

Abstract
Immune/inflammatory cells act in rheumatoid arthritis (RA)-affected patients by synthesizing several inflammatory mediators, including cytokines that initiate intracellular signaling. Recently, small molecule inhibitors of transduction and transcription signals that influence the intracellular pathways (such as the Janus kinase [JAK] family of tyrosine kinases) have been tested for RA treatment. Four members of the JAK family are known: JAK1, JAK2, JAK3, and TyK2. JAK1/JAK3 constitutively binds to the cytoplasmic portion of the cytokine receptor – the common gamma chain – that represents a common subunit of several cytokines involved in T-cell and natural killer cell development, as well as in B-cell activation. Tofacitinib is an oral JAK inhibitor that is now available and effective in RA treatment, as shown in multiple Phase II and Phase III clinical trials. However, long-term safety data and comparisons with other disease-modifying antirheumatic drugs and small molecule inhibitors are necessary to better determine the role of tofacitinib in RA.
Keywords: Janus kinase inhibitors, tofacitinib, rheumatoid arthritis, kinases, small molecules inhibitors, intracellular signaling
Targeting rheumatoid arthritis mediators
Rheumatoid arthritis (RA) is a multifactorial, systemic autoimmune disease characterized by inflammatory cell infiltration in the synovial tissue, synovial pannus formation, and it results in subsequent bone erosions, leading to joint destruction and progressive disability.1
The erosive damage reflects the systemic activity of several immune-inflammatory mediators including cytokines, growth and angiogenic factors, as well as adhesion molecules. These products are synthesized by activated immune-inflammatory cells involved in the pathogenesis of RA, such T- and B-lymphocytes, macrophages, neutrophils, as well as fibroblasts and osteoclasts. The inflammatory and degradative factors produced by these cells participate in the erosive activity on cartilage and bone.2
The natural history of RA has dramatically changed since the 1980s with the introduction of methotrexate (MTX), a potent antiproliferative drug with a good safety profile, followed by the approval of other disease-modifying antirheumatic drugs (DMARDs) that allowed for the improved management of RA progression and the easier achievement of disease activity remission.3
In the 1990s, the progressive knowledge of the pathophysiologic mechanisms of RA led to the development of monoclonal antibodies and receptor decoys that were specifically targeted (biologic DMARDs). Antibodies tailored against tumor necrosis factor (TNF)-alpha (α) and interleukin (IL)-1 were initially produced; IL-6, T- and B-cell activation, and several others cytokines (ie, IL-17) inhibitors followed these first discoveries. In addition, biological agents interact with several targets: circulating cytokines or their receptors (ie, TNF-α, IL-1, and IL-6); surface-expressed markers of immune-inflammatory cells (ie, cluster of differentiation [CD]20 on B-cells); and costimulatory molecules stuck on the cell surface (ie, CD80/CD86 on antigen-presenting cells, macrophages, and other cells such as osteoclasts), modulating the interaction between antigen-presenting cells and T-cells.4
It is, nevertheless, critical to notice that these high molecular weight biologic drugs, which bind to cell surface molecules or to secreted proteins, only inhibit or modulate the intercellular pathways, without affecting the intracellular message signaling. The efficacy of these biologic agents, which are able to target the early progression of RA and other inflammatory arthritides when compared to the nonbiologic DMARDs, made them the gold standard of treatment.
On the other hand, their potentially severe side effects and their considerable costs still limit their widespread utilization. During the last few years, a “tolerance effect” of biologic agents, which affects their efficacy over the course of treatment, has been widely observed. The scientific community thus began making an extraordinary effort to discover novel RA drugs that target crucial signals of activated cells.5
Small molecules that block the intracellular cytokine signaling pathways have recently represented a valuable pharmacological approach, alternative, or additive to the current DMARD and biologic therapies. These drugs are easy to synthesize and are available for oral administration. Several protein kinase inhibitor subgroup proteins have been tested to date in randomized clinical trials, targeting mitogen-activated protein kinase/p-38, spleen tyrosine kinase, c-Kit-activated kinases, and Janus kinase (JAK).
In particular, JAKs play an important role in both innate and adaptive immune responses. Cytokine receptors containing the common γ-chain subunit are relevant in initiating the signaling process through JAK1 and JAK3, as observed in RA.1,2 Tofacitinib is a pan-JAK inhibitor targeting the JAK/signal transducer and activator of transcription (STAT) pathway involved in RA.6
JAK/STAT pathway involvement in rheumatoid arthritis
Several kinase cascades, operating during intracellular signaling, have been tested as potential therapeutic targets. As reported previously, JAK, mitogen-activated protein kinase, spleen tyrosine kinase, and nuclear factor kappa–light-chain enhancer of activated B-cells are the better known intracellular pathway actors modulating and transmitting the messages of the proinflammatory cytokines (Figure 1).
Figure 1.
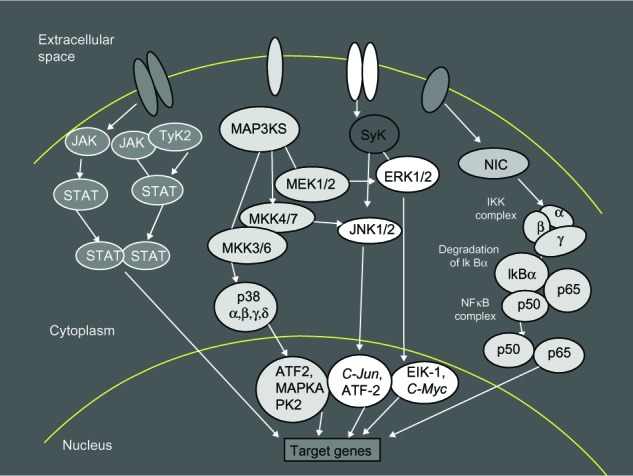
Intracellular signaling pathways.
Notes: Intracellular pathways offer many options for the inhibition of cytokine signaling. JAK/STAT pathways are one of several intracellular hubs in the inflammatory cytokine network. There are several kinase cascades operating for the intracellular signaling; these include: JAK, MAP, Syk, NF-κB, STAT, TyK2, ERK, MAP3KS that phosphorylates and activates a MKK (for example, ERK), JNK, C-Jun, ATF2, Elk-1, C-Myc, NIK, IKK, and IκBα.
Abbreviations: JAK, C-Jun N-terminal kinase; TyK2, tyrosine kinase 2; MAP3KS, mitogen-activated protein kinase kinase kinase; Syk, spleen tyrosine kinase; STAT, signal transducers and activators of transcription; MeK 1/2, dual-specificity kinase MAP kinase kinase; ERK, extracellular signal-regulated kinases; NIC, nuclear factor-kappa B inducing kinase; MKK, mitogen-activated protein kinase kinase; IKK, inhibitor kappa B kinase; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; IkBα, subunits of IKK; ATF2, activating transcription factor 2; MAPKA, mitogen-activated protein kinase A; PK2, protein kinase 2; C-Jun, transcription factor C-Jun; Elk-1, Ets-like protein 1 transcription factor; C-Myc, cellular oncogene; NIK, nuclear factor kappa B inducer kinase.
The JAK family of kinases plays an important role in cytokine-induced signal transduction. Four JAK proteins (JAK1, JAK2, JAK3, and tyrosine kinase 2 [TyK2]) and seven STAT molecules are known; they work together to spread intracellular signals that originate when a cytokine (especially IL-1, IL-2, IL-4, IL-6, IL-7, IL-9, IL-15, IL-17, and IL-21) binds to the receptor. JAKs, as intracellular enzymes, mediate biochemical signals arising from the interactions between cytokines or growth factor and cellular membrane receptors, thus influencing cellular processes like hematopoiesis and immune-mediated responses. Along this signaling process, JAKs phosphorylate and activate STATs which, in turn, modulate intracellular activity and gene expression. JAKs and STATs are critical components of several physiologic pathways, and they regulate proliferation, survival, differentiation, and pathogen action resistance of the cells. The TyK proteins represent the first step of the intracellular signal transduction cascade that follows receptor binding. Other molecules, such interferons, interleukins, colony-stimulating factors, and other cytokines share the same pathway of signal transduction.7
After the interaction receptor/ligand occurs, JAKs bind to the cytoplasmic region of the transmembrane–cytokine receptors, inducing TyK phosphorylation and subsequent STAT activation. STATs act as transcription factors; they are phosphorylated by JAK, dissociate, dimerize via their Src homology 2 domains, and move to the nucleus, where they finally initiate and modulate target gene transcription (Figure 2).8–10 Therefore, during the inflammatory cascade observed in RA in particular, type 1/2 cytokine receptors bind to JAKs, initiating the intracellular signaling and cell reactivity cascade.
Figure 2.

The JAK–STAT intracellular signaling pathway.
Notes: JAK is involved in the intracellular signal transduction pathways via STATs. JAK3 binds to the gamma chain, which is a common receptor subunit for IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21. The JAK inhibitor, tofacitinib, mainly inhibiting JAK3 and JAK1, blocks the synthesis of proinflammatory cytokines (IFN-gamma, IL-6 and, to a lesser extent, IL-12 and IL-23).
Abbreviations: IL, interleukin; JAK, Janus kinase; STAT, signal transducers and activators of transcription; TNF, tumor necrosis factor; IFN, interferon.
Among the others cytokine-binding JAK proteins, IL-1, IL-6, and IL-17 are critically involved in immune-inflammatory cell proliferation and, subsequently, in the pathogenesis of RA.11,12
Several studies, conducted on both animal models and human inflammatory cells, revealed enhanced JAK–STAT pathway gene expression, supporting the pathway’s fundamental role in inflammatory response regulation.
IL-6 is universally recognized as the main activator of the STAT1 complex.13 In another study, increased STAT1 expression has been detected in patients with RA, predominantly in T- and B-cell inflammatory infiltrates and in fibroblast-like synoviocytes, as compared to subjects with osteoarthritis and reactive arthritis.14
STAT3 plays a variety of roles due to the recruitment of distinct target genes in different cell types. It is activated by a variety of cytokines including IL-6, IL-10, and interferons-α/β. Clinical experiences with the anti-IL-6 receptor antibody suggest that the suppression of STAT3 may contrast the inflammation by blocking IL-6, whereas in activation IL-6 leads to fibroblast transformation. In vitro studies of human tissues support the role of STAT3 in contributing to the chronic trend of inflammatory arthritides.15
The JAK–STAT signal transduction pathway is proven to be altered in inflammatory arthritides due to changes in STAT1, STAT3, STAT4, and STAT6 expression.15 Therefore, the expression modulation capability makes JAK–STAT an ideal target for alternative therapeutic approaches to proinflammatory cytokines (ie, TNF-α, IL-1, and IL-6) activity, as observed in RA. RA activity suppression requires multiple actions against the variety of inflammatory cascades involved in the pathophysiology of the disease.16
The results of numerous studies that have indicated increased activation and expression of the JAK–STAT pathway in RA synovial tissue represent increasing amounts of evidence for the pathway’s role as a potential therapeutic target in RA. A new agent now available for RA treatment, belonging to the large tyrosine kinase family, is the JAK inhibitor, tofacitinib.17,18
Clinical pharmacology of tofacitinib
Tofacitinib – formerly designated as CP-690,550 and initially called tasocitinib – is a JAK inhibitor with functional selectivity for the JAK1/JAK2, JAK1/JAK3, and JAK2/JAK2 pathways. The inhibition potency is 20- to 100-fold higher for JAK3 then for JAK1 and JAK2. This drug has actually been developed and tested in different trials in citrate salt form, which is particularly soluble in water, and the commercial formulation (Xeljanz®; Pfizer, Inc., New York, NY, USA) is supplied for oral administration as 5 mg tofacitinib (which is equivalent to 8 mg of tofacitinib citrate).19
The JAK inhibitor, tofacitinib, binds to the adenosine triphosphate-binding cavity of TyK2 and forms polar and nonpolar interactions with a series of residues lining the binding cleft. Blocking the STAT activity, therefore, indirectly influences gene expression and may modulate cellular processes, including hematopoiesis and immune responses. JAK enzymes transmit cytokine signaling through the pairing of JAKs (for example, JAK1/JAK3, JAK1/JAK2, JAK1/TyK2, and JAK2/JAK2).20
Regarding the pharmacodynamics of tofacitinib, it is notable that treatment with this drug entails an early, dose-dependent depletion in the count of T-lymphocytes and B-lymphocytes, which recover 1 month after the drug administration is stopped. Interestingly, tofacitinib treatment in RA patients similarly causes a sudden decrease in C-reactive protein (CRP), which persists even after 2 weeks of drug discontinuation. In fact, peak plasma concentration is reached between 60 minutes and 1 hour after drug administration, while the drug’s half-life ranges around 3 hours at a therapeutic dose that is administered twice daily. Metabolic clearance of tofacitinib is mainly hepatic (70%) and, secondarily, renal (30%).19,20
Janus kinase inhibitor tofacitinib in rheumatoid arthritis clinical trials
After being tested on animal models and on small proof-of-concept trials among humans, and after exhibiting positive results, tofacitinib has been one of the first small molecule inhibitors to be studied in clinical trials among patients with RA.18,21
Phase II and Phase III clinical trials using the JAK inhibitor, tofacitinib, for RA treatment have concluded and are discussed in the sections that follow.
Phase II studies
The results of a number of Phase II studies are summarized in Table 1. Several Phase II (A and B dose-ranging) trials,4,22–26 lasting from 6 to 24 weeks and performed to test tofacitiinb (CP-690,550) in RA patients showed an overall significant improvement in the American College of Rheumatology (ACR)20 score as early as week 2 of treatment, and sustained at week 24.
Table 1.
Summary of the Phase II and Phase III studies conducted on tofacitinib in RA
| Author (reference) | Phase | Population – duration | Dosage of treatment – randomization | Primary endpoint |
|---|---|---|---|---|
| Kremer et al22 | IIA | Active RA inadequate response or intolerance to DMARDs – 6 weeks | Tofacitinib 5 mg, 15 mg, or 30 mg bid or placebo; monotherapy, n=61; placebo, n=65 | ACR20 at week 6 |
| Tanaka et al23 | IIB | Active RA inadequate response to MTX alone (Japanese patients only) – 12 weeks | Tofacitinib 1 mg, 3 mg, 5 mg, or 10 mg bid or placebo; stable background MTX, n=27; placebo, n=28 | ACR20 at week 12 |
| Fleischmann et al24 | IIB | Active RA inadequate response or intolerance to DMARDs – 24 weeks | Tofacitinib 1 mg, 3 mg, 5 mg, 10 mg, or 15 mg bid or placebo, or adalimumab 40 mg once every 2 weeks; monotherapy – (after week 12, nonresponder placebo/tofacitinib 1 mg or 3 mg bid advanced to tofacitinib 5 mg) – tofacitinib 5 mg bid, n=49; placebo, n=59; adalimumab, n=53 | ACR20 at week 12 |
| Tanaka et al25 | II | Active RA with an inadequate response to DMARDs – 12 weeks | Tofacitinib 1 mg, 3 mg, 5 mg, 10 mg, 15 mg or placebo; monotherapy – tofacitinib 5 mg bid, n=50; placebo, n=48 | ACR20 at week 12 |
| Kremer et al26 | IIB | Active RA with an inadequate response to MTX alone – 24 weeks | Tofacitinib 1 mg, 3 mg, 5 mg, 10 mg, or 15 mg bid or 20 mg/daily or placebo; stable background MTX (after week 12, nonresponder placebo/tofacitinib 1 mg or 3 mg bid and 20 mg/day advanced to tofacitinib 15 mg bid) – tofacitinib 5 mg bid, n=71; placebo, n=69 | ACR20 at week 12 |
| van Vollenhoven et al28 | III | Active RA with an inadequate response to MTX alone – 48 weeks | Tofacitinib 5 mg or 10 mg bid or placebo, or adalimumab 40 mg once every 2 weeks; stable background MTX (after week 12, nonresponder placebo were advanced to tofacitinib) – tofacitinib 5 mg bid, n=204; placebo, n=108; adalimumab, n=204 | ACR20 at month 6; DAS28-4 ESR <2.6 at month 6; HAQ-DI at month 3 |
| Fleischmann et al29 | III | Active RA with an inadequate response or intolerance to DMARDs – 24 weeks | Tofacitinib 5 mg or 10 mg bid or placebo; monotherapy; – (after week 12, nonresponder placebo-treated patients were advanced to tofacitinib) – tofacitinib 5 mg bid, n=243; placebo, n=122 | ACR20 response at month 3; DAS28-4 ESR <2.6 at month 3; HAQ-DI at month 3 |
| van der Heijde30 | III | Active RA with an inadequate response to MTX – 48 weeks | Tofacitinib 5 mg or 10 mg bid or placebo; stable background MTX – (after week 12, nonresponder placebo-treated patients were advanced to tofacitinib) – tofacitinib 5 mg bid, n=321; placebo, n=160 | ACR20 response at month 6; DAS28-4 ESR <2.6 at month 6; HAQ-DI at month 3 |
| Burmester et al31 | III | Active RA with an inadequate response or intolerance to TNFi and MTX – 24 weeks | Tofacitinib 5 mg or 10 mg bid or placebo; stable background MTX – (after week 12, placebo-treated patients were advanced to tofacitinib) – tofacitinib 5 mg bid, n=133; placebo, n=132 | ACR20 response at month 3; DAS28-4 ESR <2.6 at month 3; HAQ-DI at month 3 |
Abbreviations: RA, rheumatoid arthritis; DMARDs, disease-modifying antirheumatic drugs; bid, twice a day; ACR20, 20 percent improvement on the American College of Rheumatology symptoms of rheumatoid arthritis; MTX, methotrexate; DAS-28, Disease Activity Score of rheumatoid arthritis based on the assessment of 28 joints; EDC, erythrocyte sedimentation rate; HAQ-DI, Health Assessment Questionnaire–Disability Index; TNFi: tumor necrosis factor inhibitors.
In one early study, the ACR20 response rates by week 6 were 70.5%, 81.2%, and 76.8% in the 5 mg, 15 mg, and 30 mg twice daily groups, respectively, compared with 29.2% in the placebo group (P<0.001). However, the infection rate in both the 15 mg and the 30 mg twice daily group was 30.4% compared to 26.2% in the placebo group.22
Tanaka et al23 compared the efficacy, safety, and tolerability of five doses of tofacitinib monotherapy versus placebo in the treatment of RA among Japanese patients with inadequate response to DMARDs. The primary endpoint, however, was the ACR20 response rate at week 12. All tofacitinib doses were superior when compared with placebo, and a clear dose response was observed. ACR50 and ACR70 response rates also showed a dose response at week 12. The most common adverse events (AEs) were nasopharyngitis, hyperlipidemia, and increased plasma lipid levels; these were reported to be mild in severity.23
In another Phase IIB dose-ranging study,24 tofacitinib or, alternatively, adalimumab monotherapy, was tested versus placebo in patients with active RA who inadequately responded to DMARDs. In this 24-week, double-blind, Phase IIB study, RA patients (n=384) were randomized to receive either placebo; tofacitinib at 1 mg, 3 mg, 5 mg, 10 mg, or 15 mg administered orally twice daily; or 40 mg of adalimumab injected subcutaneously every 2 weeks (a total of six injections), followed by oral tofacitinib at 5 mg twice a day for 12 weeks.
Treatment with tofacitinib at a dose ≥3 mg twice daily resulted in a rapid response with significant efficacy when compared to placebo, as indicated by the primary endpoint (ACR20 response at week 12), which was achieved in 39.2% (3 mg; P<0.05), 59.2% (5 mg; P<0.0001), 70.5% (10 mg; P<0.0001), and 71.9% (15 mg; P<0.0001) of participants in the tofacitinib group. On the other hand, 35.9% of patients in the adalimumab group reached the ACR20 response at week 12 (P not significant); among patients receiving placebo, only 22.0% had the same performance (P not significant).
The most common treatment-related AEs in patients receiving tofacitinib (n=272) were urinary tract infection (7.7%), diarrhea (4.8%), headache (4.8%), and bronchitis (4.8%).24
A further randomized controlled Phase II study25 was conducted among Japanese patients with active RA, and who had an inadequate response to MTX, in order to evaluate the efficacy, safety, and tolerability of 4 mg of oral tofacitinib (CP-690,550) doses in combination with MTX and compared to placebo. ACR20 response rates at week 12 were significantly higher (P<0.0001) in all groups receiving tofacitinib; low disease activity status was achieved by 72.7% of patients, with high baseline disease activity scores (DASs) noted among those receiving tofacitinib 10 mg twice a day at week 12 (P<0.0001). The most common AEs were mild-to-moderate severity nasopharyngitis (n=13), and increased alanine aminotransferase (n=12) and aspartate aminotransferase (n=9) levels. Serious AEs were reported in five patients.25
Another Phase IIB study26 was performed to assess the efficacy, safety, and tolerability of different doses of oral tofacitinib (CP-690,550) in comparison to placebo in active RA patients receiving a stable dose of MTX, but who inadequately responded to this monotherapy. ACR20 response rates, reached at week 12 from patients receiving all tofacitinib dosages ≥3 mg twice daily (52.9% for 3 mg twice daily, 50.7% for 5 mg twice daily, 58.1% for 10 mg twice daily, 56.0% for 15 mg twice daily, and 53.8% for 20 mg/day), were significantly greater (P≤0.05) than the response rates achieved with placebo (33.3%).
Sustained improvements were noticed at week 24 for the ACR20, ACR50, and ACR70 responses, as well as for the Health Assessment Questionnaire Disability Index (HAQ-DI) scores and the three-variable DAS assessed in 28 joints using the CRP level (DAS28–CRP) permanently <2.6.
The most common treatment-related AEs observed in >10% of patients receiving tofacitinib were diarrhea, upper respiratory tract infection, and headache. In 21 patients (4.1%), serious AEs were reported. Occasionally, an increase of transaminase, cholesterol, and serum creatinine levels (which occurred parallel to a decrease in neutrophil and hemoglobin levels) was detected.26
A recent additional study27 was also conducted on patients with psoriasis; the effectiveness and safety of tofacitinib was tested in another Phase IIB, randomized, double-blind, placebo-controlled study conducted among patients with moderate-to-severe psoriasis. By considering this chronic, inflammatory skin disease with a significant impact on health-related quality of life, three tofacitinib dosage regimens and placebo were compared to characterize the efficacy and safety of tofacitinib in patients with moderate-to-severe chronic plaque psoriasis. A total of 197 patients were randomized to tofacitinib 2 mg, 5 mg, 15 mg twice daily, or placebo for 12 weeks. Six different patient-reported outcome (PRO) questionnaires were completed during the study. Treatment with tofacitinib resulted in significant, dose-dependent improvements in several PROs versus placebo from week 2 onwards. At week 12, the least squares mean change from baseline for the Dermatology Life Quality Index, Itch Severity Score, and Short Form-36 questionnaire version 2, mental component scores were significantly greater for all active drug arms versus placebo (P<0.05), and significantly greater for tofacitinib 5 mg and 15 mg for the Short Form-36 physical component scores versus placebo (P<0.05). At week 12, all dose groups had significantly greater numbers of patients reporting “Clear” or “Almost clear” on the Patient Global Assessment of psoriasis versus placebo. Finally, in patients with moderate-to-severe chronic plaque psoriasis, short-term (12-week) treatment with oral twice-daily tofacitinib improved health-related quality of life outcomes and patient assessments of disease severity and symptoms, with an early onset of efficacy.27
Phase III studies
The results of the Phase III studies are summarized in Table 1. In a recent 12-month Phase III trial,28 717 patients receiving stable doses of MTX were randomized to either take 5 mg of tofacitinib twice daily, 10 mg twice daily, 40 mg of adalimumab once every 14 days, or placebo.
ACR20 response after 6 months was significantly higher among patients receiving tofacitinib 5 mg or 10 mg (51.5% and 52.6%, respectively), and among subjects receiving adalimumab (47.2%) than among those receiving placebo (28.3%) (P<0.001 for all comparisons).
In the active-treatment groups compared to placebo, significant reductions in the HAQ-DI score at month 3, and higher percentages of patients with a DAS28–erythrocyte sedimentation rate (ESR) remission (<2.6) at month 6 were observed. Some AEs, which were more likely described among the tofacitinib group than the placebo group, included pulmonary tuberculosis diagnosed in two patients (who belonged to the 10 mg tofacitinib group).
Finally, in the RA patient population receiving a stable dose of MTX, the effectiveness of tofacitinib was similar to that of adalimumab and higher than that of the placebo.28
In a further 6-month, double-blind, placebo-controlled, parallel-group study,29 611 RA patients were randomized following a 4:4:1:1 ratio, either to 5 mg or 10 mg of tofacitinib twice daily, 3 months of placebo followed by a rescue treatment of 5 mg tofacitinib twice daily, or in a similar arm consisting of 3 months of placebo, followed by tofacitinib 10 mg twice daily.
After a 3-month observational period, tofacitinib-treated patients attained a higher ACR20 percentage than placebo (59.8% in the 5 mg tofacitinib group, and 65.7% in the 10 mg tofacitinib group, versus 26.7% in the placebo groups; P<0.001 for both comparisons).
The percentage of patients who exhibited a remission in the DAS28–ESR score, however, was not significantly higher in the tofacitinib group than in the placebo group (5.6% and 8.7% in the 5 mg and 10 mg tofacitinib groups, respectively, versus 4.4% in the placebo group; P=0.62 and P=0.10 for the two comparisons, respectively). It is important to note that serious infections developed in six patients receiving tofacitinib. The most reported AEs were headache and upper respiratory tract infections.29
More recently, two Phase III trials lasting from 6 months to 24 months were conducted to assess safety. In a double-blind, parallel-group, placebo-controlled study, RA-affected patients already treated with MTX were randomized following a 4:4:1:1 ratio to either tofacitinib 5 mg twice daily; tofacitinib 10 mg twice daily; placebo then tofacitinib 5 mg twice daily; or placebo then tofacitinib 10 mg twice daily. Regarding the last two groups, after 3 months, the nonresponder placebo patients were blindly rescued to tofacitinib active treatment, as indicated; the remaining placebo patients were advanced after 6 months of observation. The primary efficacy endpoints were all analyzed in a step-down procedure. ACR20 response rates (at month 6) for tofacitinib 5 mg and 10 mg twice daily were higher than placebo (51.5% and 61.8%, respectively, versus 25.3% for placebo; both P<0.0001); DAS28–ESR <2.6 rates, achieved after the sixth month of treatment, were 7.2% (significance not declared due to the step-down procedure employed) and 16.0% for 5 mg and 10 mg of tofacitinib, respectively, versus placebo (1.6%) (P<0.0001). The safety profile was consistent with that noted in previous studies.28,29 Data from this 12-month interim analysis demonstrated that tofacitinib inhibits the progression of structural damage and improves RA disease activity in patients on MTX baseline therapy.30
Additional data were published at the beginning of 2013 in Lancet31 regarding a 6-month, double-blind, parallel-group Phase III study conducted in 13 countries, including 399 patients with moderate-to-severe RA and exhibiting an inadequate response to TNF inhibitors. Subjects who were already treated with background MTX were randomly assigned in a 2:2:1:1 ratio to receive twice a day treatment with tofacitinib 5 mg (n=133), tofacitinib10 mg (n=134), or placebo (n=132). At month 3, patients given placebo advanced to either tofacitinib 5 mg twice a day (n=66) or 10 mg twice a day (n=66). Primary endpoints included ACR20 response rate, mean change from baseline in HAQ-DI score and rates of DAS28–ESR less than 2.6 – all of which were assessed at month 3. At that time, ACR20 response rates were 41.7% (P=0.0024) for tofacitinib 5 mg twice a day and 48.1% (P<0.0001) for tofacitinib 10 mg twice a day versus 24.4% for placebo. Improvements from baseline in HAQ-DI scores were −0.43 (P<0.0001) for the 5 mg twice a day tofacitinib and −0.46 (P<0.0001) for tofacitinib 10 mg twice a day versus −0.18 for placebo; DAS28 <2.6 rates were 6.7% (P=0.0496) for tofacitinib 5 mg twice a day and 8.8% (P=0.0105) for tofacitinib 10 mg twice a day versus 1.7% for placebo.
The safety profile of the AEs was consistent with previous Phase II and Phase III studies. The most common AEs observed in months 0–3 were diarrhea (4.9%), nasopharyngitis (4.1%), headache (4.1%), and urinary tract infections (3.0%) across the different tofacitinib groups; in addition, nausea (6.8%) was reported among the placebo group. In this treatment-refractory population, tofacitinib, along with MTX, entailed rapid and clinically meaningful improvements in RA signs and symptoms, as well as in physical function over 6 months, with an acceptable safety profile. Tofacitinib could, therefore, provide an effective treatment option in patients with an inadequate response to TNF inhibitors.31–33
Discussion
The targeted immunomodulation and disease-modifying effects exerted by tofacitinib as a JAK/STAT pathway inhibitor and an intracellular signaling modulator have been shown to be effective in RA treatment with an acceptable risk/benefit ratio, whereas the same results have not been achieved by other analogous pathway inhibitors.
Regarding efficacy, the latest clinical findings regarding tofacitinib demonstrated significant ACR20 response rates versus placebo-treated RA patients.34 In terms of safety, the most frequently observed tofacitinib AEs include liver enzyme, creatinine, and serum lipid elevations; neutropenia; a slightly increased incidence of generally benign infections (including herpes zoster, 5%), whereas most of the serious AEs included opportunistic infections, tuberculosis, cancers, gastrointestinal perforations, and lymphoma. These side effects appear due to blockade of signaling of several cytokines (some also exerting anti-inflammatory and anti-infectious actions) that is a logical consequence of the extensive intracellular signaling blockade.6
The balance between the efficacy and safety of tofacitinib, compared to standard-of-care therapy, seems to be acceptable, as better ascertained by recent Phase III clinical trials and meta-analyses that brought attention to this new oral therapy that is now available for RA patients.6,34
In November 2012, the United States Food and Drug Administration approved tofacitinib “to treat adults with moderately to severely active RA who have had an inadequate response to, or who are intolerant of, methotrexate.” The product was subsequently introduced to the market on July 15, 2013 in the US and to other countries beyond Europe, like Japan, Switzerland, Argentina, Kuwait, Russia, and the United Arab Emirates.
Once on the market, rheumatologists complained that the monthly wholesale price of the drug was too expensive, though the price is 7% less than that of related treatments.35 The Committee for Medicinal Products for Human Use of the European Medicines Agency has, nevertheless, adopted a negative view of tofacitinib citrate for the treatment of adult patients with moderate-to-severe active RA. The Committee for Medicinal Products for Human Use of the European Medicines Agency is of the opinion that tofacitinib does not demonstrate a favorable risk/benefit profile at this time (April 2013).36 Therefore, the drug carriers, as recommended by the US Food and Drug Administration, provide a boxed warning regarding these safety issues.37
Conclusion
In order to investigate the global long-term effects of tofacitinib on the human physiology, as well as on its long-term safety, postmarketing studies conducted on a larger RA patient population are required.
Footnotes
Disclosure
The authors report no conflicts of interest in this work. None of the authors has a financial relationship with a commercial entity that has an interest in the subject of the presented manuscript.
References
- 1.McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365(23):2205–2219. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- 2.Emery P, Dörner T. Optimising treatment in rheumatoid arthritis: a review of potential biological markers of response. Ann Rheum Dis. 2011;70(12):2063–2070. doi: 10.1136/ard.2010.148015. [DOI] [PubMed] [Google Scholar]
- 3.Weinblatt ME, Trentham DE, Fraser PA, et al. Long-term prospective trial of low-dose methotrexate in rheumatoid arthritis. Arthritis Rheum. 1988;31(2):167–175. doi: 10.1002/art.1780310203. [DOI] [PubMed] [Google Scholar]
- 4.Cutolo M. The kinase inhibitor tofacitinib in patients with rheumatoid arthritis: latest findings and clinical potential. Ther Adv Musculoskelet Dis. 2013;5(1):3–11. doi: 10.1177/1759720X12470753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fleischmann R. Novel small-molecular therapeutics for rheumatoid arthritis. Curr Opin Rheumatol. 2012;24(3):335–341. doi: 10.1097/BOR.0b013e32835190ef. [DOI] [PubMed] [Google Scholar]
- 6.Salgado E, Maneiro JR, Carmona L, Gomez-Reino JJ. Safety profile of protein kinase inhibitors in rheumatoid arthritis: systematic review and meta-analysis. Ann Rheum Dis. 2013 Apr 18; doi: 10.1136/annrheumdis-2012-203116. Epub. [DOI] [PubMed] [Google Scholar]
- 7.Leonard WJ. Role of Jak kinases and STATs in cytokine signal transduction. Int J Hematol. 2001;73(3):271–277. doi: 10.1007/BF02981951. [DOI] [PubMed] [Google Scholar]
- 8.Ghoreschi K, Jesson MI, Li X, et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550) J Immunol. 2011;186(7):4234–4243. doi: 10.4049/jimmunol.1003668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng H, Ross JA, Frost JA, Kirken RA. Phosphorylation of human Jak3 at tyrosines 904 and 939 positively regulates its activity. Mol Cell Biol. 2008;28(7):2271–2282. doi: 10.1128/MCB.01789-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Funakoshi-Tago M, Tago K, Kasahara T, Parganas E, Ihle JN. Negative regulation of Jak2 by its auto-phosphorylation at tyrosine 913 via the Epo signaling pathway. Cell Signal. 2008;20(11):1995–2001. doi: 10.1016/j.cellsig.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 11.Karaman MW, Herrgard S, Treiber DK, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26(1):127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 12.Ingley E, Klinken SP. Cross-regulation of JAK and Src kinases. Growth Factors. 2006;24(1):89–95. doi: 10.1080/08977190500368031. [DOI] [PubMed] [Google Scholar]
- 13.Yokota A, Narazaki M, Shima Y, et al. Preferential and persistent activation of the STAT1 pathway in rheumatoid synovial fluid cells. J Rheumatol. 2001;28(9):1952–1959. [PubMed] [Google Scholar]
- 14.Kasperkovitz PV, Verbeet NL, Smeets TJ, et al. Activation of the STAT1 pathway in rheumatoid arthritis. Ann Rheum Dis. 2004;63(3):233–239. doi: 10.1136/ard.2003.013276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levy DE, Lee CK. What does Stat3 do? J Clin Invest. 2002;109(9):1143–1148. doi: 10.1172/JCI15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’shea JJ. Targeting the Jak/STAT pathway for immunosuppression. Ann Rheum Dis. 2004;63(Suppl 2):ii67–ii71. doi: 10.1136/ard.2004.028290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanaka Y, Maeshima K, Maeshima Y, Yamaoka K. In vitro and in vivo analysis of a JAK inhibitor in rheumatoid arthritis. Ann Rheum Dis. 2012;71(Suppl 2):i70–i74. doi: 10.1136/annrheumdis-2011-200595. [DOI] [PubMed] [Google Scholar]
- 18.Riese RJ, Krishnaswami S, Kremer J. Inhibition of JAK kinases in patients with rheumatoid arthritis: scientific rationale and clinical outcomes. Best Pract Res Clin Rheumatol. 2010;24(4):513–526. doi: 10.1016/j.berh.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 19.Bannwarth B, Kostine M, Poursac N. A pharmacokinetic and clinical assessment of tofacitinib for the treatment of rheumatoid arthritis. Expert Opin Drug Metab Toxicol. 2013;9(6):753–761. doi: 10.1517/17425255.2013.789500. [DOI] [PubMed] [Google Scholar]
- 20.Rakieh C, Conaghan PG. Tofacitinib for treatment of rheumatoid arthritis. Adv Ther. 2013;30(8):713–726. doi: 10.1007/s12325-013-0047-y. [DOI] [PubMed] [Google Scholar]
- 21.Habib T, Senadheera S, Weinberg K, Kaushansky K. The common gamma chain (gamma c) is a required signaling component of the IL-21 receptor and supports IL-21-induced cell proliferation via JAK3. Biochemistry. 2002;41(27):8725–8731. doi: 10.1021/bi0202023. [DOI] [PubMed] [Google Scholar]
- 22.Kremer JM, Bloom BJ, Breedveld FC, et al. The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: Results of a double-blind, placebo-controlled phase IIa trial of three dosage levels of CP-690,550 versus placebo. Arthritis Rheum. 2009;60(7):1895–1905. doi: 10.1002/art.24567. [DOI] [PubMed] [Google Scholar]
- 23.Tanaka Y, Takeuchi T, Yamanaka H, et al. Tofacitinib (CP-690,550), an oral Janus kinase inhibitor, as monotherapy in Japanese patients with active rheumatoid arthritis: a 12-week Phase 2b study. Arthritis Rheum. 2011;63(S10):2192. [Google Scholar]
- 24.Fleischmann R, Cutolo M, Genovese MC, et al. Phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) or adalimumab monotherapy versus placebo in patients with active rheumatoid arthritis with an inadequate response to disease-modifying antirheumatic drugs. Arthritis Rheum. 2012;64(3):617–629. doi: 10.1002/art.33383. [DOI] [PubMed] [Google Scholar]
- 25.Tanaka Y, Suzuki M, Nakamura H, Toyoizumi S, Zwillich SH, Tofacitinib Study Investigators Phase II study of tofacitinib (CP-690,550) combined with methotrexate in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Care Res (Hoboken) 2011;63(8):1150–1158. doi: 10.1002/acr.20494. [DOI] [PubMed] [Google Scholar]
- 26.Kremer JM, Cohen S, Wilkinson BE, et al. A phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) versus placebo in combination with background methotrexate in patients with active rheumatoid arthritis and an inadequate response to methotrexate alone. Arthritis Rheum. 2012;64(4):970–981. doi: 10.1002/art.33419. [DOI] [PubMed] [Google Scholar]
- 27.Mamolo C, Harness J, Tan H, Menter A. Tofacitinib (CP-690,550), an oral Janus kinase inhibitor, improves patient-reported outcomes in a phase 2b, randomized, double-blind, placebo-controlled study in patients with moderate-to-severe psoriasis. J Eur Acad Dermatol Venereol. 2013 Jan 7; doi: 10.1111/jdv.12081. Epub. [DOI] [PubMed] [Google Scholar]
- 28.van Vollenhoven RF, Fleischmann R, Cohen S, et al. ORAL Standard Investigators Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med. 2012;367(6):508–519. doi: 10.1056/NEJMoa1112072. [DOI] [PubMed] [Google Scholar]
- 29.Fleischmann R, Kremer J, Cush J, et al. ORAL Solo Investigators Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med. 2012;367(6):495–507. doi: 10.1056/NEJMoa1109071. [DOI] [PubMed] [Google Scholar]
- 30.van der Heijde D, Tanaka Y, Fleischmann R, et al. ORAL Scan Investigators Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve-month data from a twenty-four-month phase III randomized radiographic study. Arthritis Rheum. 2013;65(3):559–570. doi: 10.1002/art.37816. [DOI] [PubMed] [Google Scholar]
- 31.Burmester GR, Blanco R, Charles-Schoeman C, et al. ORAL Step investigators Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet. 2013;381(9865):451–460. doi: 10.1016/S0140-6736(12)61424-X. [DOI] [PubMed] [Google Scholar]
- 32.Leah E. Clinical trials: Phase III trial results for tofacitinib bring new oral DMARD therapy a step closer for patients with rheumatoid arthritis. Nat Rev Rheumatol. 2012;8(10):561. doi: 10.1038/nrrheum.2012.145. [DOI] [PubMed] [Google Scholar]
- 33.Kremer J, Zerbini C, Lee EB, et al. Tofacitinib (CP-690,550), an oral Janus kinase inhibitor: analyses of efficacy endpoints by subgroups in a pooled phase 2 and 3 rheumatoid arthritis study population. Ann Rheum Dis. 2012;71(Suppl 3):203–207. [Google Scholar]
- 34.Kawalec P, Mikrut A, Wiśniewska N, Pilc A. The effectiveness of tofacitinib, a novel Janus kinase inhibitor, in the treatment of rheumatoid arthritis: a systematic review and meta-analysis. Clin Rheumatol. 2013;32(10):1415–1424. doi: 10.1007/s10067-013-2329-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garber K. Pfizer’s first-in-class JAK inhibitor pricey for rheumatoid arthritis market. Nat Biotechnol. 2013;31(1):3–4. doi: 10.1038/nbt0113-3. [DOI] [PubMed] [Google Scholar]
- 36.Refusal of the marketing authorization for Xeljanz (tofacitinib) EMA/460814/2013 Available from http://www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion_-_Initial_authorisation/human/002542/WC500146629.pdfAccessed November 5, 2013
- 37.Walsh N, [webpage on the Internet] Pfizer oral RA drug gets FDA approval Little Falls, NJ: MedPage Today, LLC; 2012Available from: http://www.medpagetoday.com/Rheumatology/Arthritis/35799Accessed January 11, 2013 [Google Scholar]