), whereas the [Cu(II)]-O-O⋅ ROS has the same activation barrier (SI Appendix, Fig. S9). This result suggests that the N-terminal N-methylation does not affect the LPMO catalytic activity by means ascertainable with our approach.
Discussion
The utilization of O2 to modify singlet ground state organic substrates, which are central reactions in aerobic life, is typically predicated upon the employment of metalloenzymes to circumvent spin-forbidden transitions in the triplet ground state of O2. Significant work has been conducted to understand the molecular basis of metal-activated oxidation, as these reactions are important both in cell biology and industrial chemistry (14–21). Glycosidic linkages in polysaccharides are particularly strong bonds relative to, for example, DNA or peptide bonds (35). For polysaccharide deconstruction, which is important for the global carbon cycle and renewable fuels production, glycoside hydrolases or nonspecific oxidative species using Fenton chemistry were thought to be the primary carbohydrate depolymerization paradigms until the discovery of LPMOs (4, 5). The results obtained in this study with a theozyme model of a fungal LPMO (AA9) suggest a mechanism using a copper-oxyl ROS and an oxygen-rebound mechanism based on comparison of a superoxo and an oxyl mechanism. The copper-oxyl species has been only directly detected in the gas phase to date (36), but it has been predicted in theoretical studies to be the ROS in other enzymatic and synthetic hydroxylation reactions that require significant oxidative power, such as methane (37) or arene hydroxylation (31), in dopamine-β-monooxygenase (38), and in peptidylglycine α-hydroxylating monooxygenase (39).
Phillips and coworkers (7, 9, 10) proposed an LPMO mechanism that employs hydrogen abstraction and formation of both a radical as copper-oxyl and a radical on the substrate carbon of oxidative attack, which recombine to form the hydroxylated substrate. This proposed mechanism was the starting point for the development of the superoxo mechanism hypothesized here and is equivalent in the first steps for hydrogen abstraction. However, a barrier of 34.9 kcal/mol is required for the hydrogen abstraction step for a superoxo ROS (Fig. 2). As this barrier is higher than the overall (two-step) barrier found for the oxyl mechanism of 18.8 kcal/mol (Fig. 3), it is likely that the previously proposed mechanism is unfavorable relative to the oxyl mechanism presented in this work. Additionally, the study here employs a reducing agent wherein two electrons and two protons are required in series in the catalytic cycle, which is different from the previously proposed mechanism (7, 9, 10) wherein single ET events occur at different parts of the catalytic cycle. We stress that the study here does not preclude the occurrence of single ET events, but rather just hypothesizes that they occur in series as shown in SI Appendix, Fig. S8, which is compatible with the single ET mechanisms of CDH enzymes (7).
The primary focus of this study is to predict the mechanism used by LPMOs for polysaccharide hydroxylation. Still, many questions remain for a comprehensive understanding of LPMO action, and the model used here is limited in terms of the questions able to be addressed. First, regioselectivity for the C1 or C4 carbon of cellulose has been observed (4–11), and LPMOs are specific either for cellulose or chitin (4–12). However, fungal LPMO active sites are quite conserved, and thus it is likely that motifs on the binding surface (13) distant from the active site dictate regioselectivity and substrate specificity (9, 11). Additionally, the ET step was not explicitly considered. To compute the ET barrier requires knowledge of the ET pathway from a reducing agent to the LPMO, which has not yet been elucidated. It has been previously suggested that ascorbic acid transfers a single hydrogen atom rather than via uncoupled ET and PT steps (40), and SI Appendix, Fig. S8 shows the relative energies of the system along these individual transfer steps for the oxyl mechanism. Also, the model used here does not explicitly address the order of oxygen binding to the LPMO active site relative to when the enzyme–substrate complex is formed. For example, in Figs. 2A and 3A, the substrate enters the reaction scheme after oxygen binds to the copper center, and in the latter case, after the reducing agent converts the copper-superoxo species to a copper-oxyl species. However, the potential energy landscape is not significantly altered for either the superoxo mechanism or the oxyl mechanism if the substrate enters the reaction before O2 activation (SI Appendix, Fig. S10). Lastly, the model we use here treats the orientation of the LPMO active site along the cellobiose substrate in the same putative orientation as a Family 1 CBM. This assumption stems from the similarity of LPMO and Family 1 CBM binding faces (2), as discussed previously (11).
Another question of keen interest is the difference in nonfungal LPMOs (AA10) and fungal LPMOs (AA9). Hemsworth et al. (25) described differences between these enzyme families that may lead to differences in catalytic mechanism. In particular, nonfungal LPMOs exhibit an alanine residue in the active site that may modulate the reactivity of the enzyme or the identity of the ROS. SI Appendix, Fig. S11 shows a structural alignment of the superoxide species (Fig. 1E) with two CBM33 structures [PDBs 2YOX and 4ALT, both Cu(I)] (12), which suggest that superoxide may clash with the alanine side chain, perhaps further suggesting that CBM33 oxygen activation geometry will be different from that of fungal LPMOs.
Conclusions
LPMOs represent an important unique discovery in the multitude of carbohydrate-active enzymes due to their prevalence in nature and their paramount importance to cost-effective lignocellulosic biomass deconstruction (1, 2). Here we applied DFT calculations to examine two catalytic mechanisms that suggest that fungal LPMOs exhibit a copper-oxyl mediated, oxygen rebound mechanism. The significant oxidative power of this predicted ROS for polysaccharide hydroxylation is concomitant with the extremely high bond strength of glycosidic linkages in polysaccharides (35). Certainly, many questions remain that will require myriad experimental and computational tools to fully characterize the mechanisms of these fascinating enzymes.
Methods
The ASM was taken from a T. aurantiacus LPMO structure (PDB 2YET) (5). Enzyme–substrate starting geometries were taken from molecular dynamics simulations of a similar LPMO on cellulose and the perpendicular distance between the flat faces of the enzyme and substrate was kept fixed while moving the enzyme in the parallel plane for optimal oxidative attack geometries (11). DFT calculations were performed with Gaussian 09 (41). All geometry optimizations were conducted using the hybrid GGA B3LYP functional in the spin-unrestricted Kohn–Sham approach, with the 6–31G(d) basis set for all atoms. This functional is routinely used to study copper–oxygen complexes (42, 43). Single point calculations for the mechanisms were also conducted on the geometry-optimized structures with the B3LYP functional and the 6–311++G(d,p) basis set to show the differences in the overall barriers. [Cu(I)] complexes generate open shell singlet or triplet products due to the addition of triplet O2, and thus the standard Kohn–Sham DFT is not directly applicable (44). We applied the Yamaguchi broken spin symmetry procedure (45) to compute the energy of the spin-purified low-spin (LS) state (46). We calculated two possible spin states if the complexes are open shell and found the energy of triplet products is always lower than the energy of the spin-purified LS state, unless otherwise stated. Dispersion corrected DFT results are included in SI Appendix, Fig. S12. Computations of vibrational frequencies, which include zero-point vibrational frequencies or one imaginary frequency were verified to determine the reactants and products related to each transition state. A conductor-like polarizable continuum model (CPCM) with ε = 4.3 (diethyl ether as solvent) was applied for the solvation effects. Full geometry optimizations with the meta-GGA M06-L functional have also been evaluated to compare B3LYP functional in SI Appendix, Fig. S6. Further methodological details are described in SI Appendix.
Supplementary Material
Acknowledgments
We thank Vincent Eijsink for a critical reading of the manuscript and for constructive comments; S.K. and G.T.B. thank the US Department of Energy BioEnergy Technologies Office for funding; J.S. and M.S. thank the Faculty for Natural Resources and Agriculture at the Swedish University of Agricultural Sciences through the research program MicroDrivE; and R.S.P. thanks the Oxford University Press John Fell Fund and the Royal Society (RG110617) for funding. Computer time was provided by the Texas Advanced Computing Center under the National Science Foundation Extreme Science and Engineering Discovery Environment Grant MCB-090159 and by the National Renewable Energy Laboratory Computational Sciences Center.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1316609111/-/DCSupplemental.
References
- 1.Levasseur A, Drula E, Lombard V, Coutinho PM, Henrissat B. Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol Biofuels. 2013;6(1):41. doi: 10.1186/1754-6834-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harris PV, et al. Stimulation of lignocellulosic biomass hydrolysis by proteins of glycoside hydrolase family 61: Structure and function of a large, enigmatic family. Biochemistry. 2010;49(15):3305–3316. doi: 10.1021/bi100009p. [DOI] [PubMed] [Google Scholar]
- 3.Karkehabadi S, et al. The first structure of a glycoside hydrolase family 61 member, Cel61B from Hypocrea jecorina, at 1.6 A resolution. J Mol Biol. 2008;383(1):144–154. doi: 10.1016/j.jmb.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 4.Vaaje-Kolstad G, et al. An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science. 2010;330(6001):219–222. doi: 10.1126/science.1192231. [DOI] [PubMed] [Google Scholar]
- 5.Quinlan RJ, et al. Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc Natl Acad Sci USA. 2011;108(37):15079–15084. doi: 10.1073/pnas.1105776108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forsberg Z, et al. Cleavage of cellulose by a CBM33 protein. Protein Sci. 2011;20(9):1479–1483. doi: 10.1002/pro.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phillips CM, Beeson WT, Cate JH, Marletta MA. Cellobiose dehydrogenase and a copper-dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa. ACS Chem Biol. 2011;6(12):1399–1406. doi: 10.1021/cb200351y. [DOI] [PubMed] [Google Scholar]
- 8.Langston JA, et al. Oxidoreductive cellulose depolymerization by the enzymes cellobiose dehydrogenase and glycoside hydrolase 61. Appl Environ Microbiol. 2011;77(19):7007–7015. doi: 10.1128/AEM.05815-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li X, Beeson WT, Phillips CM, Marletta MA, Cate JHD. Structural basis for substrate targeting and catalysis by fungal polysaccharide monooxygenases. Structure. 2012;20(6):1051–1061. doi: 10.1016/j.str.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beeson WT, Phillips CM, Cate JHD, Marletta MA. Oxidative cleavage of cellulose by fungal copper-dependent polysaccharide monooxygenases. J Am Chem Soc. 2012;134(2):890–892. doi: 10.1021/ja210657t. [DOI] [PubMed] [Google Scholar]
- 11.Wu M, et al. Crystal structure and computational characterization of the lytic polysaccharide monooxygenase GH61D from the Basidiomycota fungus Phanerochaete chrysosporium. J Biol Chem. 2013;288(18):12828–12839. doi: 10.1074/jbc.M113.459396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hemsworth GR, et al. The copper active site of CBM33 polysaccharide oxygenases. J Am Chem Soc. 2013;135(16):6069–6077. doi: 10.1021/ja402106e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aachmann FL, Sørlie M, Skjåk-Bræk G, Eijsink VGH, Vaaje-Kolstad G. NMR structure of a lytic polysaccharide monooxygenase provides insight into copper binding, protein dynamics, and substrate interactions. Proc Natl Acad Sci USA. 2012;109(46):18779–18784. doi: 10.1073/pnas.1208822109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klinman JP. Mechanisms whereby mononuclear copper proteins functionalize organic substrates. Chem Rev. 1996;96(7):2541–2562. doi: 10.1021/cr950047g. [DOI] [PubMed] [Google Scholar]
- 15.Himo F, Eriksson LA, Maseras F, Siegbahn PEM. Catalytic mechanism of galactose oxidase: A theoretical study. J Am Chem Soc. 2000;122(33):8031–8036. [Google Scholar]
- 16.Lewis EA, Tolman WB. Reactivity of dioxygen-copper systems. Chem Rev. 2004;104(2):1047–1076. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]
- 17.Itoh S. Mononuclear copper active-oxygen complexes. Curr Opin Chem Biol. 2006;10(2):115–122. doi: 10.1016/j.cbpa.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 18.Cramer CJ, Tolman WB. Mononuclear Cu-O2 complexes: geometries, spectroscopic properties, electronic structures, and reactivity. Acc Chem Res. 2007;40(7):601–608. doi: 10.1021/ar700008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Que L, Jr, Tolman WB. Biologically inspired oxidation catalysis. Nature. 2008;455(7211):333–340. doi: 10.1038/nature07371. [DOI] [PubMed] [Google Scholar]
- 20.Himes RA, Karlin KD. Copper-dioxygen complex mediated C-H bond oxygenation: Relevance for particulate methane monooxygenase (pMMO) Curr Opin Chem Biol. 2009;13(1):119–131. doi: 10.1016/j.cbpa.2009.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peterson RL, et al. Cupric superoxo-mediated intermolecular C-H activation chemistry. J Am Chem Soc. 2011;133(6):1702–1705. doi: 10.1021/ja110466q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horn SJ, Vaaje-Kolstad G, Westereng B, Eijsink VGH. Novel enzymes for the degradation of cellulose. Biotechnol Biofuels. 2012;5(1):45. doi: 10.1186/1754-6834-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beckham GT, et al. Molecular-level origins of biomass recalcitrance: Decrystallization free energies for four common cellulose polymorphs. J Phys Chem B. 2011;115(14):4118–4127. doi: 10.1021/jp1106394. [DOI] [PubMed] [Google Scholar]
- 24.Vaaje-Kolstad G, Horn SJ, van Aalten DMF, Synstad B, Eijsink VGH. The non-catalytic chitin-binding protein CBP21 from Serratia marcescens is essential for chitin degradation. J Biol Chem. 2005;280(31):28492–28497. doi: 10.1074/jbc.M504468200. [DOI] [PubMed] [Google Scholar]
- 25.Hemsworth GR, Davies GJ, Walton PH. Recent insights into copper-containing lytic polysaccharide mono-oxygenases. Curr Opin Struct Biol. 2013;23(5):660–668. doi: 10.1016/j.sbi.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 26.Dechancie J, et al. How similar are enzyme active site geometries derived from quantum mechanical theozymes to crystal structures of enzyme-inhibitor complexes? Implications for enzyme design. Protein Sci. 2007;16(9):1851–1866. doi: 10.1110/ps.072963707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de la Lande A, Moliner V, Parisel O. Singlet-triplet gaps in large multireference systems: Spin-flip-driven alternatives for bioinorganic modeling. J Chem Phys. 2007;126(3):035102–035107. doi: 10.1063/1.2423010. [DOI] [PubMed] [Google Scholar]
- 28.Xu X, Muller RP, Goddard WA., 3rd The gas phase reaction of singlet dioxygen with water: A water-catalyzed mechanism. Proc Natl Acad Sci USA. 2002;99(6):3376–3381. doi: 10.1073/pnas.052710099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cramer CJ, et al. Stereoelectronic effects on molecular geometries and state-energy splittings of ligated monocopper dioxygen complexes. J Phys Chem A. 2008;112(16):3754–3767. doi: 10.1021/jp800627e. [DOI] [PubMed] [Google Scholar]
- 30.Dietzel PDC, Kremer RK, Jansen M. Tetraorganylammonium superoxide compounds: Close to unperturbed superoxide ions in the solid state. J Am Chem Soc. 2004;126(14):4689–4696. doi: 10.1021/ja039880i. [DOI] [PubMed] [Google Scholar]
- 31.Huber SM, et al. Generating Cu(II)-oxyl/Cu(III)-oxo species from Cu(I)-alpha-ketocarboxylate complexes and O2: In silico studies on ligand effects and C-H-activation reactivity. Chem Eur J. 2009;15(19):4886–4895. doi: 10.1002/chem.200802338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gherman BF, Tolman WB, Cramer CJ. Characterization of the structure and reactivity of monocopper-oxygen complexes supported by beta-diketiminate and anilido-imine ligands. J Comput Chem. 2006;27(16):1950–1961. doi: 10.1002/jcc.20502. [DOI] [PubMed] [Google Scholar]
- 33.Kunishita A, et al. Aromatic hydroxylation reactivity of a mononuclear Cu(II)-alkylperoxo complex. J Am Chem Soc. 2007;129(23):7248–7249. doi: 10.1021/ja071623g. [DOI] [PubMed] [Google Scholar]
- 34.Comba P, et al. CopperII-mediated aromatic ortho-hydroxylation: A hybrid DFT and AB initio exploration. Chem Eur J. 2008;14(1):344–357. doi: 10.1002/chem.200700865. [DOI] [PubMed] [Google Scholar]
- 35.Wolfenden R, Lu X, Young G. Spontaneous hydrolysis of glycosides. J Am Chem Soc. 1998;120(27):6814–6815. [Google Scholar]
- 36.Schroder D, Holthausen MC, Schwarz H. Radical-like activation of alkanes by the ligated copper oxide cation (Phenanthroline)CuO+ J Phys Chem B. 2004;108(38):14407–14416. [Google Scholar]
- 37.Yoshizawa K, Shiota Y. Conversion of methane to methanol at the mononuclear and dinuclear copper sites of particulate methane monooxygenase (pMMO): A DFT and QM/MM study. J Am Chem Soc. 2006;128(30):9873–9881. doi: 10.1021/ja061604r. [DOI] [PubMed] [Google Scholar]
- 38.Yoshizawa K, Kihara N, Kamachi T, Shiota Y. Catalytic mechanism of dopamine beta-monooxygenase mediated by Cu(III)-oxo. Inorg Chem. 2006;45(7):3034–3041. doi: 10.1021/ic0521168. [DOI] [PubMed] [Google Scholar]
- 39.Crespo A, Martí MA, Roitberg AE, Amzel LM, Estrin DA. The catalytic mechanism of peptidylglycine α-hydroxylating monooxygenase investigated by computer simulation. J Am Chem Soc. 2006;128(39):12817–12828. doi: 10.1021/ja062876x. [DOI] [PubMed] [Google Scholar]
- 40.Njus D, Kelley PM. Vitamins C and E donate single hydrogen atoms in vivo. FEBS Lett. 1991;284(2):147–151. doi: 10.1016/0014-5793(91)80672-p. [DOI] [PubMed] [Google Scholar]
- 41.Frisch MJ, et al. Wallingford, CT: Gaussian, Inc; 2010. Gaussian 09, Revision B.01. [Google Scholar]
- 42.Gherman BF, Heppner DE, Tolman WB, Cramer CJ. Models for dioxygen activation by the CuB site of dopamine β-monooxygenase and peptidylglycine α-hydroxylating monooxygenase. J Biol Inorg Chem. 2006;11(2):197–205. doi: 10.1007/s00775-005-0066-5. [DOI] [PubMed] [Google Scholar]
- 43.Chen P, Solomon EI. Oxygen activation by the noncoupled binuclear copper site in peptidylglycine α-hydroxylating monooxygenase. Reaction mechanism and role of the noncoupled nature of the active site. J Am Chem Soc. 2004;126(15):4991–5000. doi: 10.1021/ja031564g. [DOI] [PubMed] [Google Scholar]
- 44.Cramer CJ, Truhlar DG. Density functional theory for transition metals and transition metal chemistry. Phys Chem Chem Phys. 2009;11(46):10757–10816. doi: 10.1039/b907148b. [DOI] [PubMed] [Google Scholar]
- 45.Yamaguchi K, Jensen F, Dorigo A, Houk KN. A spin correction procedure for unrestricted Hartree-Fock and Møller-Plesset wavefunctions for singlet diradicals and polyradicals. Chem Phys Lett. 1988;149(5–6):537–542. [Google Scholar]
- 46.Kunishita A, et al. Active site models for the Cu(A) site of peptidylglycine α-hydroxylating monooxygenase and dopamine β-monooxygenase. Inorg Chem. 2012;51(17):9465–9480. doi: 10.1021/ic301272h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.