

Significance
Ghrelin is a peptide released from the empty stomach into circulation, and which acts on the brain to promote appetite. Recent studies showed that ghrelin also affects cognition, but the mechanism for this effect is unknown. We show that activation of ghrelin receptors in the hippocampus enhances synaptic signaling by glutamate, the major excitatory neurotransmitter in the brain. Ghrelin promotes the synaptic accumulation of glutamate receptors of the AMPA subtype, and increases long-term potentiation, one form of synaptic plasticity that is thought to underlie learning and memory. These molecular effects of ghrelin in the hippocampus may contribute to the cognition-enhancing role of ghrelin.
Abstract
Ghrelin is a peptide mainly produced by the stomach and released into circulation, affecting energy balance and growth hormone release. These effects are guided largely by the expression of the ghrelin receptor growth hormone secretagogue type 1a (GHS-R1a) in the hypothalamus and pituitary. However, GHS-R1a is expressed in other brain regions, including the hippocampus, where its activation enhances memory retention. Herein we explore the molecular mechanism underlying the action of ghrelin on hippocampal-dependent memory. Our data show that GHS-R1a is localized in the vicinity of hippocampal excitatory synapses, and that its activation increases delivery of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic-type receptors (AMPARs) to synapses, producing functional modifications at excitatory synapses. Moreover, GHS-R1a activation enhances two different paradigms of long-term potentiation in the hippocampus, activates the phosphatidylinositol 3-kinase, and increases GluA1 AMPAR subunit and stargazin phosphorylation. We propose that GHS-R1a activation in the hippocampus enhances excitatory synaptic transmission and synaptic plasticity by regulating AMPAR trafficking. Our study provides insights into mechanisms that may mediate the cognition-enhancing effect of ghrelin, and suggests a possible link between the regulation of energy metabolism and learning.
The appetite-stimulating peptide ghrelin is a 28-aa peptide predominantly produced by X/A-like cells in the oxyntic glands of the stomach as well as in the intestine (1), and secreted into the blood stream. This peptide promotes pituitary growth hormone secretion, through activation of the growth hormone secretagogue type 1a receptor (GHS-R1a) or ghrelin receptor (2). Additionally, ghrelin is involved in the regulation of energy balance by increasing food intake and reducing fat utilization (3). Plasma ghrelin levels rise before meals and decrease thereafter (4), a pattern which is consistent with the implication of ghrelin in preprandial hunger and meal initiation. Ghrelin is secreted into the circulation and crosses the blood–brain barrier (5, 6), but there is also evidence for ghrelin synthesis locally in the brain (2, 7, 8). The GHS-R1a receptor mRNA was initially found in the hypothalamus and in the pituitary gland (9), and later detected in the hippocampus (10). GHS-R1a is a G protein-coupled seven-transmembrane domain receptor (3), which can signal through guanine nucleotide-binding protein (G protein) subunit alpha 11 (Gq class) to activate phosphatidylinositol-specific phospholipase C, generating 1,4,5-triphosphate (IP3) responsible for Ca2+ intracellular release from endoplasmic reticulum, and diacylglicerol, which in turn activates protein kinase C (PKC) (11). Ghrelin receptor activation is also coupled to the phosphatidylinositol 3 (PI3)-kinase signaling cascade in different cellular systems through a pertussis toxin-sensitive G protein (Gi/oα) (11), and to protein kinase A (PKA) in isolated hypothalamic neurons, modulating N-type Ca2+ channels (12).
The finding that GHS-R1a is expressed in the hippocampus raises the possibility that ghrelin, similarly to other appetite-regulating hormones such as leptin (13), may affect brain functions other than those related to endocrine and metabolic regulation (14). Indeed, in the last few years several studies have shown that ghrelin increases memory retention in rodents, and that the hippocampus participates in this effect (6, 15–17). Ghrelin-deficient mice exhibit decreased novel object recognition, a type of memory test dependent on hippocampal function (6), suggesting that endogenous ghrelin has a physiological role in improving learning and memory. Additionally, high-fat and high-glucose diets, which inhibit ghrelin secretion (18, 19), impair hippocampus-dependent synaptic plasticity and spatial memory (20, 21). On the other hand, caloric restriction, which results in an increase in the circulating levels of ghrelin (22), decreases aging-related deficiencies in cognitive processes (23) while increasing learning consolidation and facilitating synaptic plasticity (24). Recent evidence suggests an enhancing effect of ghrelin on long-term potentiation (LTP) in the hippocampus (6, 17), a form of activity-dependent synaptic plasticity which is the cellular correlate for learning and memory (25). However, conclusive evidence is still lacking because one study did not observe effects of ghrelin on LTP induced by theta burst stimulation (6), whereas the other only detected effects of ghrelin on a late phase of LTP [2 h after high-frequency stimulation (17)].
Although the function of ghrelin as a cognitive enhancer is well documented, the molecular mechanisms that underlie this function are still poorly understood. Here we have tested whether the activation of GHS-R1a affects the trafficking of α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptors (AMPARs), crucial for the expression of changes in synaptic strength in the hippocampus (26). We report that GHS-R1a localizes to excitatory synapses and that its activation induces the synaptic delivery of GluA1-containing AMPAR (GluA1-AMPAR) in rat hippocampal cultures and in CA1 cells in organotypic hippocampal slices. These changes enhance excitatory synaptic transmission. Furthermore, we show that ghrelin receptor activation enhances LTP expression in the CA3–CA1 synapse in organotypic hippocampal slices, and increases the synaptic and cell-surface trafficking of GluA1-AMPAR induced by chemical LTP in hippocampal cultures. Finally, we demonstrate that ghrelin receptor activation in the hippocampus increases the phosphorylation of GluA1 and stargazin. Taken together our data indicate that ghrelin receptor activation regulates AMPARs trafficking underlying synaptic plasticity and learning.
Results
GHS-R1a Localizes to Excitatory Hippocampal Synapses.
Previous evidence suggests that GHS-R1a is expressed in the hippocampus (6, 27–29), but the subcellular localization of GHS-R1a in hippocampal cells has not been studied. Primary hippocampal neurons were immunolabeled at 15 d in vitro (DIV) for endogenous GHS-R1a. GHS-R1a is distributed throughout dendrites and forms clusters that partially colocalize with the glutamatergic synapse markers PSD-95, a postsynaptic scaffold, and Vglut1, a presynaptic vesicular glutamate transporter (Fig. 1A). To evaluate the presence of GHS-R1a at excitatory synapses, we identified regions of overlap between the PSD-95 and Vglut1 signals and measured the GHS-R1a immunolabeling at these sites (Fig. 1B). We found that 31 ± 0.02% of the clusters positive for both PSD-95 and Vglut1, presumably corresponding to functional synapses, contain GHS-R1a (Fig. 1 B and C), which accounts for 36 ± 7.3% of the total GHS-R1a fluorescence intensity in dendrites. Interestingly, only 23 ± 0.01% of the PSD-95 positive sites contain GHS-R1a (Fig. 1C), suggesting that GHS-R1a is preferentially localized to the vicinity of functional synapses. This distribution pattern was confirmed in primary hippocampal neurons transfected with GFP-tagged GHS-R1a (30) and immunolabeled at 16 DIV for GFP (Fig. S1 A–C). Furthermore, we observed that GHS-R1a is enriched in crude synaptosomes (P2 fraction) purified from adult rat hippocampi (Fig. 1D), consistent with a synaptic expression of GHS-R1a in the adult rat hippocampus. Finally, we found that GHS-R1a expression levels significantly increase in cultured hippocampal neurons from 7 up to 19 DIV (Fig. 1E). Taken together, this evidence indicates that a significant fraction of GHS-R1a is localized in the vicinity of hippocampal glutamatergic synapses, and that its expression is regulated during development, thus suggesting a role for ghrelin in regulating excitatory synaptic transmission.
Fig. 1.

GHS-R1a localizes to excitatory synapses and is developmentally regulated in the hippocampus. (A) Representative immunofluorescence images of 15-DIV hippocampal neurons in culture stained for GHS-R1a, PSD-95, and Vglut1. (Scale bar: 5 µm.) (B) Quantification of the number of clusters per dendritic length that are positive for both PSD-95 and Vglut1 and of the number of glutamatergic synapses that contain GHS-R1a (PSD-95/Vglut1/GHS-R1a). n represents the total number of analyzed cells in four independent experiments. Error bars represent SEM. (C) The fraction of synapses containing GHS-R1a was calculated by evaluating the presence of GHS-R1a at regions of overlap between PSD-95 and Vglut1 clusters, or at PSD-95 clusters. n represents the total number of analyzed cells in four independent experiments. Error bars represent SEM. The statistical significance was calculated using the Mann–Whitney test (**P < 0.01). (D) GHS-R1a is enriched in purified crude synaptosomes. Synaptosomal fractions (P2) isolated from adult rat hippocampi were analyzed for the presence of GHS-R1a, PSD-95, synaptophysin (Synapt.), and actin, as indicated. The plot indicates the enrichment in each protein in the synaptosomal fraction relative to hippocampal homogenate fraction (S1), normalized to actin. Error bars represent SEM. The statistical significance was calculated using the Student t test (*P < 0.05 and **P < 0.01). n represents the number of independent experiments. (E) Developmental profile for the expression of GHS-R1a in hippocampal-cultured neurons. GHS-R1a expression levels increase from 7 to 19 DIV, remaining high up to 21 DIV. The plot represents the mean intensity of GHS-R1a bands normalized to tubulin, relative to 7 DIV. Error bars represent SEM. The statistical significance was calculated using the Student t test (*P < 0.05). n represents the number of independent experiments.
GHS-R1a Activation Enhances Excitatory Synaptic Transmission by Inducing GluA1-AMPAR Synaptic Delivery.
To explore the effect of ghrelin receptor activation on the cell surface and synaptic expression of GluA1-AMPAR, we performed quantitative immunofluorescence analysis of the expression of synaptic cell-surface GluA1 in 19 DIV hippocampal neurons treated with the nonpeptidyl GHS-R1a agonist MK-0677, the synthetic molecule initially used for expression cloning of the ghrelin receptor (9). Hippocampal neurons were treated with the GHS-R1a agonist (1 µM) for 1 h to mimic acute changes in ghrelin levels throughout the day (4), or with the agonist simultaneously with the GHS-R1a antagonist [D-Lys3]-GHRP-6 (100 µM), and live stained with an antibody against an extracellular epitope in the GluA1 N terminus. After fixation, neurons were stained for MAP2 to visualize the dendritic structure, and for PSD-95 and Vglut1 to visualize excitatory synapses (Fig. 2A). We verified that neuronal treatment with the GHS-R1a agonist for 1 h did not change the number of Vglut1 clusters per dendritic length (Fig. 2D), and have therefore used Vglut1 as a glutamatergic synaptic marker in these experimental conditions. Neurons treated with MK-0677 showed a significant increase in the fluorescence intensity of total (Fig. 2B) and Vglut1-colocalized GluA1 surface clusters (Fig. 2C), normalized to the number of Vglut1 clusters per dendritic length; this effect was abolished in the presence of the GHS-R1a antagonist (Fig. 2 A–C). The role of GHS-R1a activation on cell-surface expression of GluA1-AMPAR was further tested in acute hippocampal slices prepared from juvenile mice (8–10 wk). We found that treatment with the GHS-R1a agonist for 2 h significantly increased the cell-surface levels of GluA1, assessed using a cell-surface biotinylation assay (Fig. 2E). These observations suggest that MK-0677 increases the levels of AMPARs at the neuronal surface and at synapses through a mechanism that is specifically mediated by the ghrelin receptor. Moreover, these observations suggest that ghrelin receptor activation, in response to an acute increase in ghrelin plasma levels, modulates the synaptic trafficking of AMPARs.
Fig. 2.
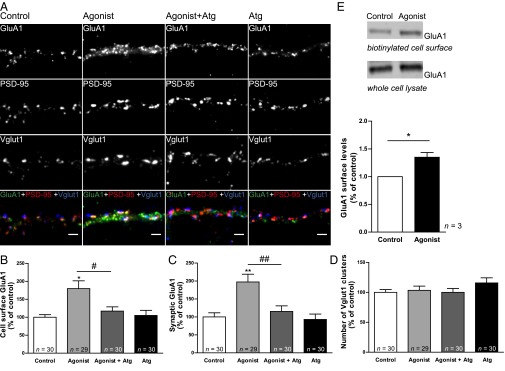
Ghrelin receptor activation promotes the synaptic expression of GluA1. (A) Hippocampal neurons 19 DIV were incubated with the ghrelin receptor agonist MK-0677 (1 µM) for 1 h, with the agonist in the presence of the ghrelin receptor antagonist [D-Lys3]-GHRP-6 (Atg; 100 µM), or with the antagonist alone. Neurons were live stained for GluA1 using an antibody against an extracellular epitope in the GluA1 N terminus. After fixation, neurons were stained for PSD-95, Vglut1, and MAP2. (Scale bars: 2 µm.) Neurons were analyzed for the total GluA1 cell-surface fluorescence intensity (B) and for the GluA1 synaptic cluster (Vglut1-colocalized) fluorescence intensity (C) per number of Vglut1 clusters per dendritic length. (D) The number of Vglut1 clusters per dendritic length was evaluated. (B–D) Results are expressed as the percentage of control cells, and are averaged from three independent experiments. Error bars represent SEM. The statistical significance was calculated using the Kruskal–Wallis test (P = 0.0045 and P = 0.0002) followed by Dunn’s multiple comparison test (*P < 0.05 and **P < 0.01). Comparisons between pairs were performed using the Mann–Whitney test (#P < 0.05 and ##P < 0.01). n represents the total number of analyzed cells in independent preparations. (E) Cell-surface expression of GluA1-AMPAR in acute hippocampal slices as determined using cell-surface biotinylation. (Upper) Representative Western blots of cell-surface and total GluA1 in control and MK-0677-treated acute slices from 8 to 10-wk mice. (Lower) Corresponding histogram of GluA1 surface levels. The plot represents the mean intensity of GluA1 bands normalized to the transferrin receptor, relative to untreated slices. Error bars represent SEM. The statistical significance was calculated using the Student t test (*P < 0.05). n represents the number of independent experiments.
To directly determine whether GHS-R1a activation induces the delivery of new AMPARs into synapses, we expressed GluA1-GFP in CA1 neurons in organotypic hippocampal slice cultures. Overexpression of GluA1, with a Sindbis virus-expression system, leads to the formation of homomeric AMPARs containing the GluA1 subunit (31). These GluA2-lacking receptors are inwardly rectifying (32–34), and therefore their recruitment to the synapse can be monitored as an increase in the ratio of the evoked postsynaptic current at –60 mV relative to the current at +40 mV (rectification index based on ref. 31). In this system it was previously shown that newly synthesized GluA1-containing AMPARs are not spontaneously inserted at synapses unless driven by strong synaptic stimulation or activation of specific signaling pathways associated with LTP induction (31, 35). Organotypic slice cultures were infected with GluA1-GFP at 1 or 2 DIV, and the electrophysiology recordings were performed at 3 or 4 DIV. Organotypic hippocampal slices were treated with 1 µM ghrelin, a concentration of ghrelin that was previously found to result in a maximal effect on the neuronal firing rate of nigral dopaminergic neurons, and in vivo dopamine release in the striatum (29). Organotypic slice cultures were treated for 20 h with the purpose of mimicking fasting, a nutritional status which has been associated with a prolonged increase in ghrelin circulating levels (4). Treatment of organotypic slices with ghrelin (1 µM, Fig. 3A) or with the GHS-R1a agonist MK-0677 (1 µM, Fig. 3B) for 20 h did not change the rectification index in uninfected cells, indicating that GHS-R1a activation does not alter the intrinsic rectification properties of endogenous AMPARs. Interestingly, we found that slice treatment with either ghrelin (Fig. 3A) or MK-0677 (Fig. 3B) increases the rectification index in neurons that express GluA1-GFP, strongly suggesting that ghrelin receptor activation induces synaptic delivery of AMPARs. Importantly, the effect of MK-0677 on the synaptic delivery of GluA1-AMPAR was blocked by the GHS-R1a antagonist [D-Lys3]-GHRP-6 (100 µM, Fig. 3B). In addition, we tested whether the observed effect requires synaptic activity, and found that in the presence of the N-methyl-D-aspartate-type receptors (NMDARs) antagonist 2-amino-5-phosphonopentanoic acid (AP5, 100 µM), or of the voltage-gated sodium channel blocker tetrodotoxin (TTX, 1 µM), the rectification index of GluA1-GFP–expressing neurons was not significantly changed upon incubation with the GHS-R1a agonist MK-0677 (Fig. 3B). Taken together these results indicate that activation of the hippocampal ghrelin receptor induces delivery of GluA1 homomeric receptors into synapses in an activity-dependent manner.
Fig. 3.

Ghrelin receptor activation induces activity-dependent GluA1-AMPAR synaptic delivery in CA1 neurons. (A and B) Voltage-clamp whole-cell recordings obtained in CA1 neurons expressing GluA1-GFP (infected cells) and adjacent nonfluorescent neurons (uninfected cells). GluA1 synaptic delivery was quantified as an increase in the rectification index (RI = I–60/I+40). (A) Ghrelin induces GluA1-AMPAR synaptic delivery in CA1 neurons. Average rectification indices for ghrelin-treated cells (1 µM, 20 h) and untreated cells. (B) The ghrelin receptor agonist MK-0677 induces activity-dependent GluA1-AMPAR synaptic delivery. Average rectification indices for untreated cells, ghrelin receptor agonist-treated cells (1 µM, 20 h), ghrelin receptor agonist-treated cells in the presence of the antagonist of the ghrelin receptor [D-Lys3]-GHRP-6 (Atg; 100 µM), the voltage-gated sodium channel inhibitor TTX, or the NMDAR antagonist AP5. (C and D) Comparison of evoked synaptic responses in control organotypic hippocampal slices and in slices treated for 20 h with the ghrelin receptor agonist MK-0677 (1 µM). (C) Average AMPA/NMDA ratios for agonist-treated and control cells. (D) Average NMDA/GABA ratios for agonist-treated and control cells. (A–D) Representative traces appear above the corresponding bars. Error bars represent SEM. The statistical significance was calculated using the Mann–Whitney test (*P < 0.05, **P < 0.01, ***P < 0.001, #P < 0.05, and ##P < 0.01). n represents the number of cells. (Scale bars: vertical, 50 pA; horizontal, 20 ms.)
To evaluate whether the induction of AMPAR synaptic delivery mediated by GHS-R1a activation produces a functional modification at excitatory CA3–CA1 synapses, 2 or 3 DIV organotypic hippocampal slices were treated with the GHS-R1a agonist MK-0677 (1 µM, 20 h) and electrophysiological recordings were performed at 3 or 4 DIV. The AMPA/NMDA ratio of synaptic responses significantly increased after MK-0677 treatment compared with control neurons (Fig. 3C), whereas the NMDA/GABA ratio was not changed (Fig. 3D). Additionally, MK-0677 treatment did not change the passive membrane properties of the neuron, indicating that cell-wide ion channel conductance was not altered (Fig. S2A). The resting membrane potential and frequency of spontaneous action potential firing were also unaltered by MK-0677, in both CA1 (Fig. S2B) and CA3 (Fig. S2C) neurons. We also measured the number of action potentials in response to current injection [frequency-intensity (f-I) curves in Fig. S2 D and E], and found no differences for CA1 neurons between control and agonist-treated slices. CA3 cells did show more action potential firing after treatment (Fig. S2E). However, this was not reflected in higher spontaneous activity in either CA3 or CA1 cells (Fig. S2 B and C). Therefore, it is not likely to contribute to the AMPAR trafficking and synaptic plasticity effects that we report in CA1 cells. These results suggest that GHS-R1a activation produces a functional change at excitatory CA1 synapses, specifically an increase in AMPARs-mediated synaptic transmission. In conclusion, this group of results suggests that a sustained GHS-R1a activation enhances excitatory synaptic transmission by inducing the insertion of new AMPARs at synapses.
GHS-R1a Activation Enhances Hippocampal LTP.
The classic paradigm for activity-dependent synaptic delivery of AMPARs is NMDAR-dependent LTP. Because our data suggest that the synaptic incorporation of AMPARs induced by GHS-R1a activation is an activity-dependent process, we hypothesized that GHS-R1a activation may facilitate LTP-like events in the hippocampus. Therefore we tested whether ghrelin receptor activation affects this form of synaptic plasticity. Hippocampal organotypic slices (2 or 3 DIV) were treated with the GHS-R1a agonist MK-0677 (1 µM, 20 h) and LTP was induced in the CA3–CA1 synapse. LTP induction significantly increased AMPAR-mediated responses in both MK-0677–treated and untreated neurons (Fig. 4 A and B). However, MK-0677 application dramatically enhanced LTP expression [5.0 ± 0.9-fold potentiation with MK-0677 vs. 2.3 ± 0.3-fold potentiation in control neurons (Fig. 4A)]. Additionally, MK-0677 treatment did not have an effect on the nonpotentiated (unpaired) pathway (Fig. S2F). This suggests that GHS-R1a activation facilitates classic NMDAR-dependent LTP. To directly test whether GHS-R1a activation affects NMDAR-triggered delivery of endogenous GluA1-containing AMPARs to synaptic sites, we used a neuronal culture model of LTP [chemical LTP (cLTP)], in which pharmacological activation of NMDARs leads to an increase in the surface expression of AMPARs (36–40). Consistent with previous results, cLTP induction caused a significant increase in the fluorescence intensity of total and Vglut1-colocalized GluA1 surface clusters in 19-DIV hippocampal-cultured neurons (Fig. 5 A–C). We found that pretreatment with the GHS-R1a agonist MK-0677 (1 µM) for 1 h before the cLTP protocol significantly increases the fluorescence intensity of total (Fig. 5 A and B) and Vglut1-colocalized GluA1 surface clusters (Fig. 5 A and C) compared with neurons subjected to cLTP only, suggesting that GHS-R1a activation increases the cLTP-induced delivery of endogenous GluA1-AMPAR to synaptic sites. The same effect was observed for neurons treated for 20 h with MK-0677 (Fig. S3 A–C). The MK-0677-induced increase in the surface expression of AMPARs upon cLTP was abolished by application of the GHS-R1a antagonist [D-Lys3]-GHRP-6 (Fig. 5 A–C and Fig. S3 A–C), in agreement with an effect specifically mediated by ghrelin receptor activation. These data suggest that pretreatment with the GHS-R1a agonist produces an alteration in the trafficking of AMPARs, which facilitates their recruitment to the synapse upon cLTP induction. Altogether, these data suggest that GHS-R1a activation in the hippocampus increases the AMPARs delivery to synapses, facilitating the expression of LTP-like events. Therefore, acute as well as chronic ghrelin receptor activation modulates the mechanisms of AMPAR synaptic trafficking that mediate synaptic plasticity in the hippocampus.
Fig. 4.

Ghrelin receptor activation enhances NMDAR-dependent LTP in the hippocampus. (A and B) The ghrelin receptor agonist MK-0677 enhances long-term synaptic potentiation in the CA3–CA1 synapse. (A, Left) Sample traces of evoked AMPAR-mediated synaptic responses recorded from CA1 neurons at –60 mV before (light gray line) and after (darker line) LTP induction. (Scale bars: vertical, 50 pA; horizontal, 20 ms.) (A, Right) Organotypic slice cultures were incubated in culture medium or in medium containing the ghrelin receptor agonist MK-0677 (1 µM, 20 h). Plot shows quantification of average synaptic potentiation from paired pathway 20–30 min after LTP induction. Error bars represent SEM. The statistical significance was calculated using the Mann–Whitney test (*P < 0.05). n represents the number of cells. (B) Time course of normalized AMPAR-mediated synaptic responses before and after LTP induction. For simplicity, each time point in the plot corresponds to the average of six consecutive stimulations (sampling rate: 0.2 Hz).
Fig. 5.

Ghrelin receptor activation increases the cell-surface trafficking of GluA1-AMPAR mediated by cLTP. (A) Nineteen-day in vitro hippocampal neurons in culture were submitted to the following stimuli: cLTP (300 µM glycine for 3 min in the absence of Mg2+) and cLTP in neurons treated with the ghrelin receptor agonist MK-0677 (1 µM, 1 h) or simultaneously with the ghrelin receptor agonist and antagonist [D-Lys3]-GHRP-6 (Atg; 100 µM). Neurons were kept at 37 °C for 20 min (without glycine) and were live stained for surface GluA1. After fixation, neurons were stained for PSD-95, Vglut1, and MAP2. (Scale bars: 2 µm.) Neurons were analyzed for the total GluA1 cell-surface fluorescence intensity (B) and for the GluA1 synaptic cluster (Vglut1-colocalized) fluorescence intensity (C) per number of Vglut1 clusters per dendritic length. (B and C) Results are expressed as the percentage of control cells, and are averaged from three to six independent experiments. Error bars represent SEM. The statistical significance was calculated using the Kruskal–Wallis test (P < 0.0001) followed by Dunn’s multiple comparison test (*P < 0.05, **P < 0.01, and ***P < 0.001). Comparisons between pairs were performed using the Mann–Whitney test (#P < 0.05 and ##P < 0.01). n represents the number of cells.
GHS-R1a Activation Induces PKC and PI3-Kinase Pathways and Increases GluA1 and Stargazin Phosphorylation in the Hippocampus.
Two distinct but interrelated mechanisms regulate AMPAR function, modulation of the receptor ion channel properties and regulation of the synaptic targeting of the receptor (31, 41), both of which are regulated by receptor phosphorylation (42). We evaluated key events linked to ghrelin receptor activation and playing an important role in the trafficking of AMPARs and consequently in LTP: PKC activation, PI3-kinase activation, GluA1 phosphorylation, and stargazin phosphorylation. These biochemical changes were evaluated in whole-cell extracts from 4-DIV organotypic hippocampal slices acutely and chronically treated with the GHS-R1a agonist MK-0677 (1 µM). The activation of the PI3-kinase pathway was evaluated by the phosphorylation of the PI3-kinase downstream effector Akt at Ser473. MK-0677 application led to rapid (30-min) significant activation of the PI3-kinase pathway (Fig. 6A). To test whether PI3-kinase activation is required for GluA1 synaptic expression upon acute GHS-R1a activation with the agonist, 19-DIV hippocampal-cultured neurons were treated with the MK-0677 agonist (1 µM, 1 h), or with the agonist simultaneously with the PI3-kinase inhibitor LY294002 (10 µM), and live stained with an antibody against an extracellular epitope in the GluA1 N terminus (Fig. 6B). The increase in the fluorescence intensity of total and Vglut1-colocalized GluA1 surface clusters in neurons incubated with MK-0677 was abolished in the presence of the PI3-kinase inhibitor (Fig. 6 B–D), thus suggesting that the rapid increase in GluA1-AMPAR synaptic surface clusters upon GHS-R1a activation (1 h) is dependent on the activation of the PI3-kinase pathway.
Fig. 6.

The ghrelin receptor-induced activation of the PI3-kinase–signaling pathway is required for increased GluA1-AMPAR synaptic trafficking in hippocampal cultured neurons. (A, Upper) Western blot analysis of protein extracts from hippocampal slices incubated with culture medium or with medium containing the ghrelin receptor agonist MK-0677 (1 µM) for 30 min. The primary antibody detected phosphorylation of Akt at Ser473, a residue targeted by PI3-kinase downstream signaling. Total Akt was also detected and tubulin was used as a loading control. (A, Lower) The graph represents the quantification of band intensities relative to control extract. Error bars represent SEM. The statistical significance was calculated using the Student t test (*P < 0.05). n represents the number of independent experiments. (B) Hippocampal neurons 19 DIV were incubated with the ghrelin receptor agonist MK-0677 (1 µM) for 1 h, with the agonist in the presence of the PI3-kinase inhibitor LY294002 (10 µM), or with the inhibitor alone. Neurons were live stained for GluA1 using an antibody against an extracellular epitope in the GluA1 N terminus. After fixation, neurons were stained for PSD-95, Vglut1, and MAP2. (Scale bars: 2 µm.) Neurons were analyzed for the total GluA1 cell-surface fluorescence intensity (C) and for the GluA1 synaptic cluster (Vglut1-colocalized) fluorescence intensity (D) per number of Vglut1 clusters per dendritic length. (C and D) Results are expressed as the percentage of control cells, and are averaged from four independent experiments. Error bars represent SEM. The statistical significance was calculated using the Kruskal–Wallis test (P = 0.0113 and P = 0.0013) followed by Dunn’s multiple comparison test (*P < 0.05). Comparisons between pairs were performed using the Mann–Whitney test (#P < 0.05). n represents the total number of analyzed cells in independent preparations.
Phosphorylation of GluA1 at Ser845 and of stargazin at Ser239/240 was evaluated with the corresponding phosphospecific antibodies. GluA1 phosphorylation at Ser845 (a PKA substrate, Fig. 7A) significantly increased after acute and prolonged treatment with MK-0677, whereas stargazin phosphorylation at Ser239/240 (a PKC and CaMKII substrate) was significantly induced after 20 h incubation with MK-0677 (Fig. 7B), an effect which was abolished by the GHS-R1a antagonist. Interestingly, neither the NMDAR antagonist AP5 (100 µM) nor the voltage-gated sodium channel blocker TTX (1 µM) affected the phosphorylation of these two substrates triggered by GHS-R1a activation (Fig. 7 A and B), whereas both compounds blocked synaptic insertion of GluA1-AMPARs (Fig. 3B). This suggests that ghrelin receptor activation acts through the phosphorylation of these substrates to prime AMPARs for synaptic incorporation, which is an activity- and NMDAR-dependent process. Finally, prolonged MK-0677 application led to an up-regulation of the PKC pathway (Fig. S4A), evaluated by monitoring the phosphorylation of multiple PKC substrates, and to GluA1 phosphorylation at Ser831 (PKC and CaMKII substrate, Fig. S4B), and did not change the activity of CaMKII (Fig. S4C). To test whether activation of PKA (as suggested by GluA1 phosphorylation at Ser845) or of PKC are required for GluA1 surface expression following GHS-R1a activation, we blocked PKA activity with the PKA inhibitor H-89 (1 μM), or PKC activity with the PKC inhibitor bisindolylmaleimide IV (BIM-IV, 1 μM), while incubating neurons with MK-0677, and live stained for cell-surface GluA1 (Fig. 7 C–F). We found that activation of PKA is required for the acute (1-h) effect of MK-0677 on cell-surface GluA1 (Fig. 7 C and D), whereas the prolonged (20-h) effect of MK-0677 on cell-surface GluA1 required PKC activity (Fig. 7 E and F).
Fig. 7.
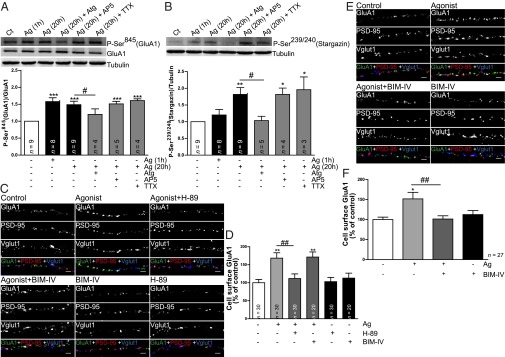
Ghrelin receptor activation triggers changes in GluA1 and stargazin phosphorylation and the PKA and PKC signaling pathways are required for increased GluA1-AMPAR surface expression induced by ghrelin receptor activation in the hippocampus. (A and B, Upper) Western blot analysis of protein extracts from hippocampal slices incubated with culture medium or with medium containing the ghrelin receptor agonist MK-0677 (1 µM) for the indicated periods of time, or with MK-0677 in the presence of the ghrelin receptor antagonist [D-Lys3]-GHRP-6 (Atg; 100 µM), the voltage-gated sodium channel inhibitor TTX or the NMDAR antagonist AP5. The primary antibodies detected phosphorylated GluA1 at Ser845 (A), and phosphorylated stargazin at Ser239/240 (B). Total GluA1 was also detected and tubulin was used as a loading control. (A and B, Lower) The graphs represent the quantification of band intensities relative to control extracts. Error bars represent SEM. The statistical significance was calculated using the one-way analysis of variance (P < 0.0001 and P = 0.0016) followed by Dunnett's multiple comparison test (*P < 0.05, **P < 0.01, and ***P < 0.001). Comparisons between pairs were performed using the Student t test (#P < 0.05). n represents the number of independent experiments. (C–E) Hippocampal neurons 19 DIV were incubated with the ghrelin receptor agonist MK-0677 (1 µM) for 1 h (C and D) or for 20 h (E and F), with the agonist in the presence of the PKA inhibitor H-89 (1 μM) or the PKC inhibitor BIM-IV (1 µM), or with the inhibitors alone. Neurons were live stained for GluA1 using an antibody against an extracellular epitope in the GluA1 N terminus. After fixation, neurons were stained for PSD-95, Vglut1 and MAP2. (Scale bars: 2 µm.) Neurons were analyzed for the total GluA1 cell-surface fluorescence intensity per number of Vglut1 clusters per dendritic length. (D and F) Results are expressed as the percentage of control cells, and are averaged from 2 to 3 independent experiments. Error bars represent SEM. The statistical significance was calculated using the Kruskal–Wallis test (P < 0.0001 and P = 0.0122) followed by Dunn’s multiple comparison test (*P < 0.05 and **P < 0.01). Comparisons between pairs were performed using the Mann–Whitney test (##P < 0.01). n represents the total number of analyzed cells in independent preparations.
Collectively, these results strongly suggest that ghrelin receptor activation initiates signaling mechanisms ultimately producing posttranslational modifications in GluA1-AMPAR and in the AMPAR-associated protein stargazin. These cellular mechanisms may be related to an enhancement of excitatory synaptic transmission, as they may prime GluA1-AMPARs to facilitate their synaptic delivery.
Discussion
The localization of the ghrelin receptor in the hippocampus raises the possibility that signaling through this receptor may control higher brain functions, and represent a molecular link between energy metabolism and learning capabilities. A decade ago Carlini et al. first suggested that ghrelin could affect cognition (15). In this study, intracerebroventricular injection of ghrelin in rats was found to increase the latency time in the step-down inhibitory avoidance test, suggesting an increase in memory retention. Diano et al. showed that ghrelin can enter the CNS and bind to hippocampal neurons, promoting dendritic spine formation. Moreover, they observed that ghrelin treatment of acute hippocampal slices enhances the rise in excitatory postsynaptic potential slope in the CA1 region after a 10-Hz stimulation (6). A recent study found that a single ghrelin infusion prolongs the expression of LTP in the dentate gyrus in vivo (17). Ghrelin knockout animals showed a decreased number of spine synapses in the hippocampus and impaired performance in novel object recognition tests, both of which were rapidly reversed by ghrelin administration (6). This evidence indicates a function for endogenous ghrelin in modulating hippocampal spines and hippocampal-dependent memory, which is further supported by the fact that ghrelin receptor knockout mice display impairments in the Morris water maze test (43). Taken together these studies strongly support a cognition enhancer effect for ghrelin through its hippocampal action. Ghrelin-mediated signaling could provide the link between metabolic requirements, feeding and memory retention, facilitating the successful search for food sources, allowing animals to remember food locations and to retain the successful approach that was used to find them (44, 45).
Despite all of the studies implicating ghrelin and ghrelin receptor-mediated signaling in hippocampal memory processes, little is known about the cellular and molecular mechanisms underlying these effects. In the present study we provide evidence that supports an effect of ghrelin receptor activation on AMPAR synaptic traffic, broadly accepted as a mechanism for the expression of the synaptic plasticity processes thought to be the cellular correlates of learning and memory (46). We found that the ghrelin receptor partially colocalizes with synaptic proteins of glutamatergic synapses in primary hippocampal neurons, and is enriched in synaptosomes purified from adult rat hippocampus, suggesting that it is appropriately localized to modulate excitatory transmission. We report that activation of hippocampal ghrelin receptors promotes the synaptic insertion of AMPARs in the hippocampus. Ghrelin receptor activation resulted in an activity-dependent increase in the inward rectification of AMPAR-mediated responses in CA1 neurons that express GluA1-GFP in hippocampal slices, and produced an increase in the synaptic levels of AMPARs in cultured hippocampal neurons. Moreover, we found that ghrelin receptor activation increases the surface expression of GluA1 in acute hippocampal slices prepared from juvenile mice. These synaptic changes in AMPAR trafficking were paralleled by an increase in the AMPA/NMDA ratio of synaptic responses from CA1 neurons upon ghrelin receptor activation in hippocampal slices. We also show that activation of the ghrelin receptor dramatically enhances LTP expression in organotypic hippocampal slices, and increases the synaptic accumulation of GluA1 triggered by cLTP in cultured hippocampal neurons. Altogether our data indicate that ghrelin receptor activation in the hippocampus, besides promoting dendritic spine formation as previously shown (6), enhances synaptic plasticity by delivering AMPARs into synapses.
How does ghrelin receptor activation affect AMPAR traffic? We found that activation of the ghrelin receptor in the hippocampus leads to changes in the phosphorylation of AMPARs and stargazin. The phosphorylation of Ser845 in GluA1, a substrate for protein kinase A (47), is increased in slices treated with MK-0677, in agreement with a previous study showing that ghrelin can activate PKA in the hippocampus (27), and our data show that the ghrelin receptor-induced increase on cell-surface GluA1 requires PKA activity. Phosphorylation of Ser845 in GluA1 is necessary but not sufficient for synaptic incorporation of GluA1-containing AMPARs (48), and was shown to prime AMPARs for synaptic delivery by trafficking them to extrasynaptic sites; subsequent synaptic incorporation requires synaptic NMDAR activation (49). In fact, we observed that ghrelin receptor-triggered phosphorylation of Ser845 in GluA1 is not affected by TTX or AP5, but the increase in AMPAR synaptic delivery mediated by ghrelin receptor activation is blocked by either TTX or AP5, suggesting an activity and NMDAR-dependent mechanism. These data suggest that activation of the ghrelin receptor promotes GluA1-Ser845 phosphorylation, thereby priming GluA1 for synaptic insertion in an activity-dependent manner. Additionally we found that short-term treatment with the ghrelin receptor agonist activates the PI3-kinase–signaling pathway, in agreement with a previous study in the dentate gyrus (17). PI3-kinase has been implicated in the induction (50) and expression (51) of LTP in the CA1 hippocampal region, and was found to be required for AMPAR insertion during LTP (52). PI3-kinase is responsible for a constant supply of phosphatidylinositol 3,4,5-triphosphate (PIP3) necessary to ensure PSD-95–mediated clustering of AMPARs at the postsynaptic membrane (53). Indeed we found that PI3-kinase–signaling activation is necessary for GluA1 synaptic expression upon 1-h stimulation with the ghrelin receptor agonist in hippocampal-cultured neurons.
The AMPAR-associated protein stargazin, a substrate for PKC and CaMKII (54), was also phosphorylated upon prolonged ghrelin receptor activation in hippocampal slices. Phosphorylation of Ser831 in GluA1, another PKC/CaMKII substrate, was also enhanced. We observed an increase in PKC activity upon ghrelin receptor activation, but could not detect activation of CaMKII, thus suggesting that stargazin and GluA1-Ser831 are phosphorylated by PKC upon ghrelin receptor activation. In fact, the effect of prolonged exposure to the ghrelin receptor agonist on GluA1 cell-surface expression depends on PKC activity. Phosphorylation of stargazin promotes synaptic trafficking of AMPARs, and is required for LTP (54). Upon phosphorylation, stargazin binds to PDZ domain-containing proteins such as PSD-95 and stops the diffusion of cell-surface AMPARs at synaptic sites (55), resulting in increased synaptic AMPAR content.
Together these results suggest that ghrelin receptor activation induces the synaptic delivery of GluA1-containg AMPARs by up-regulating the number of AMPARs that are available for synaptic incorporation, via PKA activity and phosphorylation of GluA1 at Ser845, as well as through activation of PI3K. With longer exposures, ghrelin-induced stargazin phosphorylation will ultimately lead to receptor trapping at the synapse. These observations suggest that upon sustained ghrelin receptor activation, the insertion of AMPARs into synapses is further enhanced due to their synaptic stabilization by phosphorylated stargazin. We hypothesize that in a similar way, increased levels of ghrelin upon fasting might lead to a robust potentiation of glutamatergic transmission. Interestingly, the biochemical changes triggered by ghrelin in the hippocampus do not occlude further LTP expression. In fact, LTP expression in organotypic hippocampal slices and delivery of GluA1-containing AMPARs induced by chemical LTP in hippocampal-cultured neurons are both enhanced upon ghrelin receptor activation. We thus propose that ghrelin receptor activation-triggered signaling acts on targets that facilitate rather than induce LTP expression; in fact, incubation with the ghrelin receptor agonist does not promote CaMKII activation, a key event in LTP expression. Furthermore, ghrelin-induced phosphorylation of GluA1-Ser845 or stargazin are not blocked by TTX or AP5, whereas the ghrelin receptor-triggered synaptic incorporation of GluA1-AMPARs is activity- and NMDAR-dependent, suggesting that the cellular mechanisms triggered by ghrelin act to prime AMPAR for synaptic insertion in an activity-dependent manner, as during LTP.
In conclusion, the present study has provided mechanistic insights into the synaptic events and molecular cascades that mediate enhanced cognition produced by ghrelin. Our findings suggesting that a prolonged period of stimulation of ghrelin receptor enhances the glutamatergic synaptic transmission and synaptic plasticity in the hippocampus are in line with a scenario where synaptic plasticity is enhanced under fasting (24), and are consistent with the fact that a great number of cognitive tests on laboratory rodents and nonhumans primates are routinely performed after a period of fasting.
In addition to inducing a robust feeding response through its hypothalamic action, ghrelin affects other feeding-related behaviors including olfactory function (56), motivational aspects of feeding (57, 58), and hippocampal memory retention (6, 15–17, 43, 59). Similarly to what we describe here for the hippocampal action of ghrelin, it is possible that the effect of ghrelin on other brain regions involves the modulation of glutamatergic transmission.
Materials and Methods
Electrophysiology.
Voltage-clamp whole-cell recordings were performed stimulating Schaffer collateral fibers and recording-evoked synaptic responses from CA1 pyramidal neurons at holding potentials. The AMPA/NMDA ratios were calculated by acquiring AMPAR responses at –60 mV and NMDAR responses at +40 mV at a latency at which AMPAR responses were fully decayed (60 ms after stimulation). Picrotoxin (100 µM) was present in the external solution to block the GABAAR responses. The NMDA/GABA ratios were calculated by recording NMDAR responses at –60 mV and GABAAR responses at 0 mV, in the absence of Mg2+. CNQX (10 µM) was present in the external solution to block AMPAR responses. For the rectification studies, GluA1-GFP was expressed in CA1 neurons for 48 h, and AMPAR responses were recorded at –60 mV and +40 mV in the presence of 100 µM AP5 in the external solution and 100 µM spermine in the internal solution. The rectification index was calculated as the ratio between the AMPAR synaptic response at –60 mV and +40 mV. LTP was induced using a pairing protocol by stimulating Schaffer collateral fibers at 3 Hz for 1.5 min (300 pulses) while depolarizing the CA1 postsynaptic cell to 0 mV.
Quantitative Imaging Analysis.
Imaging was performed on a Zeiss Axiovert 200M microscope and on Zeiss LSM 510 Meta or Zeiss LSM710 confocal microscopes using a 63 × 1.4 numerical aperture oil objective. Images were quantified using image analysis software (ImageJ). For quantification, sets of cells were cultured and stained simultaneously, and imaged using identical settings. The region of interest was randomly selected avoiding primary dendrites, and dendritic length was measured using MAP2 staining. Measurements were performed in two to six independent preparations, and at least five cells per condition were analyzed for each preparation. Quantitative imaging quantification was performed as previously described (60).
Statistical Analyses.
Statistical differences were calculated according to nonparametric tests for most part of the cases; when parametric tests were used, data were first converted to logarithm. Mann–Whitney or Student t tests were used to compare statistical differences between any two groups. Comparisons between multiple groups were performed with the Kruskal–Wallis analysis of variance and one-way analysis of variance followed by Dunn’s multiple comparison test and Dunnett's multiple comparison test.
Additional details are available in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Dr. Helen Wise (The Chinese University of Hong Kong) for the GHS-R1a-GFP construct, Dr. Andrew Irving (University of Dundee) for the anti-GluA1 N-terminal antibody, and Dr. Carlos B. Duarte (Center for Neuroscience and Cell Biology, University of Coimbra) for the critical reading of the manuscript. We also thank Elisabete Carvalho for assistance in the preparation of cultured hippocampal neurons. L.F.R. and T.C. were supported by Fundação para a Ciência e a Tecnologia (FCT). This work was supported by FCT and Fundo Europeu de Desenvolvimento Regional PTDC/BIA-BCM/113738/2009, PEst-C/SAU/LA0001/2013-2014, PTDC/NEU-NMC/1098/2012, and PTDC/SAU-NEU/099440/2008 (to A.L.C.), and by the Spanish Ministry of Economy (SAF2011-24730 and CSD2010-00045) and Fundacion Ramon Areces (to J.A.E.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1313798111/-/DCSupplemental.
References
- 1.Date Y, et al. Ghrelin, a novel growth hormone-releasing acylated peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans. Endocrinology. 2000;141(11):4255–4261. doi: 10.1210/endo.141.11.7757. [DOI] [PubMed] [Google Scholar]
- 2.Kojima M, et al. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402(6762):656–660. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- 3.Gao Q, Horvath TL. Neurobiology of feeding and energy expenditure. Annu Rev Neurosci. 2007;30:367–398. doi: 10.1146/annurev.neuro.30.051606.094324. [DOI] [PubMed] [Google Scholar]
- 4.Cummings DE, et al. A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes. 2001;50(8):1714–1719. doi: 10.2337/diabetes.50.8.1714. [DOI] [PubMed] [Google Scholar]
- 5.Banks WA, Tschöp M, Robinson SM, Heiman ML. Extent and direction of ghrelin transport across the blood-brain barrier is determined by its unique primary structure. J Pharmacol Exp Ther. 2002;302(2):822–827. doi: 10.1124/jpet.102.034827. [DOI] [PubMed] [Google Scholar]
- 6.Diano S, et al. Ghrelin controls hippocampal spine synapse density and memory performance. Nat Neurosci. 2006;9(3):381–388. doi: 10.1038/nn1656. [DOI] [PubMed] [Google Scholar]
- 7.Lu S, et al. Immunocytochemical observation of ghrelin-containing neurons in the rat arcuate nucleus. Neurosci Lett. 2002;321(3):157–160. doi: 10.1016/s0304-3940(01)02544-7. [DOI] [PubMed] [Google Scholar]
- 8.Cowley MA, et al. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron. 2003;37(4):649–661. doi: 10.1016/s0896-6273(03)00063-1. [DOI] [PubMed] [Google Scholar]
- 9.Howard AD, et al. A receptor in pituitary and hypothalamus that functions in growth hormone release. Science. 1996;273(5277):974–977. doi: 10.1126/science.273.5277.974. [DOI] [PubMed] [Google Scholar]
- 10.Guan XM, et al. Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Brain Res Mol Brain Res. 1997;48(1):23–29. doi: 10.1016/s0169-328x(97)00071-5. [DOI] [PubMed] [Google Scholar]
- 11.Camiña JP. Cell biology of the ghrelin receptor. J Neuroendocrinol. 2006;18(1):65–76. doi: 10.1111/j.1365-2826.2005.01379.x. [DOI] [PubMed] [Google Scholar]
- 12.Kohno D, Gao HZ, Muroya S, Kikuyama S, Yada T. Ghrelin directly interacts with neuropeptide-Y-containing neurons in the rat arcuate nucleus: Ca2+ signaling via protein kinase A and N-type channel-dependent mechanisms and cross-talk with leptin and orexin. Diabetes. 2003;52(4):948–956. doi: 10.2337/diabetes.52.4.948. [DOI] [PubMed] [Google Scholar]
- 13.Moult PR, et al. Leptin regulates AMPA receptor trafficking via PTEN inhibition. J Neurosci. 2010;30(11):4088–4101. doi: 10.1523/JNEUROSCI.3614-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andrews ZB. The extra-hypothalamic actions of ghrelin on neuronal function. Trends Neurosci. 2011;34(1):31–40. doi: 10.1016/j.tins.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 15.Carlini VP, et al. Ghrelin increases anxiety-like behavior and memory retention in rats. Biochem Biophys Res Commun. 2002;299(5):739–743. doi: 10.1016/s0006-291x(02)02740-7. [DOI] [PubMed] [Google Scholar]
- 16.Carlini VP, et al. Differential role of the hippocampus, amygdala, and dorsal raphe nucleus in regulating feeding, memory, and anxiety-like behavioral responses to ghrelin. Biochem Biophys Res Commun. 2004;313(3):635–641. doi: 10.1016/j.bbrc.2003.11.150. [DOI] [PubMed] [Google Scholar]
- 17.Chen L, et al. Local infusion of ghrelin enhanced hippocampal synaptic plasticity and spatial memory through activation of phosphoinositide 3-kinase in the dentate gyrus of adult rats. Eur J Neurosci. 2011;33(2):266–275. doi: 10.1111/j.1460-9568.2010.07491.x. [DOI] [PubMed] [Google Scholar]
- 18.Beck B, Musse N, Stricker-Krongrad A. Ghrelin, macronutrient intake and dietary preferences in long-evans rats. Biochem Biophys Res Commun. 2002;292(4):1031–1035. doi: 10.1006/bbrc.2002.6737. [DOI] [PubMed] [Google Scholar]
- 19.Lomenick JP, Melguizo MS, Mitchell SL, Summar ML, Anderson JW. Effects of meals high in carbohydrate, protein, and fat on ghrelin and peptide YY secretion in prepubertal children. J Clin Endocrinol Metab. 2009;94(11):4463–4471. doi: 10.1210/jc.2009-0949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu A, Molteni R, Ying Z, Gomez-Pinilla F. A saturated-fat diet aggravates the outcome of traumatic brain injury on hippocampal plasticity and cognitive function by reducing brain-derived neurotrophic factor. Neuroscience. 2003;119(2):365–375. doi: 10.1016/s0306-4522(03)00154-4. [DOI] [PubMed] [Google Scholar]
- 21.Stranahan AM, et al. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus. 2008;18(11):1085–1088. doi: 10.1002/hipo.20470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lutter M, et al. The orexigenic hormone ghrelin defends against depressive symptoms of chronic stress. Nat Neurosci. 2008;11(7):752–753. doi: 10.1038/nn.2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Witte AV, Fobker M, Gellner R, Knecht S, Flöel A. Caloric restriction improves memory in elderly humans. Proc Natl Acad Sci USA. 2009;106(4):1255–1260. doi: 10.1073/pnas.0808587106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fontán-Lozano A, et al. Caloric restriction increases learning consolidation and facilitates synaptic plasticity through mechanisms dependent on NR2B subunits of the NMDA receptor. J Neurosci. 2007;27(38):10185–10195. doi: 10.1523/JNEUROSCI.2757-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bliss TV, Collingridge GL. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature. 1993;361(6407):31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 26.Santos SD, Carvalho AL, Caldeira MV, Duarte CB. Regulation of AMPA receptors and synaptic plasticity. Neuroscience. 2009;158(1):105–125. doi: 10.1016/j.neuroscience.2008.02.037. [DOI] [PubMed] [Google Scholar]
- 27.Cuellar JN, Isokawa M. Ghrelin-induced activation of cAMP signal transduction and its negative regulation by endocannabinoids in the hippocampus. Neuropharmacology. 2011;60(6):842–851. doi: 10.1016/j.neuropharm.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berrout L, Isokawa M. Ghrelin promotes reorganization of dendritic spines in cultured rat hippocampal slices. Neurosci Lett. 2012;516(2):280–284. doi: 10.1016/j.neulet.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi L, et al. Peptide hormone ghrelin enhances neuronal excitability by inhibition of Kv7/KCNQ channels. Nat Commun. 2013;4:1435. doi: 10.1038/ncomms2439. [DOI] [PubMed] [Google Scholar]
- 30.Leung PK, et al. The truncated ghrelin receptor polypeptide (GHS-R1b) acts as a dominant-negative mutant of the ghrelin receptor. Cell Signal. 2007;19(5):1011–1022. doi: 10.1016/j.cellsig.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 31.Hayashi Y, et al. Driving AMPA receptors into synapses by LTP and CaMKII: Requirement for GluR1 and PDZ domain interaction. Science. 2000;287(5461):2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- 32.Boulter J, et al. Molecular cloning and functional expression of glutamate receptor subunit genes. Science. 1990;249(4972):1033–1037. doi: 10.1126/science.2168579. [DOI] [PubMed] [Google Scholar]
- 33.Verdoorn TA, Burnashev N, Monyer H, Seeburg PH, Sakmann B. Structural determinants of ion flow through recombinant glutamate receptor channels. Science. 1991;252(5013):1715–1718. doi: 10.1126/science.1710829. [DOI] [PubMed] [Google Scholar]
- 34.Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA—gated glutamate receptor channels depends on subunit composition. Science. 1991;252(5007):851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- 35.Boehm J, et al. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron. 2006;51(2):213–225. doi: 10.1016/j.neuron.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 36.Lu W, et al. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29(1):243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- 37.Passafaro M, Piëch V, Sheng M. Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat Neurosci. 2001;4(9):917–926. doi: 10.1038/nn0901-917. [DOI] [PubMed] [Google Scholar]
- 38.Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305(5692):1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- 39.Kennedy MJ, Davison IG, Robinson CG, Ehlers MD. Syntaxin-4 defines a domain for activity-dependent exocytosis in dendritic spines. Cell. 2010;141(3):524–535. doi: 10.1016/j.cell.2010.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahmad M, et al. Postsynaptic complexin controls AMPA receptor exocytosis during LTP. Neuron. 2012;73(2):260–267. doi: 10.1016/j.neuron.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Derkach V, Barria A, Soderling TR. Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci USA. 1999;96(6):3269–3274. doi: 10.1073/pnas.96.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Henley JM, Barker EA, Glebov OO. Routes, destinations and delays: Recent advances in AMPA receptor trafficking. Trends Neurosci. 2011;34(5):258–268. doi: 10.1016/j.tins.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davis JF, Choi DL, Clegg DJ, Benoit SC. Signaling through the ghrelin receptor modulates hippocampal function and meal anticipation in mice. Physiol Behav. 2011;103(1):39–43. doi: 10.1016/j.physbeh.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moran TH, Gao S. Looking for food in all the right places? Cell Metab. 2006;3(4):233–234. doi: 10.1016/j.cmet.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 45.Olszewski PK, Schiöth HB, Levine AS. Ghrelin in the CNS: From hunger to a rewarding and memorable meal? Brain Res Brain Res Rev. 2008;58(1):160–170. doi: 10.1016/j.brainresrev.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Collingridge GL, Isaac JTR, Wang YT. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci. 2004;5(12):952–962. doi: 10.1038/nrn1556. [DOI] [PubMed] [Google Scholar]
- 47.Roche KW, O’Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16(6):1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- 48.Esteban JA, et al. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6(2):136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- 49.Oh MC, Derkach VA, Guire ES, Soderling TR. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem. 2006;281(2):752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]
- 50.Opazo P, Watabe AM, Grant SGN, O’Dell TJ. Phosphatidylinositol 3-kinase regulates the induction of long-term potentiation through extracellular signal-related kinase-independent mechanisms. J Neurosci. 2003;23(9):3679–3688. doi: 10.1523/JNEUROSCI.23-09-03679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sanna PP, et al. Phosphatidylinositol 3-kinase is required for the expression but not for the induction or the maintenance of long-term potentiation in the hippocampal CA1 region. J Neurosci. 2002;22(9):3359–3365. doi: 10.1523/JNEUROSCI.22-09-03359.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Man HY, et al. Activation of PI3-kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons. Neuron. 2003;38(4):611–624. doi: 10.1016/s0896-6273(03)00228-9. [DOI] [PubMed] [Google Scholar]
- 53.Arendt KL, et al. PIP3 controls synaptic function by maintaining AMPA receptor clustering at the postsynaptic membrane. Nat Neurosci. 2010;13(1):36–44. doi: 10.1038/nn.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tomita S, Stein V, Stocker TJ, Nicoll RA, Bredt DS. Bidirectional synaptic plasticity regulated by phosphorylation of stargazin-like TARPs. Neuron. 2005;45(2):269–277. doi: 10.1016/j.neuron.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 55.Opazo P, et al. CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron. 2010;67(2):239–252. doi: 10.1016/j.neuron.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 56.Tong J, et al. Ghrelin enhances olfactory sensitivity and exploratory sniffing in rodents and humans. J Neurosci. 2011;31(15):5841–5846. doi: 10.1523/JNEUROSCI.5680-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abizaid A, et al. Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J Clin Invest. 2006;116(12):3229–3239. doi: 10.1172/JCI29867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Malik S, McGlone F, Bedrossian D, Dagher A. Ghrelin modulates brain activity in areas that control appetitive behavior. Cell Metab. 2008;7(5):400–409. doi: 10.1016/j.cmet.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 59.Atcha Z, et al. Cognitive enhancing effects of ghrelin receptor agonists. Psychopharmacology (Berl) 2009;206(3):415–427. doi: 10.1007/s00213-009-1620-6. [DOI] [PubMed] [Google Scholar]
- 60.Santos SD, et al. Contactin-associated protein 1 (Caspr1) regulates the traffic and synaptic content of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-type glutamate receptors. J Biol Chem. 2012;287(9):6868–6877. doi: 10.1074/jbc.M111.322909. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.