

Abstract
Hallervorden-Spatz disease belongs to a group of disorders characterized by predominant involvement of basal ganglia. These cases may present to the psychiatrist with symptoms of depression, nervousness and rarely other psychotic symptoms. Very few cases of this disease have been reported from India. We report a case of Hallervorden-Spatz disease that presented to the psychiatry outpatient department with catatonia. This case highlights how presentation of Hallervorden-Spatz disease may overlap with catatonic symptoms and hence a high index of suspicion is required to make an accurate diagnosis.
Keywords: Catatonia, Hallervorden-Spatz disease, tiger-eye appearance
INTRODUCTION
Hallervorden-Spatz disease is a rare neurological disorder characterized by progressive degeneration of central nervous system, especially the basal ganglia, globus pallidus and substantia nigra. This degeneration is produced by abnormal iron accumulation, with normal levels of this metal in blood and cerebrospinal fluid.[1] Julius Hallervorden and Hugo Spatz first described this disease in 1922 as a form of familial brain degeneration characterized by pyramidal and extrapyramidal symptoms, dysarthria, and dementia. As far as psychiatric manifestations are concerned, these patients usually present with cognitive impairment, depressive symptoms and occasionally psychotic symptoms. We present the case report of a patient with late-onset Hallervorden-Spatz disease, who presented to the psychiatry outpatient department with catatonic symptoms that preceded the onset of Hallervorden-Spatz disease.
CASE REPORT
A 28-year-old married man presented to the psychiatry outpatient department with complaints of staring spells, maintaining odd postures for long time, decreased sleep, not eating or drinking, tremulousness in the body, stiffness of hands and legs and deviation of the angle of mouth. All these complaints were present since 10-15 days. His illness started 4 years back when, without any precipitating stressors, he developed gradual onset suspiciousness on neighbors, irrelevant talk, muttering and gesticulating to self with occasional history of aggressiveness. Although he was taking treatment from some psychiatrist, details of this treatment were not known and moreover he was not maintained on regular treatment. He was brought to the psychiatry outpatient department for acute deterioration in his symptoms.
There was no history of affective features, substance use, any other psychiatric illness and other medical or surgical illness. He was born full-term and had attained childhood milestones normally. He had poor scholastic performance from 5th grade, after which he left studies. Even after his school he did not seem very keen on doing some work. He would frequently change his jobs for no apparent reasons. He was temperamentally quiet, introvert and shy.
His general examination was within normal limits but nervous system examination revealed rigidity and diminished reflexes in all four limbs. Systemic examination did not reveal anything substantial.
In view of his past psychiatric history of four years and increased presenting complaints since 10-15 days, he was provisionally given a diagnosis of catatonic schizophrenia with neuroleptic induced extrapyramidal reaction (EPR). He was admitted to the psychiatry inpatient unit.
Course in the ward
In the psychiatric ward, he was started on clozapine (25 mg HS), trihexyphenidyl (6 mg) and diazepam (5 mg) along with monitoring of vitals and adequate hydration. Since the patient was suspected to have drug induced EPR and clinically he had catatonia, we wanted to make sure that he does not develop drug induced EPR that may have complicated the picture, hence clozapine was the best choice in this case. His routine blood investigations (such as complete blood count, renal and liver function tests) and fundus examination were within normal limits. Brain CT scan revealed bilateral basal ganglia calcification [Figure 1].
Figure 1.
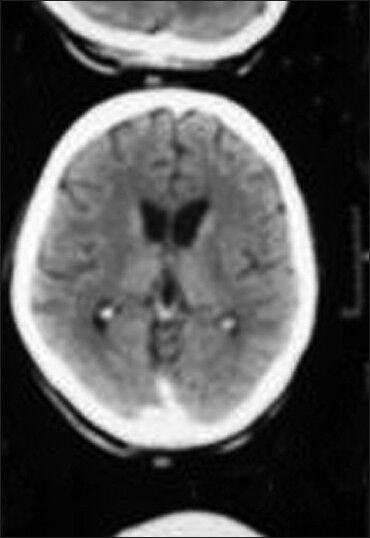
CT scan brain image of patient showing bilateral basal ganglia calcification
As there was no improvement in his catatonic features, he was started on electroconvulsive therapy (ECT) on fourth day of his admission (ECT being the best treatment for catatonia). However, even after 3 ECTs there was no improvement in his rigidity; instead he started remaining confused. Hence all his medications and ECTs were stopped. His serum electrolytes were within normal limits but serum creatine phosphokinase (CPK) levels were elevated to 3493 U/L (Normal limit: 35-232 U/L). A neuromedicine reference was taken and he was subsequently diagnosed as neuroleptic malignant syndrome (NMS) and started on bromocriptine 2.5 mg thrice daily.
After 3 days of bromocriptine, there was improvement in his condition; his confusion and rigidity had decreased considerably. By day 15, his serum CPK levels had decreased (220 U/L) after which he was restarted on clozapine 12.5 mg for psychosis (clozapine being the least D2 antagonist is the safest choice in such cases). However, within the first day of clozapine re-dosing, his confusion increased and condition deteriorated. Hence clozapine was stopped. In view of his increasing psychosis, ECTs were restarted by day 17th. His repeat serum CPK levels on days 18 and 29 showed an increasing level (386 U/L and 563 U/L). His rigidity had also increased by 7th ECT. Valproate was added as he developed affective features in the form of excessive cheerfulness and jocularity, singing songs, and over-familiarity in the ward. Along with this, baclofen and injection magnesium sulfate (4 gm once a day for 3 days) were also added. However, patient's condition further deteriorated and he developed urinary and fecal incontinence. Again both ECTs and valproate were stopped for nearly 15 days during which he was given bromocriptine (2.5 mg thrice daily; dopamine agonist), baclofen (30 mg at bedtime; muscle relaxant) and diazepam (5 mg at bedtime).
Despite these medications, his early morning rigidity, urinary and fecal incontinence did not improve. He was again referred for a neuromedicine opinion in view of persistently rising serum CPK levels and rigidity. The patient was given a differential diagnosis of Hallervorden-Spatz Disease, Wilson's disease, neuroleptic malignant syndrome, Huntington's disease and hypoparathyroidism. Wilson's disease was ruled out by serum ceruloplasmin level (49.23 mg/dl), serum copper (94 μg/dl) and 24 hrs urine copper level (32 μg/24 hrs) and ophthalmic examination for KF ring (which was negative). Huntington's disease was ruled out by genetic testing for CAG repeats. Hypoparathyroidism was ruled out when serum calcium (9.1 mg/dl), serum phosphate (3.6 mg/dl), serum magnesium (2.4 mg/dl), alkaline phosphatase (98 U/L) and PTH (42 pg/ml) levels were reported within normal limits. MRI brain (T2W images) revealed bilaterally symmetric hyperintense signal changes in globus pallidus with surrounding hypointensity [Figure 2] suggestive of tiger-eye appearance found in Hallervorden-Spatz disease. Genetic testing could not be done due to non-availability of the test in our set up. Finally he was given a diagnosis of Hallervorden-Spatz disease by day 49 of admission to psychiatric ward. He was maintained on baclofen 10 mg four times a day and bromocriptine 5 mg thrice a day. Patient showed a gradual improvement, with his psychosis, rigidity and serum CPK level (50 U/L) decreasing. He was discharged at request by 50th day.
Figure 2.
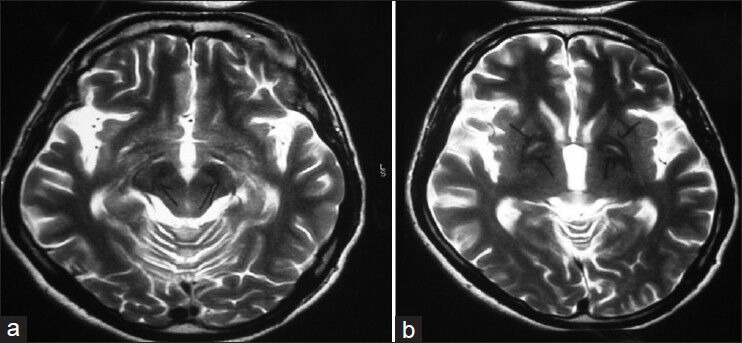
MRI brain image of patient showing the classical ‘eye-of-the-tiger’ appearance
After a fortnightly follow-up, patient was much improved in so far as his psychotic or catatonic symptoms were concerned. He did not have any rigidity, dystonia or neurodeficits. His last CPK levels were within normal limits (67 IU). However the patient was subsequently lost to follow-up after a month.
DISCUSSION
Hallervorden-Spatz Disease (HSD) is a rare genetic disorder with progressive neuronal degeneration and accumulation of iron in the brain. It is also known as Martha-Alma disease, pantothenate kinase associated neurodegeneration (PKAN),[2] neurodegeneration with brain iron accumulation type 1 (NBIA-1),[3] Gilman-Barrett neuro-axonal dystrophy type 1, and pigmentary pallidal degeneration. The disorder has its onset either in late childhood or early adolescence. It may occur as a familial (autosomal recessive) or sporadic disorder. The familial form is associated with mutations in pantothenate kinase gene (PANK2) on chromosome 20 (20p13)[4,5,6] that causes iron storage in the basal ganglia.
Patients may present with psychiatric symptoms such as nervousness, irritability, depressive symptoms, impulsiveness, behavior disorders and cognitive deficits.[7] Cases presenting with psychotic symptoms have been rarely reported in the literature.[8,9,10] Our case was unique in two aspects. First, it presented with psychotic symptoms in the beginning for around four years that worsened to a catatonic state, after which the patient was diagnosed as Hallervorden-Spatz disease. It was interesting that our case presented to us initially with catatonia and there were no symptoms that suggested atypicality in presentation to raise the index of suspicion of coarse brain disease. Secondly, our patient had a normal development of milestones in his childhood, whereas patients of Hallervorden-Spatz disease usually have a history of childhood psychomotor delay.[11,12]
The typical MRI appearance is that of bilaterally symmetric hyperintense signal changes in anterior medial globus pallidus with surrounding hypointensity on T2-weighted images. These imaging features are fairly diagnostic of HSD, especially in the late stages of the disease[12] and have been termed the “eye-of-the-tiger” sign. The hyperintensity represents various pathological changes including gliosis, demyelination, neuronal loss, and axonal swelling, and the surrounding hypointensity is due to loss of signal secondary to iron deposition based on which the diagnosis is made.[13]
There is no treatment for this disease, although a few preliminary studies suggest the safety and tolerability of deferiprone as a chelating agent for both intra- and extra-neuronal iron accumulation[14,15] and also that of bilateral sub-thalamic nucleus deep brain stimulation;[16] the only option that remains is symptomatic treatment of psychiatric and neurological symptoms.
The overall prognosis of this disorder is very poor with the affected individual dying by 2nd to 3rd decade or within one to ten years of onset of severe symptoms.[17,18] Unfortunately, we lost follow-up of our patient within a month of his discharge from our center.
Many medical and neurological disorders may present with psychiatric symptomatology at their outset. It is important to understand that in such situations apart from active management of psychiatric symptoms clinicians need to look out actively for any organic causation of such symptoms.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Pascual GJ. Enfermedades degenerativas del sistema nervioso. In: Rodes J, Guardia J, editors. Medicina Interna. Tomo II. Barcelona: Masson; 1997. [Google Scholar]
- 2.Schneider SA, Hardy J, Bhatia KP. Iron accumulation in syndromes of neurodegeneration with brain iron accumulation 1 and 2: Causative or consequential? J Neurol Neurosurg Psychiatry. 2009;80:589–90. doi: 10.1136/jnnp.2008.169953. [DOI] [PubMed] [Google Scholar]
- 3.Neumann M, Adler S, Schluter O, Kremmer E, Benecke R, Kretzschmar HA. Alpha-synuclein accumulation in a case of neurodegeneration with brain iron accumulation type 1 (NBIA-1, formerly Hallervorden-Spatz syndrome) with widespread cortical and brainstem-type Lewy bodies. Acta Neuropathol. 2000;100:568–74. doi: 10.1007/s004010000224. [DOI] [PubMed] [Google Scholar]
- 4.Taylor TD, Litt M, Kramer P, Pandolfo M, Angelini L, Nardocci N, et al. Homozygosity mapping of Hallervorden-Spatz syndrome to chromosome 20p12.3-p13. Nat Genet. 1996;14:479–81. doi: 10.1038/ng1296-479. [DOI] [PubMed] [Google Scholar]
- 5.Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet. 2001;28:345–9. doi: 10.1038/ng572. [DOI] [PubMed] [Google Scholar]
- 6.Hayflick SJ. First scientific workshop on Hallervorden-Spatz syndrome: Executive summary. Pediatr Neurol. 2001;25:99–101. doi: 10.1016/s0887-8994(00)00273-3. [DOI] [PubMed] [Google Scholar]
- 7.Morphy MA, Feldman JA, Kilburn G. Hallervorden-Spatz disease in a psychiatric setting. J Clin Psychiatry. 1989;50:66–8. [PubMed] [Google Scholar]
- 8.Öner Ö, Öner P, Deda G, Içağasioğlu D. Psychotic disorder in a case with Hallervorder- Spatz disease. Acta Psychiatr Scand. 2003;108:394–7. doi: 10.1034/j.1600-0447.2003.00159.x. [DOI] [PubMed] [Google Scholar]
- 9.Panas M, Spengos K, Koutsis G, Tsivgoulis G, Sfagos K, Kalfakis N, et al. Psychosis as presenting symptoms in adult onset Hallervorden-Spatz syndrome. Acta Neuropsychiatr. 2007;19:122–4. doi: 10.1111/j.1601-5215.2006.00163.x. [DOI] [PubMed] [Google Scholar]
- 10.Sunwoo YK, Lee JS, Kim WH, Shin YB, Lee MJ, Cho IH, et al. Psychiatric disorder in two siblings with Hallervorden-Spatz disease. Psychiatry Investig. 2009;6:226–9. doi: 10.4306/pi.2009.6.3.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rossi D, De Grandis E, Barzaghi C, Mascaretti M, Garavaglia B, Zanotto E, et al. Early-onset neurodegeneration with brain iron accumulation due to PANK2 mutation. Brain Dev. 2012;34:536–8. doi: 10.1016/j.braindev.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 12.Chiapparini L, Savoiardo M, D’Arrigo S, Reale C, Zorzi G, Zibordi F, et al. The “eye-of-the-tiger” sign may be absent in the early stages of classic pantothenate kinase associated neurodegeneration. Neuropediatrics. 2011;42:159–62. doi: 10.1055/s-0031-1285925. [DOI] [PubMed] [Google Scholar]
- 13.Sethi KD, Adams RJ, Loring DW, el Gammal T. Hallervorden-Spatz syndrome: Clinical and magnetic resonance imaging correlations. Ann Neurol. 1988;24:692–4. doi: 10.1002/ana.410240519. [DOI] [PubMed] [Google Scholar]
- 14.Abbruzzese G, Cossu G, Balocco M, Marchese R, Murgia D, Melis M, et al. A pilot trial of deferiprone for neurodegeneration with brain iron accumulation. Haematologica. 2011;96:1708–11. doi: 10.3324/haematol.2011.043018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zorzi G, Zibordi F, Chiapparini L, Bertini E, Russo L, Piga A, et al. Iron-related MRI images in patients with pantothenate kinase-associated neurodegeneration (PKAN) treated with deferiprone: Results of a phase II pilot trial. Mov Disord. 2011;26:1756–9. doi: 10.1002/mds.23751. [DOI] [PubMed] [Google Scholar]
- 16.Ge M, Zhang K, Ma Y, Meng FG, Hu WH, Yang AC, et al. Bilateral subthalamic nucleus stimulation in the treatment of neurodegeneration with brain iron accumulation type 1. Stereotact Funct Neurosurg. 2011;89:162–6. doi: 10.1159/000323374. [DOI] [PubMed] [Google Scholar]
- 17.Hickman SJ, Ward NS, Surtees RA, Stevens JM, Farmer SF. How broad is the phenotype of Hallervorden-Spatz disease? Acta Neurol Scand. 2001;103:201–3. doi: 10.1034/j.1600-0404.2001.103003201.x. [DOI] [PubMed] [Google Scholar]
- 18.Gillberg C. 1st ed. Cambridge: Cambridge University Press; 2003. Clinical Child Neuropsychiatry; p. 210. [Google Scholar]