

Significance
How receptor for hyaluronan-mediated motility (RHAMM) expression is regulated, how statins exert anticancer effects, and what roles mevalonate and Hippo pathways play in tumors are important issues in cancer biology. We find that the two pathways converge onto Yes-associated protein (YAP)/TEAD to control RHAMM transcription leading to ERK activation and cancer metastasis, which is inhibited by simvastatin. YAP/TEAD binds RHAMM promoter at specific sites to activate RHAMM transcription, and mevalonate/simvastatin affects RHAMM transcription by modulating YAP phosphorylation and nuclear-cytoplasmic distribution. These in vitro and in vivo findings identify a mechanism regulating RHAMM expression and cancer metastasis wherein RHAMM is a downstream effector of mevalonate/Hippo pathways, and a YAP/TEAD-transcription and simvastatin-inhibition target, revealing interesting interplay of the pathways and potential targets for cancer therapeutic agents.
Keywords: metabolism, actin assembly, oncogene, tumor suppressor, crosstalk
Abstract
Expression of receptor for hyaluronan-mediated motility (RHAMM), a breast cancer susceptibility gene, is tightly controlled in normal tissues but elevated in many tumors, contributing to tumorigenesis and metastases. However, how the expression of RHAMM is regulated remains elusive. Statins, inhibitors of mevalonate metabolic pathway widely used for hypercholesterolemia, have been found to also have antitumor effects, but little is known of the specific targets and mechanisms. Moreover, Hippo signaling pathway plays crucial roles in organ size control and cancer development, yet its downstream transcriptional targets remain obscure. Here we show that RHAMM expression is regulated by mevalonate and Hippo pathways converging onto Yes-associated protein (YAP)/TEAD, which binds RHAMM promoter at specific sites and controls its transcription and consequently breast cancer cell migration and invasion (BCCMI); and that simvastatin inhibits BCCMI via targeting YAP-mediated RHAMM transcription. Required for ERK phosphorylation and BCCMI, YAP-activated RHAMM transcription is dependent on mevalonate and sensitive to simvastatin, which modulate RHAMM transcription by modulating YAP phosphorylation and nuclear-cytoplasmic localization. Further, modulation by mevalonate/simvastatin of YAP-activated RHAMM transcription requires geranylgeranylation, Rho GTPase activation, and actin cytoskeleton rearrangement, but is largely independent of MST and LATS kinase activity. These findings from in vitro and in vivo investigations link mevalonate and Hippo pathways with RHAMM as a downstream effector, a YAP-transcription and simvastatin-inhibition target, and a cancer metastasis mediator; uncover a mechanism regulating RHAMM expression and cancer metastases; and reveal a mode whereby simvastatin exerts anticancer effects; providing potential targets for cancer therapeutic agents.
Breast cancer is by far the most frequent cancer in women worldwide, ranking second among all cancers, and is one of the most deadly cancers (1). Unraveling the molecular and cellular mechanisms underlying breast cancer progression and metastasis is critical for development of therapeutic agents to treat this deadly disease. Receptor for hyaluronan (HA)-mediated motility (RHAMM), also known as HMMR, IHABP, or CD168, has been identified as a breast cancer susceptibility gene (2, 3), with dual oncogenic functions as HA receptor and mitotic spindle binding protein (4, 5). RHAMM is generally not detected in homeostatic tissues, and is transiently produced during wound repair, but its hyperexpression is associated with tumor development, progression, and metastasis (2, 6). Overexpression of RHAMM causes transformation and promotes breast cancer cell migration and invasion (BCCMI), and its expression is up-regulated in a variety of human tumors, including breast and endometrial carcinomas (6–8), gastrointestinal cancers (9, 10), prostate cancer (11), aggressive fibromatosis (i.e., desmoid tumor) (12), lung and liver cancer (13, 14), glioma (15), and B-cell malignancies (16, 17). RHAMM binds to mitotic spindles and promotes interphase microtubule instability and mitotic spindle integrity (18, 19). Uniquely, it is also unconventionally exported onto extracellular surface to partner with CD44, thereby enhancing CD44-mediated tumor progression via ERK1/2 association, and maintaining high proliferative activities and motility of invasive cancer cells (20, 21). Thus, expression of RHAMM is critical for its normal and oncogenic functions, but how it is regulated remains obscure.
Statins are specific inhibitors of the 3-hydroxy-methylglutaryl CoA reductase (HMGCR), the enzyme catalyzing the rate-limiting, mevalonate-making step in the mevalonate pathway for the biosynthesis of isoprenoids and downstream products. The mevalonate pathway is biologically very important because the isoprenoids it produces play vital roles in multiple cellular functions, including protein posttranslational modifications such as geranylgeranylation and farnesylation, cell signaling, cell membrane integrity, cell cycle progression, and cholesterol synthesis (22). As potent blockers of the mevalonate pathway and biosynthesis of cholesterol, statins have long been used to treat hypercholesterolemia and prevent cardiovascular diseases (22). Remarkably, statins have recently been found to also have multiple anticancer effects such as antiproliferative, proapoptotic, antiinvasive, and radiosensitizing properties, making them promising therapeutic agents against many cancers, including mammary carcinoma (22, 23). Several potential mechanisms have been suggested to explain the anticancer activities of statins. Statins could trigger tumor-specific apoptosis by blocking protein geranylgeranylation, leading to disrupted membrane localization and function of the Ras superfamily including CDC42 and Rac and Rho GTPase (22, 23), as well as disorganization of actin stress fibers (24). Simvastatin was shown to foster enhanced expression of mutant p53 to down-regulate CD44 expression, therefore preventing breast cancer cell metastasis to bone (25). Furthermore, simvastatin inactivated NF-κB, leading to derepression of PTEN and repression of Bcl-xl to prevent breast cancer cell growth (26). Notably, it was recently demonstrated that the mevalonate pathway is necessary and sufficient to maintain the malignant state of breast cancer cells in 3D culture (27). However, specific antitumor targets and mechanisms of statins are poorly understood.
The Hippo pathway, with the transcriptional coactivator Yes-associated protein (YAP) as its downstream effector, is highly conserved from Drosophila to mammals and critical in controlling organ size, tissue regeneration, and stem cell self-renewal (28, 29). Recently, the Hippo–YAP pathway has been demonstrated to be involved in tumorigenesis and tumor progression (30, 31). Regulated by cell density, shape, and actin cytoskeleton, the mammalian Hippo pathway consists of a core kinase cascade in which Mst1 or Mst2 forms a complex with the adaptor protein WW45 and phosphorylates the LATS1/2 kinases and another adaptor protein MOB (32–34). The LATS/MOB complex subsequently phosphorylates YAP (at Ser127) and its paralog TAZ (at Ser89), leading to their cytoplasmic retention and repression (28, 30, 35). YAP protein level and activity are regulated at multiple levels by several additional regulators, including the FERM domain proteins Merlin/NF2 (neurofibromatosis 2) and FRMD6 and protein phosphatases PP2A and ASPP1 (28, 30, 35). YAP can promote metastasis through interacting with the TEAD/TEF transcription factors, and increased YAP/TEAD activity plays a causal role in cancer progression and metastasis (36). YAP has been demonstrated as a driving oncogene on amplicon 11q22, which is amplified in several human tumors (37). Up-regulation of YAP and its nuclear localization strongly correlate with poor prognosis and tumor progression in multiple cancers, including breast (38), lung, colorectal, ovarian, and liver carcinomas (39). Overexpression of YAP in a conditional YAP transgenic mouse model led to tissue overgrowth and tumorigenesis (40). Furthermore, mutation or epigenetic silencing of several components of the Hippo pathway, including NF2, LATS1/2, MST1/2, WW45, MOB, and KIBRA, have been associated with several human cancers (30, 35, 41). Together, these studies highlight a pivotal role of the Hippo–YAP pathway in cancer development and progression, but the transcriptional targets remain unclear. Moreover, it is unknown how the roles of the Hippo pathway, mevalonate pathway, statin action, and RHAMM are related in tumor development and progression.
Thus, how the expression of the oncogene RHAMM is regulated, whether and how the mevalonate metabolic pathway and Hippo signaling pathway interact in cancer metastasis, and how the statin drugs exert anticancer effects are important outstanding issues in cancer biology and therapy. In the present study, we have sought to address these and related issues in the context of breast cancer. We have focused on molecular and cellular investigations by using a couple of highly metastatic breast cancer cell lines and a nontumor cell line, and conducted selective and relevant in vivo investigations with human breast tumor tissues and its xenografts mouse model, as previous animal model studies had established that the metastasis of breast cancer was inhibited by simvastatin (25). We find that the mevalonate pathway promotes, and simvastatin inhibits, the expression of RHAMM, which is necessary for ERK phosphorylation and BCCMI, via a transcriptional mechanism mediated directly by YAP. With two previously unrecognized TEAD binding sites in its promoter, RHAMM is demonstrated as a unique transcriptional target of YAP-TEAD. Mevalonate or simvastatin modulates RHAMM transcription through regulating the phosphorylation and nuclear or cytoplasmic localization of YAP, the downstream effector of the Hippo pathway. In vivo experiments and analysis show that RHAMM and YAP are overexpressed in human breast invasive ductal carcinoma, and that simvastatin inhibits expression of RHAMM and activation of YAP and ERK in human breast tumor xenografts in mice. Our findings therefore identify RHAMM as a direct transcriptional target of YAP and a downstream action target of simvastatin, and reveal interesting interplay between the mevalonate metabolic pathway and the Hippo signaling pathway, in breast cancer metastasis.
Results
RHAMM Expression and RHAMM-Mediated ERK Activation and BCCMI Are Promoted by the Mevalonate Pathway and Attenuated by Simvastatin.
BCCMI has been shown to be inhibited by simvastatin (25), and our experiments of transwell migration assays using MDA-MB-231 human mammary tumor cells and 4T1 mouse mammary tumor cells confirmed this observation (Fig. S1). As simvastatin is a specific inhibitor of the rate-limiting enzyme HMGCR in the mevalonate pathway, we tested whether the inhibition of migration is caused by disturbance of the mevalonate pathway. Indeed, this inhibition was reversed by addition of mevalonate (Fig. S1 A and C). In vitro invasion assay using Matrigel-coated Transwell inserts further confirmed the effects of simvastatin and mevalonate (Fig. S1 B and D), showing that the mevalonate pathway plays an essential role in regulating BCCMI, which is blocked by simvastatin.
As the oncogenic protein RHAMM is reported to overexpress and act as a cell surface receptor to promote BCCMI through forming complexes with CD44 and ERK1/2 (20), we investigated the direct relationship between RHAMM protein and the migration and invasion in the MDA-MB-231 cell line. Two independent lentivirus-mediated shRNAs directed toward RHAMM down-regulated the expression of RHAMM protein (Fig. 1A), and significantly inhibited the migration (Fig. 1B) and invasion (Fig. 1C) of MDA-MB-231 cells in transwell assays. In the same time frame, knockdown of RHAMM did not significantly affect cell growth of MDA-MB-231. These results demonstrate that RHAMM is essential in promoting the motility and aggression of the breast cancer cells, and, taken together with the preceding results, raised the possibility that the mevalonate pathway and RHAMM are connected, as they are both involved in the regulation of BCCMI.
Fig. 1.

RHAMM is essential for BCCMI, and the mevalonate pathway regulates RHAMM expression, ERK activation, and BCCMI. (A) Western blot showing lentivirus-mediated shRNAs against RHAMM markedly knock down RHAMM expression in MDA-MB-231 cells. (B and C) Knockdown of RHAMM inhibited the migration (B) and invasion (C) of MDA-MB-231 cells. (D and E) The mevalonate pathway inhibitor simvastatin (Sim) significantly inhibited RHAMM mRNA (D) and protein (E) expression, whereas adding mevalonate (Meva) abolished the inhibitory effect. MDA-MB-231 cells were incubated with or without 250 μM mevalonate for 6 h before treatment with DMSO or 5 μM simvastatin for 24 h. (F and G) As in D and E but with 4T1 cells. (H) Simvastatin markedly inhibited RHAMM promoter activity, whereas adding mevalonate abolished the inhibition. MDA-MB-231 cells were transfected with RHAMM-Luc reporter plasmid for 6 h, and then treated with or without 250 μM mevalonate for 6 h before treatment with DMSO or 5 μM simvastatin for 24 h. Luciferase activity was measured and normalized to GAPDH. (I) Knockdown of RHAMM decreased ERK phosphorylation in MDA-MB-231 cells. (J) Simvastatin significantly inhibited ERK activity, whereas adding mevalonate abolished the inhibition. MDA-MB-231 cells were incubated with or without 250 μM mevalonate for 6 h before treatment with DMSO or 5 μM simvastatin for 24 h. Data are shown as the mean ± SD of three independent experiments. Student two-tailed t test was used for statistical analysis (*P < 0.05).
To test this hypothesis, we measured the mRNA and protein levels of RHAMM by using real-time RT-PCR and Western blot, respectively, with and without the interference of the mevalonate pathway. Both mRNA and protein levels of RHAMM were markedly reduced after the cells were treated by simvastatin, whereas adding mevalonate abolished the inhibitory effect of simvastatin on RHAMM expression in MDA-MB-231 (Fig. 1 D and E) and 4T1 (Fig. 1 F and G) cells. Moreover, inhibition of the mevalonate pathway by simvastatin markedly down-regulated the activity of RHAMM promoter as indicated by reporter gene expression (Fig. 1H), whereas adding mevalonate restored the promoter activity of RHAMM. These results indicate that RHAMM expression is positively regulated by the mevalonate pathway and targeted by simvastatin inhibition through a transcriptional mechanism.
That RHAMM expression is regulated by mevalonate pathway suggests that RHAMM is involved in mevalonate pathway-mediated regulation of BCCMI. RHAMM has been shown to form a complex with ERK1/2 to maintain the activity of ERK1/2 in breast cancer cells, which promoted the motility of invasive breast cancer cells (20). Consistently, we found that down-regulation of RHAMM expression by lentivirus-mediated shRNA inhibited ERK activity by reducing the phosphorylation level of ERK (Fig. 1I). In addition, simvastatin reduced the phosphorylation level of ERK, whereas mevalonate rescued the inhibition by simvastatin (Fig. 1J and Fig. S2). These results suggest that RHAMM-mediated ERK activation is required for the mevalonate pathway-promoted, simvastatin-inhibited BCCMI. That down-regulation of RHAMM expression by shRNA and by simvastatin had equivalent effect on ERK activation and BCCMI indicates that the effect of mevalonate pathway or simvastatin on BCCMI is indeed mediated through RHAMM expression and ERK activation.
RHAMM Is a Direct Transcriptional Target Gene of YAP-TEAD of the Hippo Pathway.
As the mevalonate pathway and simvastatin appeared to modulate RHAMM expression at the transcriptional level, we next tried to identify the transcription factor(s) that mediated this effect. We analyzed the RHAMM promoter region by using MatInspector (Genomatix) and found it harbors two putative binding sites (“GGAATG”) for the transcription factor TEAD at the nucleotide positions of −291 to −285 (TEAD binding site 1, TB1) and −103 to −97 (TEAD binding site 2, TB2) of the transcription start site (Fig. 2A). TEAD-mediated transcription is activated upon binding to the transcription coactivator YAP when the latter is not phosphorylated at Ser127 (28, 29). To verify physical association of endogenous YAP and TEAD with the RHAMM promoter sequence in MDA-MB-231 cells, ChIP assay was performed. As shown in Fig. 2 B and C, RHAMM promoter is occupied by YAP and TEAD4 in MDA-MB-231 cells. To further test for the binding and direct control of RHAMM transcription by YAP/TEAD, we separately cloned the WT RHAMM promoter (−1830 to 1) and the RHAMM promoter mutants with single (ΔTB1 and ΔTB2) or double (ΔTB1/2) TEAD-binding site deletion into the luciferase reporter plasmid. Then, we cotransfected WT or one of the ΔTB1, ΔTB2, or ΔTB1/2 RHAMM reporters with V5-YAP (human YAP expression plasmid; provided by Makiko Fujii, Aichi Cancer Center Research Institute, Nagoya, Japan) into HEK 293T cells, a cell line with minimal level of active YAP expressed. Thus, this cell line allows us to observe the effect of increased active YAP on RHAMM transcription more directly. As shown in Fig. 2D, deletion of single or both TEAD-binding sites at nucleotide positions −291 to −285 and −103 to −97 relative to the transcriptional start site dramatically decreased the transcriptional activity of the RHAMM promoter, and the WT RHAMM luciferase promoter was significantly activated by the exogenous V5-YAP, whereas the mutant reporters were essentially unaffected (Fig. 2D), indicating that the TEAD-binding sites are required for YAP-TEAD binding and RHAMM promoter activity, and RHAMM reporter transcription is activated directly by YAP-TEAD. To further elucidate the contribution of endogenous YAP and TEAD in controlling the RHAMM promoter activity, YAP or TEAD1/3/4 were individually knocked down in MDA-MB-231 cells by shRNA (Fig. 2E). As shown in Fig. 2E, down-regulation of YAP or TEAD1/3/4 repressed RHAMM promoter activity markedly, showing that endogenous YAP and TEAD are required for RHAMM promoter activity, i.e., transcriptional activation of RHAMM reporter gene.
Fig. 2.

YAP and TEAD bind to RHAMM promoter at specific sites and regulate RHAMM transcription. (A) The human RHAMM promoter region contains two putative TEAD-binding sites (TB; boxed). (B and C) YAP (B) and TEAD4 (C) bound to the RHAMM promoter by ChIP assay. ChIP from fragmented chromatins of MDA-MB-231 cells were incubated with IgG, anti-YAP or anti-TEAD4, or anti-RNA polymerase II (positive control; Con) antibody. (D) The putative TEAD-binding sites were important for YAP-mediated RHAMM promoter activity. (Upper) The two TEAD-binding sites were deleted individually or in combination. HEK 293T cells were cotransfected with control vector or V5-YAP in combination with WT or mutant RHAMM-Luc reporter plasmid. At 24 h after transfection, the luciferase activity was measured. (Lower) Western blot showing YAP protein level. (E) Knockdown of YAP or TEAD1/3/4 decreased RHAMM promoter activity. MDA-MB-231 cells were infected with the indicated shRNA lentiviruses, and luciferase activity (Upper) and protein levels (Lower) are shown. Data are shown as mean ± SD of three independent experiments. Student two-tailed t test was used for statistical analysis (*P < 0.05 and **P < 0.01).
We next tested whether YAP/TEAD directly regulates the endogenous RHAMM gene expression at the mRNA and protein levels in MDA-MB-231 cells. Lentivirus-mediated shRNA was used to knockdown YAP or TEAD1/3/4, and RNAi specificity and efficiency were confirmed by Western blot (Fig. 3 B, 2 and 3). Knockdown of YAP or TEAD1/3/4 expression resulted in a dramatic reduction in RHAMM mRNA (Fig. 3A) and protein levels (Fig. 3 B, 1) in MDA-MB-231 cells. As a positive control, the transcriptional activation activity of YAP and TEAD was confirmed by the measurement of the mRNA of CTGF, whose expression is known to depend on YAP-TEAD. Knockdown of YAP or TEAD1/3/4 caused a significant decrease in CTGF mRNA in MDA-MB-231 cells (Fig. 3C). In addition, we overexpressed V5-YAP in HEK 293T cells and found that it markedly up-regulated both RHAMM mRNA (Fig. 3D) and protein (Fig. 3E). Taken together, these results demonstrate that the transcription coactivator YAP, in partner with the transcription factor TEAD, directly regulate and are required for transcription of RHAMM.
Fig. 3.

YAP and TEAD are required for RHAMM expression, ERK activity, and BCCMI, whereas ectopic expression of RHAMM bypasses the requirement. (A and B) Knockdown of YAP or TEAD1/3/4 decreased RHAMM transcription (A) and protein (B) levels. MDA-MB-231 cells were infected with the indicated shRNA lentiviruses, and RHAMM mRNA (A) and protein (B) levels were determined by real-time RT-PCR and Western blot, respectively. (B) (Right) Quantification of the protein bands. (C) Knockdown of YAP or TEAD1/3/4 reduced CTGF mRNA levels (experiments as in A). (D and E) Overexpression of YAP increased RHAMM mRNA (D) and protein (E) levels. HEK 293T cells were transiently transfected with the indicated YAP plasmids, and, 48 h later, total RNA was extracted to detect RHAMM mRNA by using real-time RT-PCR (D). (E) Cell lysates were immunoblotted with the indicated antibodies (Left), and the protein bands were quantified (Right). (F) Knockdown of YAP or TEAD1/3/4 decreased ERK phosphorylation levels. Experiments were as in B except Western blot was performed with the indicated antibodies. (G and H) Knockdown of YAP or TEAD1/3/4 decreased MDA-MB-231 cell migration (G, Left) and invasion (H, Left), whereas reexpression of RHAMM rescued the effects (G and H, Right). Data are shown as mean ± SD of three independent experiments. Student two-tailed t test was used for statistical analysis (*P < 0.05 and ***P < 0.001).
YAP and TEAD Regulate BCCMI Through Control of RHAMM Transcription.
As YAP and TEAD depletion repressed transcription of RHAMM, and knockdown of RHAMM expression inhibited ERK phosphorylation and migration and invasion in MDA-MB-231 breast cancer cells, we next investigated the involvement of endogenous YAP and TEAD in ERK activity and the cell migration and invasion by using lentivirus-mediated shRNA targeting YAP or TEAD1/3/4. As shown in Fig. 3, knockdown of YAP or TEAD1/3/4, by short hairpin RNA of YAP (shYAP) or short hairpin RNA of TEAD1/3/4 (shTEAD1/3/4), resulted in down-regulated ERK activity (Fig. 3F). Transwell assay showed dramatic inhibition of migration (Fig. 3G) and invasion (Fig. 3H) in shYAP- or shTEAD1/3/4-infected MDA-MB-231 breast cancer cells. Reexpression of RHAMM, as confirmed in Fig. S3, largely rescued the inhibited migration (Fig. 3G) and invasion (Fig. 3H) caused by YAP or TEAD1/3/4 knockdown. These results show that YAP and TEAD are essential for ERK activity and BCCMI, in accordance with their role of directly regulating transcription of RHAMM necessary for ERK activation and BCCMI. In other words, YAP/TEAD-controlled transcription of RHAMM is responsible for the regulation of BCCMI, i.e., the regulation of BCCMI by YAP/TEAD goes through RHAMM expression.
Mevalonate Pathway and Simvastatin Regulates YAP Phosphorylation and Cellular Localization to Control RHAMM Transcription.
Having shown that simvastatin attenuates RHAMM transcription via the mevalonate pathway and that RHAMM transcription is controlled by YAP, we next tried to delineate the mechanism whereby YAP-mediated RHAMM transcription is regulated by the mevalonate pathway and simvastatin. YAP activity is known to depend on its cellular localization which, in turn, is determined by its phosphorylation status. Phosphorylation of YAP at Ser127 induces translocation of YAP from the nucleus to cytoplasm, thus reducing the transcriptional activity of TEAD. Based on these, the phosphorylation status of YAP at Ser127 was detected by using a specific antibody with and without the inhibition of the mevalonate pathway by simvastatin. As shown in Fig. 4, simvastatin induced an increase in phosphorylation level of YAP at Ser127 in MDA-MB-231 and 4T1 cells, and this effect was abolished by adding mevalonate (Fig. 4 A and B), which suggested that the mevalonate pathway or simvastatin modulates the transcriptional activity of YAP via regulating its phosphorylation at Ser127. In fact, simvastatin robustly repressed the transcription of a YAP target gene CTGF, which contained three putative YAP-TEAD binding sites, whereas the repression was reversed by mevalonate (Fig. 4C), indicating specificity of the effect of simvastatin or mevalonate on YAP. We then examined the localization of YAP in the cells treated with simvastatin. Consistent with the increased phosphorylation level of YAP at Ser127, simvastatin treatment induced translocation of YAP from the nucleus to cytoplasm as visualized by immunofluorescent staining, whereas adding mevalonate prevented the YAP translocation (Fig. 4 D and E). Nucleocytoplasmic separation assay further confirmed that simvastatin induced the nucleocytoplasmic translocation of YAP through blockage of the mevalonate pathway (Fig. 4F). Surprisingly, although immunofluorescent images (and their quantification) (Fig. 4 D and E) and Western blot of nucleocytoplasmic fractionations (Fig. 4F) show that simvastatin caused the nuclear YAP protein to decrease dramatically, that was accompanied by a marginal increase in cytoplasmic YAP protein, which suggests that YAP may be degraded upon translocation to the cytoplasm. Indeed, YAP was significantly polyubiquitinated after the cells were treated by simvastatin, indicating that YAP is degraded upon translocation to the cytoplasm through the ubiquitin–proteasome pathway, whereas adding mevalonate abolished the effect of simvastatin (Fig. S4). These results indicate that simvastatin promotes YAP phosphorylation and cytoplasmic sequestration and degradation to suppress RHAMM transcription. In addition, simvastatin significantly reduced the binding of YAP to RHAMM promoter (Fig. 4G), consistent with YAP being sequestered by simvastatin treatment. Taken together, our results reveal possibly a key mechanism for simvastatin to inhibit the metastasis of human breast cancer, in which simvastatin acts on the mevalonate pathway, which converges with Hippo pathway onto YAP and its target gene RHAMM transcription.
Fig. 4.

YAP phosphorylation, cytoplasmic localization, and transactivating activity are modulated by mevalonate pathway and simvastatin (Sim). (A and B) Simvastatin significantly inhibited YAP activity (enhanced YAP phosphorylation), whereas adding mevalonate (Meva) abolished the effect in MDA-MB-231 (A) and 4T1 (B) cells. Cells were treated with or without 250 μM Meva for 6 h before incubation with DMSO or 5 μM simvastatin for 24 h and preparation of lysates, which were immunoblotted with p-YAP (Ser-127) and YAP antibodies, respectively (Left), and the protein bands quantified (Right). (A, Left) L, long exposure; S, short exposure. (C) Simvastatin significantly inhibited CTGF mRNA expression, whereas adding mevalonate abolished the inhibition in MDA-MB-231 cells. (D and E) Simvastatin induced YAP translocation from the nucleus to cytoplasm, whereas adding mevalonate prevented the YAP translocation in MDA-MB-231 (D) and 4T1 (E) cells. Immunofluorescent staining of YAP in the cells in the absence and presence of simvastatin and mevalonate. Cells were fixed and stained with YAP antibody (green) and DAPI (blue). (Right) Quantification of immunofluorescent images (ratio of nuclear YAP). (F) Western blot of nucleocytoplasmic fractionation indicated that simvastatin induced YAP translocation from the nucleus to cytoplasm, whereas adding mevalonate prevented the effect. CE, cytoplasmic extract; NE, nuclear extract. (G) Simvastatin prevented binding of YAP to RHAMM promoter. Chromatins were prepared from MDA-MB-231 cells incubated with or without 5 μM simvastatin. ChIP was carried out by using anti-YAP antibody, and the amplification was performed by using SYBR Green real-time PCR. Data are shown as the mean ± SD of three independent experiments. Student two-tailed t test was used for statistical analysis (*P < 0.05, **P < 0.01, and ***P < 0.001).
RHAMM and YAP Are Overexpressed in Human Invasive Breast Tumors, and Simvastatin Attenuates RHAMM Expression and YAP and ERK Activation in Human Breast Tumor Xenografts in Mice.
Given the role of YAP and RHAMM in BCCMI, we investigated the expression profile of YAP and RHAMM in human invasive ductal breast carcinoma. Immunohistochemistry were performed on tissue sections from patients with breast cancer. Compared with cancer-adjacent normal breast tissue, YAP and RHAMM are expressed at much higher levels in the invasive breast tumors, with YAP nuclear staining also observed (Fig. 5A), which is in agreement with the in vitro data and the role of and relation between YAP and RHAMM. Having shown in cells that simvastatin regulates YAP activity to inhibit RHAMM expression and ERK activity, we further investigated the in vivo relevance of the in vitro findings through testing the effects of simvastatin on the YAP phosphorylation, RHAMM expression, and ERK activity in MDA-MB-231–induced tumors in nude mice. Compared with tumors from control mice, mRNA (Fig. 5B) and protein (Fig. 5C) levels of RHAMM were markedly reduced in tumors from simvastatin-treated mice whereas the phosphorylation of YAP increased and the phosphorylation of ERK decreased (Fig. 5C). These in vivo results are in agreement with and extend our results from cellular and molecular investigations, and establish the physiological and pathological significance for the role of YAP-controlled RHAMM expression, the interplay between the mevalonate and Hippo pathways, and the inhibition by simvastatin in intervention of BCCMI.
Fig. 5.
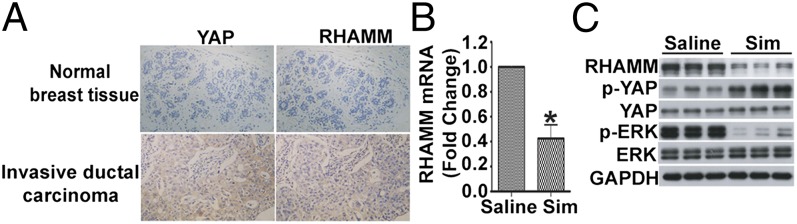
YAP and RHAMM are co-overexpressed in human breast invasive tumors, and simvastatin (Sim) modulates YAP phosphorylation, RHAMM expression, and ERK activity in the MDA-MB-231 human breast tumor xenografts in mice. (A) YAP and RHAMM are both overexpressed in human breast invasive ductal carcinoma. Tissue sections from breast invasive ductal carcinoma and cancer-adjacent normal breast tissue were immunostained with anti-YAP and anti-RHAMM antibody, and analyzed by light microscopy. (B and C) Simvastatin modulated YAP phosphorylation, RHAMM expression, and ERK activity in the MDA-MB-231 human breast tumor xenografts. (B) Simvastatin inhibited RHAMM mRNA expression in vivo. Total RNA was extracted from tumors from control (saline) or simvastatin-treated mice and then RHAMM mRNA levels were determined by real-time RT-PCR. (C) Simvastatin inhibited YAP activity, RHAMM protein expression, and ERK activity in vivo. Lysates of tumors from control (saline) or simvastatin-treated mice were immunoblotted with the indicated antibodies. Data are shown as the mean ± SD of three independent experiments. Student two-tailed t test was used for statistical analysis (*P < 0.05).
Geranylgeranylation Downstream of Mevalonate Is Essential for the Regulation of YAP Activity, RHAMM Transcription, and ERK Activation.
As simvastatin blocks at an upstream step in the mevalonate pathway, which encompasses multiple additional downstream components as well as branches, we next investigated which of those participate in regulating YAP-mediated RHAMM transcription leading to BCCMI. Geranylgeranyl pyrophosphate (GGPP) lies downstream of mevalonate in the pathway and is a key precursor molecule for protein prenylation including geranylgeranylation. Preventing geranylgeranylation of RhoA has been suggested as a mechanism for statin-induced blockade of tumor cell migration and invasion (42). We used a selective geranylgeranyl transferase inhibitor GGTI-2133 to test whether GGPP-dependent geranylgeranylation is involved. Inhibition of geranylgeranyl transferase by GGTI-2133 had a profound negative impact on RHAMM mRNA (Fig. 6 A and C) and protein expression levels (Fig. 6 B and D, Upper), YAP and ERK phosphorylation (Fig. 6 B and D), and cytoplasmic location of YAP (Fig. 6 E and F). These results suggest that geranylgeranylation is required to regulate YAP activity, RHAMM transcription, and ERK activation. Moreover, to the cells treated with simvastatin, addition of GGPP, which is downstream of mevalonate in the pathway, was sufficient to reverse the effects of simvastatin on YAP activity (Fig. 6 H and J) and nuclear localization (Fig. 6 K and L), RHAMM expression (Fig. 6 G–J), and ERK activity (Fig. 6 H and J). These results suggest that geranylgeranylation is indeed an essential step downstream of mevalonate in the pathway leading to YAP-dependent RHAMM transcription and BCCMI.
Fig. 6.

Geranylgeranylation downstream of mevalonate pathway mediates regulation of YAP activity, RHAMM expression, and ERK activity. (A and B) GGTI-2133 inhibited RHAMM mRNA expression (A) and RHAMM protein expression and YAP and ERK activation (B). MDA-MB-231 cells were incubated with or without 1 μM GGTI-2133 for 24 h, and analyzed by real-time RT-PCR (A) and Western blot with the indicated antibodies (B). (C and D) As in A and B but with 4T1 cells. (E and F) GGTI-2133 induced translocation of YAP from the nucleus to the cytoplasm in MDA-MB-231 (E) and 4T cells (F). Cells were incubated with or without 1 μM GGTI-2133 for 24 h and fixed and stained with YAP antibody (green) and DAPI (blue) for immunofluorescent imaging. (G and H) GGPP rescued inhibition by simvastatin (Sim) of RHAMM mRNA expression (G) and RHAMM protein expression and YAP and ERK activity (H) in MDA-MB-231 cells. Cells were incubated with or without 25 μM GGPP for 2 h before treatment with DMSO or 5 μM simvastatin for 24 h, and analyzed by real-time RT-PCR (G) and Western blot with the indicated antibodies (H). (I and J) As in G and H but with 4T1 cells. (K and L) GGPP blocked translocation of YAP from the nucleus to the cytoplasm induced by simvastatin in MDA-MB-231 (K) and 4T1 cells (L). Cells were incubated with or without 25 μM GGPP for 2 h before treatment with DMSO or 5 μM simvastatin, and fixed and stained with YAP antibody (green) and DAPI (blue) for immunofluorescent imaging. Data are shown as the mean ± SD of three independent experiments. Student two-tailed t test was used for statistical analysis (*P < 0.05, **P < 0.01, and ***P < 0.001).
Mevalonate Pathway Modulates YAP Phosphorylation via Rho GTPase Activation and Actin Cytoskeleton Rearrangement Independent of MST and LATS Kinase Activity.
Rho GTPase and actin cytoskeleton rearrangement have recently been reported to play a role in regulating YAP of the Hippo pathway (33, 34), which bypasses the core kinase cascade, and the mevalonate pathway has been shown to control Rho GTPase activity and actin cytoskeleton organization (24). We next tested whether Rho GTPase and cytoskeleton are involved in the mevalonate pathway-mediated regulation of YAP activity and RHAMM expression. Similar to previous work (24), we also observed that simvastatin inhibited Rho activity and disrupted actin cytoskeleton, and these effects could be rescued by adding mevalonate or GGPP (Fig. S5 A and C). Furthermore, GGTI-2133 inhibited Rho activity and disrupted actin cytoskeleton (Fig. S5 B and C). To investigate whether mevalonate pathway regulated YAP activity through Rho GTPase, lentivirus-mediated constitutively active RhoA (Q63L) was used to infect the cells. Indeed, RhoA (Q63L)-EGFP largely restored the levels of YAP activity, RHAMM expression, and ERK activity, which had been markedly brought down by simvastatin (Fig. 7 A and B). Furthermore, a specific Rho inhibitor, C3 transferase, could also mimic simvastatin effect on YAP activity, RHAMM expression, and ERK activity (Fig. 7 C–E). To test whether the actin cytoskeleton is involved in the regulation of YAP by the mevalonate pathway, we used chemical (cytoskeleton disrupting reagent cytochalasin D) and physical (hydrogel) methods to induce actin cytoskeleton change. Similar to simvastatin treatment, disruption of actin cytoskeleton by cytochalasin D and hydrogel increased YAP phosphorylation (Fig. 7E), induced YAP cytoplasmic translocation (Fig. 7C), inhibited RHAMM expression (Fig. 7 D and E), and ERK activity (Fig. 7E). These data support that Rho GTPase and cytoskeleton organization downstream of geranylgeranylation are required to regulate YAP activity, RHAMM transcription, and ERK activation.
Fig. 7.

Mevalonate pathway regulates YAP activity, RHAMM expression, and ERK activity through Rho GTPase activity and actin cytoskeleton rearrangement largely independent of MST and LATS. (A and B) Constitutively active RhoA (Q63L) rescued RHAMM mRNA expression (A) and RHAMM protein expression, and YAP and ERK activity (B) decreased by simvastatin (Sim). MDA-MB-231 cells were infected with lentivirus-mediated constitutively active RhoA (Q63L)-EGFP, and, 24 h later, treated with simvastatin for another 24 h and analyzed by real-time RT-PCR (A) and Western blot with the indicated antibodies (B). (C–E) Disruption of actin cytoskeleton by various methods induced translocation of YAP from the nucleus to the cytoplasm (C), decrease in RHAMM mRNA level (D), and inhibition of RHAMM protein expression and YAP and ERK activation (E). MDA-MB-231 cells were treated with cytochalasin D or C3 transferase for 6 h or cultured on 1 kPa hydrogel for 24 h to disrupt actin cytoskeleton assembly, and then fixed and stained with phalloidin (green), YAP antibody (red) and DAPI (blue) for immunofluorescent imaging (C), or analyzed by real-time RT-PCR (D) or Western blot with the indicated antibodies (E). (F and G) MST phosphorylation (F) or LATS phosphorylation (G) was not modulated by mevalonate pathway/simvastatin, or actin cytoskeleton-disrupting cytochalasin D, C3 transferase, or culturing on 1 kPa hydrogel in MDA-MB-231 cells. (F) Cells were transfected with Flag-MST2 for 24 h and then treated with the indicated reagents, and cell lysates were immunoblotted with p-MST and Flag antibodies. The specificity of the p-MST antibody was confirmed by λ-phosphatase (PPase) treatment (lane 3). (G) Cell lysates were immunoblotted with p-LATS1 and LATS1 antibodies. The specificity of the p-LATS antibody was confirmed by λ-phosphatase treatment (lane 2). (H and I) Knockdown of LATS did not affect simvastatin-induced inhibition of RHAMM mRNA expression (H) or RHAMM protein expression and YAP and ERK activation (I). MDA-MB-231 cells were infected with the indicated shRNA lentiviruses, treated with or without simvastatin for 24 h, and then analyzed by real-time RT-PCR to determine RHAMM mRNA levels (H) or Western blot to detect the proteins (I). Data are shown as the mean ± SD of three independent experiments. Student two-tailed t test was used for statistical analysis (*P < 0.05, **P < 0.01, and ***P < 0.001).
The core kinases of the Hippo pathway, MST1/2 and LATS1/2, are known to control the phosphorylation level and activity of YAP. We next investigated whether MST and LATS kinases were involved in mevalonate pathway-mediated regulation of YAP. MDA-MB-231 cells express minimal level of phosphorylated MST as detected by Western blot (Fig. 7F). The cells were transfected with Flag-MST2 to boost its MST in the phosphorylated form. However, the phosphorylation level of MST in the MDA-MB-231 cells transfected with Flag-MST2 was not affected by the treatment of mevalonate, simvastatin, and simvastatin plus mevalonate (Fig. 7F, lanes 4–6 vs. lane 2). The specificity of the phospho (p)-MST antibody was confirmed by λ-phosphatase treatment (Fig. 7F, lane 3 vs. lane 2). We also examined the phosphorylation of endogenous LATS in these treatment settings, similar to MST, the phosphorylation level of LATS was basically unchanged (Fig. 7G, lanes 3–5 vs. lane 1). Also, the specificity of the p-LATS antibody was confirmed by λ-phosphatase treatment (Fig. 7G, lane 2 vs. lane 1). In addition, Rho inhibitor C3, cytochalasin D, or hydrogel showed no effect on MST phosphorylation in the MDA-MB-231 cells transfected with Flag-MST2 (Fig. 7F) or the phosphorylation of endogenous LATS (Fig. 7G), which suggests that the mevalonate pathway modulates YAP phosphorylation and activity via actin cytoskeleton rearrangement independent of MST and LATS kinase activity.
To further confirm the results, lentivirus-mediated shRNAs against LATS were used. As shown in Fig. 7 H and I, depletion of LATS1 or/and LATS2 had marginal effect on YAP phosphorylation (Fig. 7I), RHAMM mRNA (Fig. 7H) and protein expression (Fig. 7I), and ERK activity (Fig. 7I) induced by simvastatin. Taken together, these data suggest that the core kinase cascade of the Hippo pathway is not affected by the mevalonate pathway, and vice versa, and the mevalonate pathway modulates YAP phosphorylation and activity largely independent of MST and LATS kinase activity.
Discussion
In this article, we describe the findings that, in breast cancer cells, the mevalonate metabolic pathway, or its inhibitor simvastatin, exerts regulation upon the Hippo signaling pathway via controlling the YAP phosphorylation and thus its activity. YAP acts directly on the promoter and regulates the transcription of RHAMM, which modulates the ERK activity and cell motility in the breast cancer cells. In addition, the molecular and cellular findings are corroborated by results of in vivo investigations.
The mevalonate pathway produces isoprenoids that are vital for many cellular functions, including cholesterol synthesis and growth control (22). Tumor cell metabolism is regulated by the mevalonate pathway as a number of components or metabolic products in this pathway, including HMGCR, mevalonate, cholesterol, and isoprenoids, have been linked to tumor progression (23). HMGCR, the rate-limiting enzyme of the mevalonate pathway, is a candidate metabolic oncogene reportedly to deregulate in several types of cancers (43). In patients with breast cancer, high messenger RNA levels of HMGCR, among other four mevalonate pathway genes, are found to correlate with poor prognosis (23). Mutant p53 can significantly up-regulate mevalonate metabolism and protein prenylation through promoting transcription of genes encoding mevalonate pathway enzymes, including HMGCR in carcinoma cells, which contribute to the maintenance of the malignant phenotype (27). In addition, HMGCR apparently cooperates with RAS in promoting the transformation of WT primary mouse embryonic fibroblasts (43). We found that the mevalonate pathway affects the migration and invasion of MDA-MB-231 and 4T1 breast cancer cells (Fig. 1 and Fig. S1). Inhibiting the mevalonate pathway by simvastatin down-regulates RHAMM expression and suppress BCCMI, whereas adding mevalonate reverses the suppression of RHAMM as well as the migration and invasion caused by the simvastatin, demonstrating a role of the mevalonate pathway in controlling breast cancer metastasis via RHAMM.
It has long been recognized that intervening with the mevalonate pathway may be useful for certain cancer treatments (44). Statins, a class of HMGCR inhibitors that block the mevalonate pathway at its rate-limiting step, are clinically used to treat cardiovascular and cerebrovascular diseases. They have gained increasing recognition as anticancer agents and exhibited anticancer activity in a variety of in vitro and in vivo preclinical models (22, 23). Beside preclinical and some clinical observations, epidemiologic data suggest that statins can lower the risk of certain cancers by as much as 50%. Those promising preclinical results have led to at least 18 reported phase I and I/II clinical trials (23). In breast cancer, statins prevented metastasis by inhibiting CD44 expression through promoting p53 expression (25). Statins enhance the effects of a number of chemotherapeutic drugs with only a few exceptions, and also increase the chemotherapeutic sensitivity of multidrug resistance cells. In tumor-bearing mice, statins used in combination treatments show the additional advantageous effects of attenuated cardiotoxicity and reduced kidney or liver damage, although these effects are still waiting to be verified in clinical trials (43). Our present results further show that simvastatin is able to down-regulate key factors in tumor formation and progression, particularly metastasis, and thus has valuable therapeutic benefits for cancer treatment.
As our results showed that RHAMM is a downstream target regulated by the mevalonate pathway, we then set out to search for the factors that may regulate the promoter activity of RHAMM. Surprisingly, we found that key components of the Hippo signaling pathway, namely YAP and TEAD, directly control the transcription of RHAMM. Moreover, we found that the mevalonate pathway regulates the expression of RHAMM through impacting on the Hippo pathway and its downstream effectors YAP and TEAD.
Highly conserved in mammals, the Hippo pathway is known to play a key role in organ size control and tissue regeneration, and to be involved in embryonic development, stem cell differentiation, and control of proliferation, epithelial–mesenchymal transition, and tumorigenesis. Deregulation of the Hippo pathway induces tumors in model organisms and occurs in many human carcinomas, including lung, colorectal, ovarian, and liver cancer (39). The core mammalian Hippo pathway is a kinase cascade consisting of MST1/2, Sav1, Lats1/2, and Mob1, which phosphorylates and inhibits YAP/TAZ. YAP/TAZ in conjunction with TEAD1–4 mediates major physiological functions of the Hippo pathway. TEADs contain in the N-terminal region a conserved TEA domain for recognizing and binding DNA elements such as GGAATG in the promoter region of target genes (45).
We examined the promoter sequence of RHAMM and found two putative TEAD binding sites (TB1 and TB2; Fig. 2A). YAP and its target transcription factor TEAD bind to the TEAD binding sites in RHAMM promoter (Fig. 2 B and C), and YAP/TEAD binding to TEAD-binding sites in RHAMM promoter is required for YAP-mediated RHAMM transcriptional activation (Fig. 2 D and E), consistent with the previous report that YAP/TEAD interaction and activity plays causal roles in cancer progression and metastasis (36). Moreover, knockdown of YAP or TEAD in MDA-MB-231 cells induced a dramatic reduction of RHAMM expression in mRNA and protein levels (Fig. 3 A and B). Taken together, these data demonstrated RHAMM as a direct transcriptional target of YAP.
We have further demonstrated that the mevalonate pathway controls YAP phosphorylation at Ser127 and cytoplasmic sequestration (Fig. 4). Meanwhile, YAP/TEAD is essential to sustain the motility of breast cancer cells, as knockdown of YAP or TEAD significantly suppressed migration and invasion of MDA-MB-231 cells which could be largely rescued by reexpression of RHAMM (Fig. 3 G and H). These and our other results establish an important and specific role of YAP (Hippo pathway)-regulated RHAMM in BCCMI, of YAP in RHAMM expression, and of RHAMM in YAP biology.
Furthermore, immunohistochemistry indicated that YAP and RHAMM are overexpressed in human invasive ductal breast carcinoma (Fig. 5A), and in vivo data also revealed that simvastatin modulates YAP phosphorylation, RHAMM expression, and ERK activity in the MDA-MB-231–generated tumors in mice (Fig. 5 B and C). These findings corroborate our results from in vitro investigations and provide mechanistic insights into how mevalonate pathway or simvastatin mediates its pro- or anticancer actions via regulating YAP activity, RHAMM transcription, and subsequent migration and invasion in breast cancer cells. More importantly, our findings reveal a direct link between the mevalonate and the Hippo pathway in controlling tumor progression.
In cancers, Hippo pathway is often perturbed through cross-talk with a number of key signaling pathways that frequently harbor oncogenic mutations. These major pathways include Wnt, TGF-β, bone morphogenetic protein, G protein-coupled receptor (GPCR), Hedgehog, insulin–mammalian target of rapamycin, and Notch pathways (39). Our present results suggest that, in addition to these major pathways, the mevalonate pathway interacts with and regulates the Hippo pathway by directly controlling the YAP phosphorylation, cellular localization, and thus its activity. These findings added another layer of complexity to the networks that can act upon the Hippo pathway, and underscore the vital role of Hippo pathway as a tumor suppressor that needs to succumb for tumor advancement.
We further investigated how the mevalonate pathway interplays with the Hippo pathway in regulating YAP activity and the downstream RHAMM transcription activity. The isoprenoids farnesyl pyrophosphate and GGPP are key end products of the mevalonate pathway. The isoprenylation process by the aforementioned products can posttranslationally modify proteins with C-terminal CAAX, CXC, or CC motifs (23). Isoprenylation is essential for proper localization and activity of the RAS superfamily of small guanosine triphosphatases (GTPases). Among these, the Rho family has been closely linked to cancer, as their deregulated activation drives transformation and their elevated expression is associated with several human tumors. RhoA and RhoC are thought to have a role in metastasis. Recent evidence has linked Rho GTPases to tumor metabolism through activation of glutaminase, which catalyzes the conversion of glutamine to glutamate and ammonia, a key step in glutamine metabolism that contributes to the Warburg phenomenon (23). The activity of these small GTPases depends on the mevalonate pathway, as they require isoprenylation to function properly. The mevalonate pathway has been reported to lead to geranylgeranylation of RhoA and its subsequent membrane tethering and activation (22). The Hippo pathway is regulated by GPCR signaling, as cell-surface GPCRs also couple to RhoA GTPase activation, leading to Lats1/2 inhibition and induction of YAP/TAZ; and RhoA activation induces actin cytoskeleton reorganization, which plays important roles in the Hippo–YAP cascade (34). Indeed, we found that geranylgeranylation downstream of the mevalonate pathway is required and sufficient to activate YAP and RHAMM expression and ERK activation, as selective geranylgeranyl transferase inhibitor significantly inhibited YAP activity, RHAMM expression, and ERK1/2 phosphorylation (Fig. 6 A–F), and simvastatin-induced inhibition was reversed by supplementing with GGPP (Fig. 6 G–L). We have further demonstrated that simvastatin disrupted actin cytoskeleton rearrangement, which is required for YAP activation and RHAMM transcription (Fig. 7). RhoA has been reported to activate YAP via inhibition of LATS1/2 but independent of MST1/2 (31) or independent of both MST1/2 and LATS1/2 activities (33). We found that the mevalonate pathway modulates YAP phosphorylation via actin cytoskeleton rearrangement independent of MST and LATS kinase activity (Fig. 7 and Fig. S5).
RHAMM is poorly expressed in most normal human tissues. RHAMM promotes wound repair, and its expression is increased during wound repair in response to hypoxia and fibrogenic factors (5). Its expression is also elevated during the neoplastic progression of a variety of human tumors. Overexpression of RHAMM is transforming and required for maintaining RAS transformation (6). Consistently, RHAMM is considered a novel breast cancer susceptibility gene, and a significant association between homozygous variation in this gene and early-onset breast cancer has been found (2). Overexpression of RHAMM in patients with primary breast cancer was prognostic of poor outcome in cancer progression. In particular, RHAMM expression contributes to the motility and invasiveness of a tumor cell subpopulation in breast cancers (7). RHAMM overexpression occurred within subsets of tumor cells in the primary tumor that was associated with lymph node metastases. The metastatic tumors showed a significantly higher level of RHAMM than did the primary tumor. RHAMM is also a tumor-associated antigen found in solid and blood tumors. RHAMM functions as cell-surface HA receptor and cytoplasmic mitotic spindle binding protein (4, 5). It mediates tumor progression through CD44 partnership and promotes genomic instability via regulating the mitotic spindle/centrosome integrity (18, 19). This RHAMM-regulated activation process results in increased cell-surface expression of CD44 and enhanced activation of ERK1/2. In breast cancer, activation of extracellular HA binding by CD44–RHAMM complexes confers malignant potential (20). Importantly, RHAMM expression is down-regulated by the tumor suppressor p53, and the RHAMM promoter, including the first exon and intron, mediates the repression by p53 (46). Our present results show that YAP, a key component of the Hippo signaling pathway, exerts direct regulation on the RHAMM promoter activity. In addition, the mevalonate pathway exerts regulation on the transcription activity of RHAMM by modulating the phosphorylation level and the cytoplasmic localization and degradation of YAP (Fig. 8). Further, we show that RHAMM and YAP expression correlate with each other—both are elevated in human breast tumors, and our findings at molecular and cellular levels are confirmed in the human tumor mouse model (Fig. 5) and are thus physiologically and pathologically significant.
Fig. 8.

Model of mevalonate and Hippo pathways regulating YAP-controlled RHAMM transcription and cancer cell motility. Mevalonate pathway regulates activity of YAP, the downstream effector of Hippo pathway, which activates transcription of RHAMM that is required for ERK activation and cancer cell migration and invasion. YAP/TEAD activates RHAMM transcription by binding to RHAMM promoter at two specific TEAD-binding sites. Mevalonate pathway promotes and simvastatin inhibits YAP activity, and consequently RHAMM transcription, ERK activation, and cancer metastasis, via modulating YAP phosphorylation and nuclear-cytoplasmic distribution. The regulation of YAP-mediated RHAMM transcription and cancer metastasis depends on geranylgeranylation, Rho GTPase activity, and actin cytoskeleton assembly, but not the canonical MST/LATS cascade.
In summary, we have found that the mevalonate pathway and the Hippo pathway interact and converge onto YAP and TEAD to regulate RHAMM transcription and subsequent ERK activation and cancer metastasis. YAP/TEAD activates RHAMM transcription via binding to RHAMM promoter at two TEAD-binding sites; mevalonate promotes whereas simvastatin attenuates RHAMM transcription by modulating YAP phosphorylation and nuclear-cytoplasmic distribution; and geranylgeranylation, Rho GTPase, and actin cytoskeleton rearrangement are required for YAP activity and RHAMM transcription. These findings uncover a mechanism regulating RHAMM expression and cancer metastasis, wherein RHAMM functions as a downstream effector of mevalonate and Hippo pathways, a YAP transcriptional target, a simvastatin inhibition downstream target, and a mediator of cancer cell motility and invasiveness; and also identify a mode whereby simvastatin exerts anticancer metastasis efficacy. These insights are of scientific significance and therapeutic and clinical potentials.
Materials and Methods
Detailed materials and methods and associated references are described in SI Materials and Methods, which include the following: cell culture, chemicals, plasmids, cell migration and invasion assays, real-time RT-PCR, Western blot, nuclear and cytoplasmic protein extraction, immunofluorescence microscopy, ChIP, luciferase reporter gene, lentiviral shRNA, breast tumor animal model, immunohistochemistry, and statistical analysis. Transwell cell migration and invasion assays and generation of the mouse model of MDA-MB-231 cell-derived tumor and treatment with simvastatin were performed essentially as described previously (25). All animal studies were carried out according to the protocols approved by the Administrative Committee on Animal Research of the Graduate School at Shenzhen, Tsinghua University.
Supplementary Material
Acknowledgments
We thank Dr. Volker Assmann for providing RHAMM antibody, Dr. Makiko Fujii for plasmid V5-YAP, Dr. Kunliang Guan for plasmid Flag-MST2, and other members of the laboratory of L.H. for discussions and assistance. This work was supported by China Ministry of Science and Technology National Key Basic Research 973 Program 2005CCA03500 (to L.H.), National Natural Science Foundation of China 30570960 (to L.H.), Guangdong Province Natural Science Foundation 05010197 (to L.H.), and Shenzhen Municipal Science and Technology Programs for Building State and Shenzhen Key Laboratories 2006464, 200712, SG200810150043A, CXB201005260070A, CXB201104220043A, and ZDSY20120616222747467 (to L.H.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1319190110/-/DCSupplemental.
References
- 1.Ferlay J, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Pujana MA, et al. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat Genet. 2007;39(11):1338–1349. doi: 10.1038/ng.2007.2. [DOI] [PubMed] [Google Scholar]
- 3.Maxwell CA, et al. Interplay between BRCA1 and RHAMM regulates epithelial apicobasal polarization and may influence risk of breast cancer. PLoS Biol. 2011;9:e1001199. doi: 10.1371/journal.pbio.1001199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Telmer PG, Tolg C, McCarthy JB, Turley EA. How does a protein with dual mitotic spindle and extracellular matrix receptor functions affect tumor susceptibility and progression? Commun Integr Biol. 2011;4(2):182–185. doi: 10.4161/cib.4.2.14270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maxwell CA, McCarthy J, Turley E. Cell-surface and mitotic-spindle RHAMM: Moonlighting or dual oncogenic functions? J Cell Sci. 2008;121(pt 7):925–932. doi: 10.1242/jcs.022038. [DOI] [PubMed] [Google Scholar]
- 6.Wang C, et al. The overexpression of RHAMM, a hyaluronan-binding protein that regulates ras signaling, correlates with overexpression of mitogen-activated protein kinase and is a significant parameter in breast cancer progression. Clin Cancer Res. 1998;4(3):567–576. [PubMed] [Google Scholar]
- 7.Assmann V, et al. The pattern of expression of the microtubule-binding protein RHAMM/IHABP in mammary carcinoma suggests a role in the invasive behaviour of tumour cells. J Pathol. 2001;195(2):191–196. doi: 10.1002/path.941. [DOI] [PubMed] [Google Scholar]
- 8.Rein DT, et al. Expression of the hyaluronan receptor RHAMM in endometrial carcinomas suggests a role in tumour progression and metastasis. J Cancer Res Clin Oncol. 2003;129:161–164. doi: 10.1007/s00432-003-0415-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H, et al. Expression of hyaluronan receptors CD44 and RHAMM in stomach cancers: relevance with tumor progression. Int J Oncol. 2000;17:927–932. [PubMed] [Google Scholar]
- 10.Lugli A, et al. Overexpression of the receptor for hyaluronic acid mediated motility is an independent adverse prognostic factor in colorectal cancer. Mod Pathol. 2006;19:1302–1309. doi: 10.1038/modpathol.3800648. [DOI] [PubMed] [Google Scholar]
- 11.Gust KM, et al. RHAMM (CD168) is overexpressed at the protein level and may constitute an immunogenic antigen in advanced prostate cancer disease. Neoplasia. 2009;11(9):956–963. doi: 10.1593/neo.09694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tolg C, Poon R, Fodde R, Turley EA, Alman BA. Genetic deletion of receptor for hyaluronan-mediated motility (Rhamm) attenuates the formation of aggressive fibromatosis (desmoid tumor) Oncogene. 2003;22(44):6873–6882. doi: 10.1038/sj.onc.1206811. [DOI] [PubMed] [Google Scholar]
- 13.Teder P, Bergh J, Heldin P. Functional hyaluronan receptors are expressed on a squamous cell lung carcinoma cell line but not on other lung carcinoma cell lines. Cancer Res. 1995;55(17):3908–3914. [PubMed] [Google Scholar]
- 14.Du YC, Chou CK, Klimstra DS, Varmus H. Receptor for hyaluronan-mediated motility isoform B promotes liver metastasis in a mouse model of multistep tumorigenesis and a tail vein assay for metastasis. Proc Natl Acad Sci USA. 2011;108:16753–16758. doi: 10.1073/pnas.1114022108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akiyama Y, et al. Hyaluronate receptors mediating glioma cell migration and proliferation. J Neurooncol. 2001;53:115–127. doi: 10.1023/a:1012297132047. [DOI] [PubMed] [Google Scholar]
- 16.Maxwell CA, Keats JJ, Belch AR, Pilarski LM, Reiman T. Receptor for hyaluronan-mediated motility correlates with centrosome abnormalities in multiple myeloma and maintains mitotic integrity. Cancer Res. 2005;65(3):850–860. [PubMed] [Google Scholar]
- 17.Turley EA, Belch AJ, Poppema S, Pilarski LM. Expression and function of a receptor for hyaluronan-mediated motility on normal and malignant B lymphocytes. Blood. 1993;81:446–453. [PubMed] [Google Scholar]
- 18.Tolg C, et al. RHAMM promotes interphase microtubule instability and mitotic spindle integrity through MEK1/ERK1/2 activity. J Biol Chem. 2010;285(34):26461–26474. doi: 10.1074/jbc.M110.121491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joukov V, et al. The BRCA1/BARD1 heterodimer modulates ran-dependent mitotic spindle assembly. Cell. 2006;127(3):539–552. doi: 10.1016/j.cell.2006.08.053. [DOI] [PubMed] [Google Scholar]
- 20.Hamilton SR, et al. The hyaluronan receptors CD44 and Rhamm (CD168) form complexes with ERK1,2 that sustain high basal motility in breast cancer cells. J Biol Chem. 2007;282(22):16667–16680. doi: 10.1074/jbc.M702078200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hatano H, et al. RHAMM/ERK interaction induces proliferative activities of cementifying fibroma cells through a mechanism based on the CD44-EGFR. Lab Invest. 2011;91(3):379–391. doi: 10.1038/labinvest.2010.176. [DOI] [PubMed] [Google Scholar]
- 22.Demierre MF, Higgins PD, Gruber SB, Hawk E, Lippman SM. Statins and cancer prevention. Nat Rev Cancer. 2005;5(12):930–942. doi: 10.1038/nrc1751. [DOI] [PubMed] [Google Scholar]
- 23.Clendening JW, Penn LZ. Targeting tumor cell metabolism with statins. Oncogene. 2012;31(48):4967–4978. doi: 10.1038/onc.2012.6. [DOI] [PubMed] [Google Scholar]
- 24.Liao JK. Isoprenoids as mediators of the biological effects of statins. J Clin Invest. 2002;110(3):285–288. doi: 10.1172/JCI16421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mandal CC, Ghosh-Choudhury N, Yoneda T, Choudhury GG, Ghosh-Choudhury N. Simvastatin prevents skeletal metastasis of breast cancer by an antagonistic interplay between p53 and CD44. J Biol Chem. 2011;286(13):11314–11327. doi: 10.1074/jbc.M110.193714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghosh-Choudhury N, Mandal CC, Ghosh-Choudhury N, Ghosh Choudhury G. Simvastatin induces derepression of PTEN expression via NFkappaB to inhibit breast cancer cell growth. Cell Signal. 2010;22(5):749–758. doi: 10.1016/j.cellsig.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Freed-Pastor WA, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012;148(1-2):244–258. doi: 10.1016/j.cell.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao B, Tumaneng K, Guan KL. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol. 2011;13(8):877–883. doi: 10.1038/ncb2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pan D. Hippo signaling in organ size control. Genes Dev. 2007;21(8):886–897. doi: 10.1101/gad.1536007. [DOI] [PubMed] [Google Scholar]
- 30.Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19(4):491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harvey K, Tapon N. The Salvador-Warts-Hippo pathway - an emerging tumour-suppressor network. Nat Rev Cancer. 2007;7(3):182–191. doi: 10.1038/nrc2070. [DOI] [PubMed] [Google Scholar]
- 32.Zhao B, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21(21):2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dupont S, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474(7350):179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 34.Yu FX, et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell. 2012;150(4):780–791. doi: 10.1016/j.cell.2012.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bao Y, Hata Y, Ikeda M, Withanage K. Mammalian Hippo pathway: From development to cancer and beyond. J Biochem. 2011;149(4):361–379. doi: 10.1093/jb/mvr021. [DOI] [PubMed] [Google Scholar]
- 36.Lamar JM, et al. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc Natl Acad Sci USA. 2012;109(37):E2441–E2450. doi: 10.1073/pnas.1212021109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Overholtzer M, et al. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci USA. 2006;103(33):12405–12410. doi: 10.1073/pnas.0605579103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X, Su L, Ou Q. Yes-associated protein promotes tumour development in luminal epithelial derived breast cancer. Eur J Cancer. 2012;48(8):1227–1234. doi: 10.1016/j.ejca.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 39.Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer. 2013;13(4):246–257. doi: 10.1038/nrc3458. [DOI] [PubMed] [Google Scholar]
- 40.Dong J, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130(6):1120–1133. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hill VK, et al. Frequent epigenetic inactivation of KIBRA, an upstream member of the Salvador/Warts/Hippo (SWH) tumor suppressor network, is associated with specific genetic event in B-cell acute lymphocytic leukemia. Epigenetics. 2011;6(3):326–332. doi: 10.4161/epi.6.3.14404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Denoyelle C, et al. Cerivastatin, an inhibitor of HMG-CoA reductase, inhibits the signaling pathways involved in the invasiveness and metastatic properties of highly invasive breast cancer cell lines: an in vitro study. Carcinogenesis. 2001;22(8):1139–1148. doi: 10.1093/carcin/22.8.1139. [DOI] [PubMed] [Google Scholar]
- 43.Clendening JW, et al. Dysregulation of the mevalonate pathway promotes transformation. Proc Natl Acad Sci USA. 2010;107(34):15051–15056. doi: 10.1073/pnas.0910258107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343(6257):425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 45.Hong W, Guan KL. The YAP and TAZ transcription co-activators: Key downstream effectors of the mammalian Hippo pathway. Semin Cell Dev Biol. 2012;23(7):785–793. doi: 10.1016/j.semcdb.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sohr S, Engeland K. RHAMM is differentially expressed in the cell cycle and downregulated by the tumor suppressor p53. Cell Cycle. 2008;7(21):3448–3460. doi: 10.4161/cc.7.21.7014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.