

Abstract
Loss of lung-fluid homeostasis is the hallmark of acute lung injury (ALI). Association of catenins and actin cytoskeleton with vascular endothelial (VE)-cadherin is generally considered the main mechanism for stabilizing adherens junctions (AJs), thereby preventing disruption of lung vascular barrier function. The present study identifies endothelial focal adhesion kinase (FAK), a nonreceptor tyrosine kinase that canonically regulates focal adhesion turnover, as a novel AJ-stabilizing mechanism. In wild-type mice, induction of ALI by intraperitoneal administration of lipopolysaccharide or cecal ligation and puncture markedly decreased FAK expression in lungs. Using a mouse model in which FAK was conditionally deleted only in endothelial cells (ECs), we show that loss of EC-FAK mimicked key features of ALI (diffuse lung hemorrhage, increased transvascular albumin influx, edema, and neutrophil accumulation in the lung). EC-FAK deletion disrupted AJs due to impairment of the fine balance between the activities of RhoA and Rac1 GTPases. Deletion of EC-FAK facilitated RhoA's interaction with p115-RhoA guanine exchange factor, leading to activation of RhoA. Activated RhoA antagonized Rac1 activity, destabilizing AJs. Inhibition of Rho kinase, a downstream effector of RhoA, reinstated normal endothelial barrier function in FAK−/− ECs and lung vascular integrity in EC-FAK−/− mice. Our findings demonstrate that EC-FAK plays an essential role in maintaining AJs and thereby lung vascular barrier function by establishing the normal balance between RhoA and Rac1 activities.
Keywords: focal adhesion kinase, adherens junctions, acute lung injury, endothelial barrier
the vascular endothelium controls the passage of macromolecules and fluid between the blood and interstitial space and thereby plays a vital role in maintaining tissue-fluid homeostasis (20). It is known that loss of endothelial barrier function results in tissue edema, the hallmark of acute lung injury (ALI), which induces ∼40% mortality in affected patients (18, 20, 28). Homotypic interaction between vascular endothelial (VE)-cadherin from contiguous endothelial cells forms adherens junctions (AJs) that primarily maintain endothelial barrier function (20). While VE-cadherin linkage with intracellular catenins (α, β, and p120) and actin cytoskeleton is a well-accepted mechanism for stabilizing AJs, additional cellular molecules may be required.
Focal adhesion kinase (FAK), a nonreceptor tyrosine kinase, regulates endothelial cell-matrix attachment (20, 27). FAK may also maintain AJs by converging on multiple signaling pathways, but this remains controversial (20). We showed that FAK mediates the interaction of p120-catenin with actin-binding machinery by phosphorylating neural-Wiscott Aldrich syndrome protein that stabilizes AJs (15, 25). FAK also suppressed the activity of the small GTPase RhoA to restrict endothelial contraction (13). FAK thereby maintained basal endothelial barrier function and induced resealing of endothelial junctions following the increase in endothelial permeability by thrombin (13, 15, 21). Similarly, Quadri et al. showed that FAK prevents oxidant-induced barrier dysfunction by regulating AJs function (23, 24). Also small-interfering RNA (siRNA)-induced depletion of FAK or expression of dominant-negative FAK impaired AJ formation and enhancement of barrier function by sphingosine-1-phosphate and lysophosphatidic acid (12, 30). However, other studies showed that dominant-negative FAK or a kinase dead FAK mutant prevented AJ disruption in response to VEGF or oxidants, indicating that FAK in fact disrupts barrier function (6, 41, 46). Endothelial-specific deletion of FAK in mice induced embryonic lethality (3, 29). Thus, it remains enigmatic whether FAK preserves lung vascular barrier function and how FAK attains it.
In the present study, we conditionally induced FAK deletion in the endothelium of mice, referred to as EC-FAK−/− mice hereafter, to address this question fundamental to regulation of vascular barrier function. We demonstrate that endothelial FAK deletion spontaneously disrupts lung endothelial barrier function. We show that loss of FAK results in disruption of AJs due to impairment of the fine balance between the activities of RhoA and Rac1 GTPases.
MATERIALS AND METHODS
Materials.
Human pulmonary arterial endothelial cells (HPAECs), endothelial growth medium (EBM-2), Nucleofector kit, and Amaxa electroporation kit were obtained from Lonza. Secondary fluorescent antibodies (Abs) and ProLong Gold antifade were from Invitrogen. FAK, actin, RhoA, VE-cadherin, MLC, and phospho-MLC Abs were purchased from Santa Cruz Biotechnology. p115RhoGEF Ab was a gift from Drs. Tohru Kozasa and Christina Chow (University of Illinois). Anti-Rac1 Ab was purchased from Cell Signaling Technology. GST-rhotekin and GST-PAK binding beads were purchased from Cytoskeleton. Adenoviral β-Gal and Cre recombinase were purchased from Eton Bioscience. Y-27632, tamoxifen, and lipopolysaccharide (LPS, from Escherichia coli strain 055:B5) were from Sigma. Hematoxylin and eosin stain was purchased from Fisher. Control and FAK siRNA were purchased from Dharmacon.
Animals.
Mice were bred and maintained in a pathogen-free animal facility at the University of Illinois (UIC). Mouse studies were approved by the Institutional Animal Care and Use Committee of the UIC. FAKfl/fl mice were generated by inserting two loxP sites flanking the third codon of FAK exon as described (29). Stem cell leukemia (SCL) Cre-ERT mice were generously provided by Drs. David Cheresh and Sarah Weis (Univ. of California at San Diego). Six- to eight-week-old male mice in C57BLk/6J background were used for all experiments. Genotyping primers were as follows: FAK forward: 5′-CGTGATGTCCCAAGCTATTCC-3′, reverse: 5′-AGGCTGGTCTGCGTGACAGG-3′. PCR conditions were as follows: 55° for 20 min, 94°C for 3 min; 25 cycles of 94°C for 30 s, 57°C for 30 s, and 72°C for 105 s; then 72°C for 5 min. Scl-Cre forward: 5′-TCGATGCAACGAGTGATGAG-3′, reverse: 5′-TTCGGCTATACGTAACAGGG-3′. PCR conditions were as follows: 55° for 20 min, 94°C for 3 min; 30 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s; then 72°C for 5 min.
Induction of ALI in mice.
A single dose of 30 mg/kg body wt LPS (L2880, Strain 055:B5, 500,000 EU/mg; Sigma) was injected intraperitoneally in mice to induce lung injury. Cecal ligation and puncture (CLP) was induced by ligating and puncturing cecum as described (38). Control mice were subjected to cecal ligation without puncture.
Immunohistochemistry and immunofluorescence.
Formalin-fixed paraffin-embedded lung sections (5 μM) were stained with hematoxylin and eosin or indicated Abs as described (15, 38). Digital images were collected using a ×63 objective in a Zeiss LSM510 Meta confocal microscope equipped with a three-line laser. Sequential image acquisition (multitrack) minimized potential cross talk between the fluorophores, and images were processed using LSM Image Browser software (Zeiss). Quantitative analysis of FAK deletion in endothelial cells (ECs) was carried out by colocalization of FAK with CD31 using Pearson's correlation coefficients using LSM510 software. Images were further processed with Adobe Photoshop software. Pearson correlation coefficients were calculated across similar regions of CD31 staining between six and nine images. Interendothelial gap area in FAK null and control cells was quantified using Image J software (15).
Endothelial cell culture.
Mouse lung endothelial cells (LECs) were isolated and used following their characterization (38, 40). FAK deletion in FAKfl/fl LECs was induced by infecting them with 25 plaque-forming units/cell of β-Gal (control) or Cre recombinase adenovirus in 0.1% serum and antibiotic-free media for 6 h after which cells were refreshed with complete media containing growth factors, 20% serum, and antibiotics. Experiments were performed between 36 and 48 h postinfection when maximal FAK deletion was achieved. HPAECs cultured as described were used between passages 6 and 8 (15).
FAK depletion.
HPAECs were transfected with scrambled or FAK siRNA (5′-GCAUGUGGCCUGCUAUGGA-5′ sense and 5′-UCCAUAGCAGGCCACAUGC-3′ antisense) (31). Cells were used after 72 h, since at this time maximum depletion of FAK was observed.
Heme quantification.
Heme from lung supernatants was quantified using a QuantiChrom heme assay kit (Bioxys/Gentaur).
Assessment of lung capillary leakage.
Evans blue-labeled albumin extravasation in lung parenchyma and lung wet-to-dry weight ratio were used as indexes of lung vascular barrier function (15, 37).
Myeloperoxidase assay.
Myeloperoxidase activity was measured as described (1). Data are represented as change in absorbance at 450 nm over a 5-min period after addition of H2O2 per gram lung weight.
RhoGTPase activities and immunoblotting.
Lysates were incubated with either PAK-PBD or Rhotekin-PBD beads to determine RhoGTPase activities (13, 37). Where indicated, cell monolayers were incubated with 20 μM SB-203580, 10 μM Y-27632, or DMSO (vehicle) for 30 min before experiment.
Endothelial monolayer permeability.
Monolayer permeability was determined by measuring transendothelial influx of Evans blue-labeled albumin (37).
Statistical analysis.
Statistical comparisons were made using ANOVA followed by two-tailed Student's t-test. Differences were considered significant at P < 0.05.
RESULTS
Lung injury is associated with decreased FAK expression in murine lungs.
To assess whether increased endothelial permeability is associated with decreased FAK expression, we used two well-characterized mouse models of ALI (1, 38). We injected a sublethal dose (30 mg/kg) of LPS intraperitoneally or performed CLP. Intriguingly, both LPS and CLP induced an ∼40% decrease in FAK protein expression (Fig. 1, A and B), indicating that FAK expression is decreased during lung injury.
Fig. 1.

Focal adhesion kinase (FAK) protein expression is decreased during acute lung injury in mice. Lung lysates from wild-type mice exposed to vehicle or lipopolysaccharide (LPS, 30 mg/kg, 6 h) (A) or subjected to cecal ligation (control) or cecal ligation and puncture (CLP, 22 h) (B) were immunoblotted with anti-FAK or anti-β-actin (loading control) antibodies (Abs). Bar graphs show the densitometric analysis of FAK expression from 4 lungs/group (A and B); *statistically significant difference from vehicle or control lungs (P < 0.05).
Endothelial cell-specific FAK deletion impairs lung fluid balance.
To determine whether the decreased endothelial expression of FAK seen in the above studies is causally related to impairment of lung vascular barrier function, we generated mice in which FAK deletion was conditionally induced in endothelial cells. We crossed FAK-floxed mice (FAKfl/fl) (29) with SCL-Cre-ERT mice. Scl-Cre-ERT mice express Cre recombinase in endothelial cells driven by the 5′-enhancer region of the SCL locus in a tamoxifen-inducible manner (10). FAK deletion was induced by tamoxifen administration at 4 wk of age (Fig. 2A). We also subjected aged-matched FAKfl/fl mice to identical tamoxifen treatment to control for its nonspecific effects on endothelium. Recombination of the FAK gene was detected in Cre-expressing FAKfl/fl mice (EC-FAK−/−), whereas no recombination was detected in mice lacking the Cre transgene (FAKfl/fl) (Fig. 2B). Tamoxifen-induced Cre recombinase activity markedly decreased FAK mRNA and protein expression in EC-FAK−/− lung lysates (Fig. 2C). Next, we coimmunostained lung sections obtained from EC-FAK−/− or FAKfl/fl mice with anti-FAK and anti-CD31 (an endothelial cell marker) Abs to determine specific deletion of FAK in the endothelium. As expected, FAKfl/fl lung sections revealed homogenous FAK staining in CD31+ cells (Fig. 2, D and E). However, EC-FAK−/− lungs barely showed FAK staining in CD31+ cells (Fig. 2, D and E). In EC-FAK−/− lungs, tamoxifen did not alter FAK expression in epithelial or smooth muscle cells (Fig. 2F) or in hematopoietic cells (Fig. 2G). Detectable FAK expression in EC-FAK−/− lung lysates reflected FAK expression in nonendothelial cells, since Cre recombinase activation completely deleted FAK in LECs isolated from FAKfl/fl mice (Fig. 2H).
Fig. 2.

Inducible deletion of endothelial FAK. A: schematic of tamoxifen-induced deletion of FAK in CRE-expressing FAKfl/fl mice. Four-week-old FAKfl/fl (control)- and FAKfl/fl-expressing Cre mice were injected with 2 mg tamoxifen ip for 5 consecutive days, followed by a rest period of 5 days. Mice were used for experiments on the 11th day. B: genotype showing tamoxifen deletion of FAK. Tails from FAKfl/fl or endothelial cell (EC)-FAK−/− mice were digested, and FAK and Cre expression was determined using specific primers. Cre induces FAK deletion in Cre-expressing EC-FAK−/− mice that is absent in FAKfl/fl mice. C–E: assessment of endothelial FAK expression. Lungs harvested from FAKfl/fl or EC-FAK−/− mice were homogenized, and FAK mRNA and protein expression was quantified using FAK primers and FAK antibody (C). The bar graph shows mean ± SD of FAK expression normalized against the housekeeping gene GAPDH (for RNA) or β-actin (for protein). *Statistically significant difference from FAKfl/fl lungs (P < 0.05, n = 4–5). D: FAKfl/fl and EC-FAK−/− lung sections were immunostained with anti-FAK (red) and anti-CD31 (green) Abs, followed by appropriate Alexa-fluor-conjugated secondary Abs to assess FAK expression in CD31+ endothelial cells. Scale bar, 20 μM. Bar graph shows %CD31+ endothelial cells that express FAK in EC-FAK null and FAKfl/fl lungs from 6–7 individual vessels from multiple lung sections (E). F: lung sections from FAKfl/fl and EC-FAK−/− mice were either coimmunostained with anti-FAK (green) and anti-E-cadherin (red) or anti-FAK and anti-α-smooth muscle actin (α-SMA; red) Abs to confirm FAK expression in epithelial and smooth muscle cells, respectively. Arrows show similar FAK colocalization with E-cadherin or α-SMA in FAKfl/fl and EC-FAK−/− mice lungs. Scale bars, 10 μM. Images represent 3–4 mice/group. G: macrophages were isolated from bronchoalveloar lavage (BAL) of FAKfl/fl and EC-FAK−/− mice after which RNA was extracted. FAK expression was quantified using suitable primers as described in materials and methods. GAPDH was used as an internal control. Plot represents the mean ± SD from 4 mice. H: lung endothelial cells (LECs) isolated from FAKfl/fl mice infected with β-gal (control, FAK+/+) or Cre adenovirus (FAK−/−) were lysed to assess FAK expression using actin as a loading control. Blot is representative of five individual isolations.
Intriguingly, we observed diffuse hemorrhage in EC-FAK−/− lungs as indicated by the threefold increase in heme levels (Fig. 3A). Additionally, endothelial FAK deletion spontaneously increased lung edema formation as demonstrated by significant increases in transvascular albumin influx and lung wet-to-dry weight ratio in EC-FAK−/− lungs (Fig. 3, B and C). We confirmed that tamoxifen injection alone had no effect on lung vascular permeability or lung wet-to-dry weight ratio in wild-type (WT), Cre, or FAKfl/fl mice (Fig. 3, D and E). Hematoxylin and eosin staining of EC-FAK−/− lungs revealed a threefold increase in lung leukocyte infiltration and a twofold increase in tissue myeloperoxidase activity (Fig. 3, F and G). Also, deletion of FAK did not alter the mRNA expression of related nonreceptor tyrosine kinases, including Fyn, Src, and Pyk2 (Fig. 3H).
Fig. 3.


Endothelial FAK deletion disrupts lung vascular barrier function. A: loss of EC-FAK leads to diffuse lung hemorrhage. Top, representative images of buffer-perfused lungs harvested from EC-FAK−/− and FAKfl/fl mice. Bottom, mean ± SD of heme concentration from four FAKfl/fl and EC-FAK−/− mouse lungs. *Values significantly different from FAKfl/fl lungs (P < 0.05). B and C: EC-FAK deletion increases lung microvascular permeability. Transendothelial albumin influx into lung parenchyma (B) and lung wet-to-dry weight ratio (C) were determined as described in materials and methods to quantify lung vascular protein permeability and lung edema formation, respectively. *Significant difference from FAKfl/fl lungs (P < 0.05; n = 7–8 mice/group). D and E: tamoxifen does not affect lung vascular permeability or lung edema formation in wild-type (WT), CRE, or FAKfl/fl mice. Tamoxifen was injected into WT, CRE, or FAKfl/fl mice following the protocol described in Fig. 1A. Mice were killed at day 11 to determine lung wet-to-dry weight ratios (D) or transendothelial albumin influx into lung parenchyma (E). Data represent means ± SD from three groups of four lungs. F and G: EC-FAK deletion induces lung leukocyte infiltration and activation. Representative images of hematoxylin and eosin (H & E)-stained lung sections from multiple FAKfl/fl and EC-FAK−/− mice (F). Scale bars, 20 μm. G: top, plot shows mean ± SD leukocyte infiltration in the lungs. *Significant difference from FAKfl/fl lungs (P < 0.05, n = 4 mice/group). Bottom, lungs from FAKfl/fl or EC-FAK−/− were harvested, and myeloperoxidase (MPO) activity was quantified as indicated in materials and methods. *Significant difference from FAKfl/fl lungs (P < 0.05, n = 4 mice/group). H: endothelial FAK deletion does not alter mRNA expression of related kinases. RNA was extracted from FAKfl/fl and EC-FAK−/− mouse lungs. The expression of cSrc, Fyn, Pyk2, and FAK was analyzed by RT-PCR using specific primers. GAPDH was used as a loading control. Results represent data from three individual lungs. I–K: FAK deletion disrupts adherens junctions. FAK+/+ and FAK−/− LECs were immunostained with anti-vascular endothelial (VE)-cadherin (green) antibody or rhodamine phallodin (red) as described in materials and methods (I). Immunoblot from FAK+/+ and FAK−/− lysates was probed with indicated Abs (J). *Significant difference between FAK+/+ vs. FAK−/− LECs (n = 3; P < 0.05). K: plot shows mean ± SD of interendothelial gap area in FAK+/+ and FAK−/− monolayers. *Significant difference from FAK+/+ monolayers (P < 0.05, n = 5).
To corroborate the above studies in lungs, we assessed the integrity of FAK−/− endothelial monolayers by determining VE-cadherin and actin organization. FAK deletion impaired cell-surface VE-cadherin localization (Fig. 3I, top) and increased actin stress fiber formation (Fig. 3I, bottom) and MLC phosphorylation (Fig. 3J), resulting in a sixfold elevation in interendothelial gap area (Fig. 3K). Total expression of VE-cadherin, p120-catenin, and MLC proteins was not altered in FAK−/− ECs (Fig. 3J).
FAK deletion disrupts balance between RhoA and Rac1 GTPase activities.
It is well known that RhoA and Rac1 play a key role in maintaining AJ strength (2, 4, 43–45). Thus, we asked whether deletion of FAK compromises endothelial barrier function by modulating RhoA and Rac1 activities. We observed that FAK null endothelial cells showed a twofold increase in RhoA activity (Fig. 4A), but, crucially, the activity of Rac1 was concomitantly decreased by a factor of approximately five (Fig. 4A). Similarly, reduction of FAK levels in HPAECs by 90% using siRNA inactivated Rac1 while inducing RhoA activity (Fig. 4B).
Fig. 4.
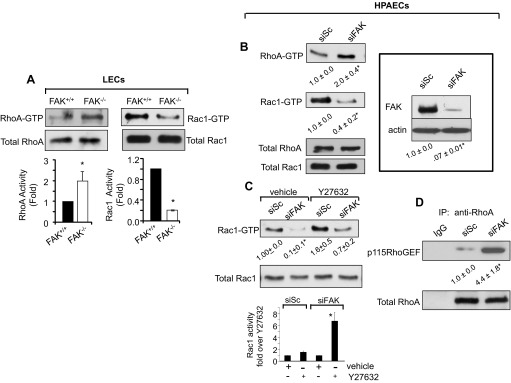
FAK deletion impairs normal balance between RhoA and Rac1 activities. A and B: effect of FAK deletion on RhoA and Rac1 activities. FAK−/− and FAK+/+ LECs were lysed, and the activity of RhoA or Rac1 was determined using rhotekin or PAK pull down beads (A). Data represent means ± SD of GTPase activities compared with those in FAK+/+ LECs. *Significant difference from FAK+/+ LECs (P < 0.05; n = 3). Human pulmonary artery endothelial cells (HPAECs) transfected with scrambled (siSc) or FAK (siFAK) small-interfering RNA (siRNA) were lysed to determine RhoA and Rac1 activities (B). In parallel, lysates were immunoblotted with anti-FAK and anti-actin Abs to assess FAK depletion in ECs using actin as a loading control (inset). Blot is representative of findings from three individual experiments. *Significant difference from siSc-transfected HPAECs (P < 0.05; n = 3). C: RhoA inhibits Rac1 activity. Scrambled and FAK siRNA-expressing HPAECs were incubated with 10 μM Y-27632 to inhibit Rho kinase (ROCK), a downstream effector of RhoA. After 30 min, cells were lysed, and Rac1 activity was determined to assess whether inhibition of ROCK restores normal Rac1 activity. *Significant difference from siSc-transfected HPAECs (P < 0.05; n = 3). In bar graph, Rac1 activity in Y-27632-pretreated siSc- or siFAK-transfected cells is plotted relative to their respective vehicle-treated values to better discern the level of restoration seen in siFAK cells after inhibiting ROCK. *Significant difference from siSc-transfected HPAECs or vehicle-treated siFAK cells (P < 0.05; n = 3). D: FAK depletion promotes RhoA interaction with p115RhoGEF. Lysates from scrambled and FAK siRNA-expressing HPAECs were immunoprecipitated with either control IgG or anti-RhoA antibody followed by immunoblotting with anti-p115RhoGEF and anti-RhoA Abs. The blot shown is representative of data from multiple experiments. *Significant difference from siSc-transfected HPAECs (P < 0.05; n = 4).
Next, we inhibited RhoA signaling using the Rho kinase (ROCK) inhibitor Y-27632 (9, 44) to assess whether activated RhoA was responsible for suppression of Rac1 activity in FAK-depleted ECs. Whereas inhibition of ROCK, which we confirmed by determining the phosphorylation of myosin-binding subunit of myosin phosphatase, modestly increased Rac1 activity in ECs transfected with scrambled siRNA (Fig. 4C), it resulted in an eightfold increase in Rac1 activity in FAK-depleted ECs (Fig. 4C).
RhoA activity is regulated by the GDP-GTP exchange cycle induced by guanine exchange factors (GEFs) such as p115RhoGEF and p190Rho guanine-activating protein (p190RhoGAP) (14, 20, 35). Because p190RhoGAP was shown to require FAK for full guanine-activating protein (GAP) activity (13), we tested the hypothesis if loss of FAK facilitated the complex formation between p115RhoGEF and RhoA, required for RhoA activation (14). Cell lysates from FAK-knockdown cells were immunoprecipitated with anti-RhoA Ab followed by immunoblotting with anti-p115RhoGEF Ab to assess their interaction. We found that RhoA barely interacted with p115RhoGEF in control cells, but, importantly, the interaction of p115RhoGEF with RhoA markedly increased in FAK knockdown ECs (Fig. 4D). No interaction of RhoA was observed with control IgG (Fig. 4D).
Inhibition of RhoA reinstates lung-fluid balance in EC-FAK−/− mice.
We transduced WT-FAK cDNA in FAK null lungs using liposomes (15, 38) to assess whether restoration of FAK expression in EC-FAK null mice suppressed edema formation. WT mice injected with liposomes containing vector were used as controls. Restoration of FAK in EC-FAK−/− mice significantly reduced edema formation (Fig. 5A).
Fig. 5.
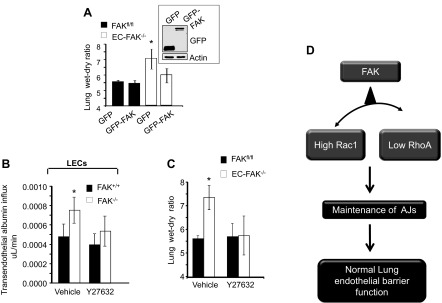
Blockade of RhoA reinstates lung vascular permeability. A: restoration of FAK expression in EC-FAK−/− mice rescues lung vascular permeability. FAKfl/fl or EC-FAK−/− mice expressing indicated constructs were assessed for lung edema formation by determining lung wet-to-dry weight ratios. Graph represents means ± SD. *Significance from GFP or GFP-FAK transducing FAKfl/fl lungs or GFP-FAK transducing EC-FAK−/− lungs (P < 0.05, n = 4). Inset shows representative immunoblots of free GFP or GFP-FAK fusion protein found in lung lysates from EC-FAK−/− mice with the aid of an anti-GFP antibody. A representative immunoblot from 4 mouse lungs/group is shown. All experiments were performed 48 h after injection of liposome-encapsulated mutants. B and C: inhibition of RhoA signaling rescues endothelial barrier function in FAK-depleted ECs and lungs. B: FAK+/+ or FAK−/− LEC monolayers were pretreated with vehicle or 10 μM ROCK inhibitor Y-27632 to inhibit ROCK. After 30 min, transendothelial albumin influx was determined using Evans blue-conjugated albumin. Experiments were performed in duplicate at least three times. *Significant difference from FAK+/+ LECs (P < 0.05). C: FAKfl/fl or EC-FAK−/− mice were retro-orbitally injected with either vehicle or ROCK inhibitor Y-27632 (10 mg/kg). Mice were killed after 30 min, and lung wet-to-dry weight ratio was calculated. *Significance from PBS- or Y-27632-exposed FAKfl/fl lungs or EC-FAK−/− lungs receiving Y-27632 (P < 0.05; n = 3–4 mice/group). D: model of endothelial FAK regulation of lung vascular permeability described in text.
To address whether increased RhoA activity was required and sufficient for increasing lung vascular leak in EC-FAK null mice, we inhibited RhoA signaling in FAK null ECs and EC-FAK null mice and assessed changes in endothelial permeability. RhoA inhibition in FAK−/− ECs restored basal endothelial permeability (Fig. 5B) and reinstated lung-fluid balance in EC-FAK−/− mice (Fig. 5C).
DISCUSSION
We have demonstrated in these studies that endothelial FAK is required for stabilizing AJs and thereby maintains lung-vascular barrier function. We showed that induction of ALI in mice decreases FAK protein expression in the lungs. Importantly, conditional deletion of FAK in endothelial cells facilitated the interaction of p115RhoGEF with RhoA enabling RhoA antagonism of Rac1 activity which in turn disrupted AJs leading to an increase in endothelial permeability. Therefore, our findings identify for the first time a novel role of endothelial FAK in maintaining AJs in pulmonary endothelium by determining the normal balance between RhoA and Rac1 activities.
ALI is a leading cause of death after sepsis (18, 28). Increased endothelial permeability is known to be the primary cause of ALI (18, 20, 28). FAK plays a key role in regulating endothelial permeability in response to several edemagenic mediators (20, 39). However, whether sepsis induces ALI by altering FAK expression remains unclear. We showed that FAK protein expression is decreased following sepsis in murine lungs. Intriguingly, conditional deletion of FAK in mouse endothelial cells induced lung vascular barrier disruption and leukocyte infiltration in the interstitium, recapitulating key features of ALI. Decreased endothelial FAK expression is therefore a likely factor responsible for the pulmonary vascular hyperpermeability and edema formation seen during ALI. The mechanism by which FAK is degraded during ALI in the endothelium remains to be parsed out. LPS activates caspases and calpain (36). FAK contains caspase- and calpain-binding sites (5, 16), making FAK susceptible to degradation during ALI, thereby leading to vascular dysfunction.
AJs formed by intercellular interaction of VE-cadherin primary regulate endothelial permeability (20). Association of catenins and actin cytoskeleton with VE-cadherin is generally considered the main mechanism for maintaining AJ stability (20), thereby preventing disruption of lung vascular barrier function. FAK has been shown to regulate several endothelial cell functions, such as migration, proliferation, and angiogenesis, by regulating the turnover of focal adhesions (20). Our findings showing that FAK deletion markedly impairs cell-surface VE-cadherin expression and elevates actin stress fiber as well as interendothelial gap formation subsequently leading to lung vascular leak in EC-FAK null mice provide unequivocal evidence that FAK is required to maintain AJs. Consistently, studies in endothelial cells have shown that impairment of FAK function also disrupted AJs (13, 15, 21, 23–25, 30, 34). Thus, we conclude that FAK prevented the loss of lung vascular barrier by maintaining AJs.
The monomeric RhoGTPases RhoA and Rac1 play a central role in regulating the integrity of AJs (20). Permeability-increasing mediators rapidly activate RhoA, which mediates stress fiber formation and disruption of AJs (4, 13, 15, 44). In contrast, Rac1 induces AJ formation by stimulating lamellipodia formation and through interaction with the AJ protein p120-catenin (19, 26). Inhibition of Rac1 therefore disorganizes AJs, leading to increased endothelial permeability in cultured cells and mesenteric microvessels (43, 44). These studies indicate that a fine balance between the activities of Rac1 and RhoA is a critical determinant of a stable endothelial barrier. However, until recently, there has been a paucity of information regarding the cellular mechanisms that maintain this crucial balance between RhoA and Rac1 activities in vivo, enabling attainment of lung-fluid homeostasis. Intriguingly, we showed that RhoA activity was markedly elevated in FAK null ECs while Rac1 activity was suppressed, demonstrating FAK to be the cellular mechanism balancing normal RhoA and Rac1 activities. We surmised that FAK directly regulates both RhoA and Rac1 activities (20). However, we showed that inhibition of RhoA was sufficient to restore normal Rac1 activity, suggesting that RhoA antagonizes Rac1 activity. The concept that RhoA can downregulate the activity of Rac1 is supported by recent findings that RhoA suppresses Rac1 activity by activating FILGAP, a specific GTPase for Rac1 (22).
RhoA activity is regulated by the GDP-GTP cycle induced by GEFs and GAPs (20, 35). p190RhoGAP inhibits RhoA activity (17, 20, 32, 33, 35). We showed that p190RhoGAP require FAK for full GAP activity (13). Moreover, p190RhoGAP acts on a GTP-bound RhoA (active RhoA) (17, 20, 32, 33, 35). Thus, we surmised that, in the absence of FAK, another mechanism such as RhoGEF may be involved. In this regard, we focused on p115 RhoGEF, since it binds with GDP-bound RhoA to exchange GTP (11) and may be active at the level of AJs (14, 42). In fact, we observed that RhoA immunoprecipitated with p115RhoGEF in FAK-depleted ECs but failed to do so in control cells. These data are consistent with a model in which endothelial FAK suppresses p115RhoGEF's interaction with RhoA, limiting activation of RhoA and thus its antagonism of Rac1 activity and thereby the loss of lung endothelial barrier function (Fig. 5D). However, details on how FAK regulates the interaction between p115RhoGEF and RhoA remains to be parsed out. Evidence indicates that the activity of several GEFs, including p115RhoGEF, is regulated by phosphorylation-dependent homo-oligomerization of the COOH-terminal region (7, 8). Because p115RhoGEF contains several tyrosine residues, FAK may induce p115RhoGEF homo-oligomerization, inhibiting its interaction with RhoA. Our findings that depletion of FAK promoted p115RhoGEF interaction with RhoA support this concept.
In summary, we showed that FAK expression is significantly decreased in the setting of two well-established murine models of ALI. Conditional loss of FAK in the mouse endothelium leads to hyperactivation of RhoA, which antagonizes Rac1 activity, leading to disruption of AJs and loss of lung vascular barrier function, a characteristic feature of ALI. In this context, normalizing the level of FAK expression in endothelial cells after sepsis may be an attractive pharmacological approach to preventing sepsis-induced lung injury.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-71794, HL-84153, and HL-060678. T. Thennes was supported by HL-007829 and by an American Heart Association predoctoral fellowship (10PRE2610268).
DISCLOSURES
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Author contributions: T.T.S. and D.M. conception and design of research; T.T.S., M.T., and L.Y. performed experiments; T.T.S., M.T., L.Y., and R.T.S. analyzed data; T.T.S., M.T., L.Y., M.G.B., and D.M. interpreted results of experiments; T.T.S., M.T., and L.Y. prepared figures; T.T.S. and D.M. drafted manuscript; T.T.S. and D.M. edited and revised manuscript; T.T.S., M.T., L.Y., M.G.B., J.G., T.-L.S., J.-L.G., S.A.P., R.T.S., and D.M. approved final version of manuscript.
ACKNOWLEDGMENTS
We acknowledge Drs. A. B. Malik, S. Vogel, and R. D. Minshall (University of Illinois at Chicago, IL) for constructive criticism during preparation of this manuscript.
REFERENCES
- 1. Bachmaier K, Toya S, Gao X, Triantafillou T, Garrean S, Park GY, Frey RS, Vogel S, Minshall R, Christman JW, Tiruppathi C, Malik AB. E3 ubiquitin ligase Cblb regulates the acute inflammatory response underlying lung injury. Nat Med 13: 920–926, 2007 [DOI] [PubMed] [Google Scholar]
- 2. Birukov KG. Small GTPases in mechanosensitive regulation of endothelial barrier. Microvasc Res 77: 46–52, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Braren R, Hu H, Kim YH, Beggs HE, Reichardt LF, Wang R. Endothelial FAK is essential for vascular network stability, cell survival, and lamellipodial formation. J Cell Biol 172: 151–162, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carbajal JM, Schaeffer RC., Jr RhoA inactivation enhances endothelial barrier function. Am J Physiol Cell Physiol 277: C955–C964, 1999 [DOI] [PubMed] [Google Scholar]
- 5. Chan KT, Bennin DA, Huttenlocher A. Regulation of adhesion dynamics by calpain-mediated proteolysis of focal adhesion kinase (FAK). J Biol Chem 285: 11418–11426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen XL, Nam JO, Jean C, Lawson C, Walsh CT, Goka E, Lim ST, Tomar A, Tancioni I, Uryu S, Guan JL, Acevedo LM, Weis SM, Cheresh DA, Schlaepfer DD. VEGF-induced vascular permeability is mediated by FAK. Dev Cell 22: 146–157, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chikumi H, Barac A, Behbahani B, Gao Y, Teramoto H, Zheng Y, Gutkind JS. Homo- and hetero-oligomerization of PDZ-RhoGEF, LARG and p115RhoGEF by their C-terminal region regulates their in vivo Rho GEF activity and transforming potential. Oncogene 23: 233–240, 2004 [DOI] [PubMed] [Google Scholar]
- 8. Chikumi H, Fukuhara S, Gutkind JS. Regulation of G protein-linked guanine nucleotide exchange factors for Rho, PDZ-RhoGEF, and LARG by tyrosine phosphorylation: evidence of a role for focal adhesion kinase. J Biol Chem 277: 12463–12473, 2002 [DOI] [PubMed] [Google Scholar]
- 9. Essler M, Amano M, Kruse HJ, Kaibuchi K, Weber PC, Aepfelbacher M. Thrombin inactivates myosin light chain phosphatase via Rho and its target Rho kinase in human endothelial cells. J Biol Chem 273: 21867–21874, 1998 [DOI] [PubMed] [Google Scholar]
- 10. Gothert JR, Gustin SE, van Eekelen JA, Schmidt U, Hall MA, Jane SM, Green AR, Gottgens B, Izon DJ, Begley CG. Genetically tagging endothelial cells in vivo: bone marrow-derived cells do not contribute to tumor endothelium. Blood 104: 1769–1777, 2004 [DOI] [PubMed] [Google Scholar]
- 11. Hart MJ, Jiang X, Kozasa T, Roscoe W, Singer WD, Gilman AG, Sternweis PC, Bollag G. Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by Galpha13. Science 280: 2112–2114, 1998 [DOI] [PubMed] [Google Scholar]
- 12. He D, Su Y, Usatyuk PV, Spannhake EW, Kogut P, Solway J, Natarajan V, Zhao Y. Lysophosphatidic acid enhances pulmonary epithelial barrier integrity and protects endotoxin-induced epithelial barrier disruption and lung injury. J Biol Chem 284: 24123–24132, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Holinstat M, Knezevic N, Broman M, Samarel AM, Malik AB, Mehta D. Suppression of RhoA activity by focal adhesion kinase-induced activation of p190RhoGAP: role in regulation of endothelial permeability. J Biol Chem 281: 2296–2305, 2006 [DOI] [PubMed] [Google Scholar]
- 14. Holinstat M, Mehta D, Kozasa T, Minshall RD, Malik AB. Protein kinase Calpha-induced p115RhoGEF phosphorylation signals endothelial cytoskeletal rearrangement. J Biol Chem 278: 28793–28798, 2003 [DOI] [PubMed] [Google Scholar]
- 15. Knezevic N, Tauseef M, Thennes T, Mehta D. The G protein betagamma subunit mediates reannealing of adherens junctions to reverse endothelial permeability increase by thrombin. J Exp Med 206: 2761–2777, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Levkau B, Herren B, Koyama H, Ross R, Raines EW. Caspase-mediated cleavage of focal adhesion kinase pp125FAK and disassembly of focal adhesions in human endothelial cell apoptosis. J Exp Med 187: 579–586, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Manchinelly SA, Miller JA, Su L, Miyake T, Palmer L, Mikawa M, Parsons SJ. Mitotic down-regulation of p190RhoGAP is required for the successful completion of cytokinesis. J Biol Chem 285: 26923–26932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Matthay MA, Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol 6: 147–163, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mehta D, Konstantoulaki M, Ahmmed GU, Malik AB. Sphingosine 1-phosphate-induced mobilization of intracellular Ca2+ mediates rac activation and adherens junction assembly in endothelial cells. J Biol Chem 280: 17320–17328, 2005 [DOI] [PubMed] [Google Scholar]
- 20. Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev 86: 279–367, 2006 [DOI] [PubMed] [Google Scholar]
- 21. Mehta D, Tiruppathi C, Sandoval R, Minshall RD, Holinstat M, Malik AB. Modulatory role of focal adhesion kinase in regulating human pulmonary arterial endothelial barrier function. J Physiol 539: 779–789, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ohta Y, Hartwig JH, Stossel TP. FilGAP, a Rho- and ROCK-regulated GAP for Rac binds filamin A to control actin remodelling. Nat Cell Biol 8: 803–814, 2006 [DOI] [PubMed] [Google Scholar]
- 23. Quadri SK, Bhattacharjee M, Parthasarathi K, Tanita T, Bhattacharya J. Endothelial barrier strengthening by activation of focal adhesion kinase. J Biol Chem 278: 13342–13349, 2003 [DOI] [PubMed] [Google Scholar]
- 24. Quadri SK, Bhattacharya J. Resealing of endothelial junctions by focal adhesion kinase. Am J Physiol Lung Cell Mol Physiol 292: L334–L342, 2007 [DOI] [PubMed] [Google Scholar]
- 25. Rajput C, Kini V, Smith M, Yazbeck P, Chavez A, Schmidt T, Zhang W, Knezevic N, Komarova Y, Mehta D. Neural Wiskott-Aldrich syndrome protein (N-WASP)-mediated p120-catenin interaction with Arp2-Actin complex stabilizes endothelial adherens junctions. J Biol Chem 288: 4241–4250, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reynolds AB, Carnahan RH. Regulation of cadherin stability and turnover by p120ctn: implications in disease and cancer. Semin Cell Dev Biol 15: 657–663, 2004 [DOI] [PubMed] [Google Scholar]
- 27. Romer LH, Birukov KG, Garcia JG. Focal adhesions: paradigm for a signaling nexus. Circ Res 98: 606–616, 2006 [DOI] [PubMed] [Google Scholar]
- 28. Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med 353: 1685–1693, 2005 [DOI] [PubMed] [Google Scholar]
- 29. Shen TL, Park AY, Alcaraz A, Peng X, Jang I, Koni P, Flavell RA, Gu H, Guan JL. Conditional knockout of focal adhesion kinase in endothelial cells reveals its role in angiogenesis and vascular development in late embryogenesis. J Cell Biol 169: 941–952, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shikata Y, Birukov KG, Birukova AA, Verin A, Garcia JG. Involvement of site-specific FAK phosphorylation in sphingosine-1 phosphate- and thrombin-induced focal adhesion remodeling: role of Src and GIT. FASEB J 17: 2240–2249, 2003 [DOI] [PubMed] [Google Scholar]
- 31. Shiratsuchi H, Basson MD. Extracellular pressure stimulates macrophage phagocytosis by inhibiting a pathway involving FAK and ERK. Am J Physiol Cell Physiol 286: C1358–C1366, 2004 [DOI] [PubMed] [Google Scholar]
- 32. Su L, Agati JM, Parsons SJ. p190RhoGAP is cell cycle regulated and affects cytokinesis. J Cell Biol 163: 571–582, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Su L, Pertz O, Mikawa M, Hahn K, Parsons SJ. p190RhoGAP negatively regulates Rho activity at the cleavage furrow of mitotic cells. Exp Cell Res 315: 1347–1359, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sun X, Shikata Y, Wang L, Ohmori K, Watanabe N, Wada J, Shikata K, Birukov KG, Makino H, Jacobson JR, Dudek SM, Garcia JG. Enhanced interaction between focal adhesion and adherens junction proteins: involvement in sphingosine 1-phosphate-induced endothelial barrier enhancement. Microvasc Res 77: 304–313, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev 81: 153–208, 2001 [DOI] [PubMed] [Google Scholar]
- 36. Tang PS, Tsang ME, Lodyga M, Bai XH, Miller A, Han B, Liu M. Lipopolysaccharide accelerates caspase-independent but cathepsin B-dependent death of human lung epithelial cells. J Cell Physiol 209: 457–467, 2006 [DOI] [PubMed] [Google Scholar]
- 37. Tauseef M, Kini V, Knezevic N, Brannan M, Ramchandaran R, Fyrst H, Saba J, Vogel SM, Malik AB, Mehta D. Activation of sphingosine kinase-1 reverses the increase in lung vascular permeability through sphingosine-1-phosphate receptor signaling in endothelial cells. Circ Res 103: 1164–1172, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tauseef M, Knezevic N, Chava KR, Smith M, Sukriti S, Gianaris N, Obukhov AG, Vogel SM, Schraufnagel DE, Dietrich A, Birnbaumer L, Malik AB, Mehta D. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J Exp Med 209: 1953–1968, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thennes T, Mehta D. Heterotrimeric G proteins, focal adhesion kinase, and endothelial barrier function. Microvasc Res 83: 31–44, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tiruppathi C, Freichel M, Vogel SM, Paria BC, Mehta D, Flockerzi V, Malik AB. Impairment of store-operated Ca2+ entry in TRPC4(−/−) mice interferes with increase in lung microvascular permeability. Circ Res 91: 70–76, 2002 [DOI] [PubMed] [Google Scholar]
- 41. Usatyuk PV, Parinandi NL, Natarajan V. Redox regulation of 4-hydroxy-2-nonenal-mediated endothelial barrier dysfunction by focal adhesion, adherens, and tight junction proteins. J Biol Chem 281: 35554–35566, 2006 [DOI] [PubMed] [Google Scholar]
- 42. Vandenbroucke E, St. Amant E, Tauseef M, Vogel SM, Gao XP, Mehta D, Komarova YA, Malik AB. PKCalpha activation of p120-catenin serine 879 phospho-switch disassembles VE-cadherin junctions and disrupts vascular integrity. Circ Res 111: 739–749, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Waschke J, Baumgartner W, Adamson RH, Zeng M, Aktories K, Barth H, Wilde C, Curry FE, Drenckhahn D. Requirement of Rac activity for maintenance of capillary endothelial barrier properties. Am J Physiol Heart Circ Physiol 286: H394–H401, 2004 [DOI] [PubMed] [Google Scholar]
- 44. Wojciak-Stothard B, Potempa S, Eichholtz T, Ridley AJ. Rho and Rac but not Cdc42 regulate endothelial cell permeability. J Cell Sci 114: 1343–1355, 2001 [DOI] [PubMed] [Google Scholar]
- 45. Wojciak-Stothard B, Ridley AJ. Rho GTPases and the regulation of endothelial permeability. Vascul Pharmacol 39: 187–199, 2002 [DOI] [PubMed] [Google Scholar]
- 46. Wu MH, Guo M, Yuan SY, Granger HJ. Focal adhesion kinase mediates porcine venular hyperpermeability elicited by vascular endothelial growth factor. J Physiol 552: 691–699, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]