

Abstract
Exposing preterm infants or newborn mice to high concentrations of oxygen disrupts lung development and alters the response to respiratory viral infections later in life. Superoxide dismutase (SOD) has been separately shown to mitigate hyperoxia-mediated changes in lung development and attenuate virus-mediated lung inflammation. However, its potential to protect adult mice exposed to hyperoxia as neonates against viral infection is not known. Here, transgenic mice overexpressing extracellular (EC)-SOD in alveolar type II epithelial cells are used to test whether SOD can alleviate the deviant pulmonary response to influenza virus infection in adult mice exposed to hyperoxia as neonates. Fibrotic lung disease, observed following infection in wild-type (WT) mice exposed to hyperoxia as neonates, was prevented by overexpression of EC-SOD. However, leukocyte recruitment remained excessive, and levels of monocyte chemoattractant protein (MCP)-1 remained modestly elevated following infection in EC-SOD Tg mice exposed to hyperoxia as neonates. Because MCP-1 is often associated with pulmonary inflammation and fibrosis, the host response to infection was concurrently evaluated in adult Mcp-1 WT and Mcp-1 knockout mice exposed to neonatal hyperoxia. In contrast to EC-SOD, excessive leukocyte recruitment, but not lung fibrosis, was dependent upon MCP-1. Our findings demonstrate that neonatal hyperoxia alters the inflammatory and fibrotic responses to influenza A virus infection through different pathways. Therefore, these data suggest that multiple therapeutic strategies may be needed to provide complete protection against diseases attributed to prematurity and early life exposure to oxygen.
Keywords: lung fibrosis, monocyte chemoattractant protein-1, respiratory viral infection, superoxide dismutase
the lungs of infants born prematurely, especially those with low birth weight, are often physiologically and structurally immature, thus increasing the risk for development of neonatal lung disease, such as bronchopulmonary dysplasia (BPD) (22, 23). Although milder ventilation strategies, the use of exogenous surfactant, and the administration of antenatal steroids have markedly improved the survival of even the most premature infants, the prevalence of lung disease among survivors of prematurity has not decreased. In fact, reduced pulmonary function and lung capacity have been reported in young adolescents who survived premature birth, suggesting that disrupted lung development may be permanent in these individuals (12–14, 36). Moreover, there is an increased incidence of respiratory viral infection, asthma, retinopathy of prematurity, and neurodevelopmental delay in children who are born prematurely (38, 46). Because these children often require additional medical care to treat diseases now attributed to being born too early, there is an urgent need to understand the developmental influence of premature birth on health later in life.
Similar to findings observed in infants born prematurely who have died from complications of BPD, the prolonged exposure of newborn rodents to elevated levels of oxygen (hyperoxia) at birth causes a disruption in the rate at which the lung grows and ultimately develops (17). Even when returned to room air, the lungs of these animals remain simplified and are functionally impaired, suggesting that the developmental changes resulting from early life exposure to oxygen may be permanent (7, 29, 50, 51). Moreover, mice exposed to 100% oxygen during postnatal days (PND) 0–4 show increased sensitivity to influenza virus infection later in life, as defined by increased leukocyte recruitment to the lungs, a selective increase in the proinflammatory chemokine monocyte chemoattractant protein (MCP)-1 in bronchoalveolar lavage (BAL) fluid, and evidence of lung fibrosis (5, 33, 34).
Changes in health associated with early life exposure to oxygen have often been attributed to the generation of reactive oxygen species (ROS), which can injure or reprogram development of the lungs, eyes, and brain. In particular, infants born prematurely are at increased risk for sustaining lung injury attributable to oxidative stress, since antioxidant defenses do not mature until late in gestation and the ability to increase antioxidant synthesis in response to hyperoxia is often impaired (45, 47). Interestingly, infants born prematurely and treated with recombinant human copper-zinc superoxide dismutase (CuZn-SOD) had improved pulmonary function and reduced hospitalization rates at one year of corrected age (8). Furthermore, recombinant human CuZn-SOD and overexpression of extracellular (EC)-superoxide dismutase (SOD) in alveolar type II epithelial cells have been shown to preserve lung development in preterm lambs and newborn mice, respectively, that had been exposed to high levels of oxygen at birth (1, 2, 24, 27). Lung-specific overexpression of EC-SOD has also been shown to prevent influenza-induced lung injury in adult mice (40). These studies underscore the impact of oxidant injury on the developing lung. Moreover, they provide a rationale for continued investigation of antioxidants as a potential therapeutic strategy to reduce the incidence of diseases attributed to premature birth and early life exposure to oxygen. Here, we test the hypothesis that lung-specific overexpression of EC-SOD can alleviate the deviant pulmonary response to influenza virus infection in adult mice that had been previously exposed to hyperoxia as neonates. Understanding how antioxidants protect lung development and the host response to respiratory viral infection is important because it may clarify how individuals prematurely exposed to oxygen and treated with SOD at birth will respond to respiratory challenges later in life.
MATERIALS AND METHODS
Exposure of mice to hyperoxia and influenza A virus.
C57BL/6J mice [wild-type (WT) control], transgenic mice overexpressing EC-SOD (EC-SOD Tg) under control of the human surfactant protein C promoter (16), and Mcp-1 knockout mice (KO) (The Jackson Laboratory, Bar Harbor, ME) were used for this study. Newborn mice were exposed to room air or 100% oxygen (hyperoxia) between PND 0 and 4. Dams were cycled between litters exposed to room air and hyperoxia every 24 h to protect them from acute oxygen toxicity. On PND 4, oxygen-exposed mouse pups were returned to room air. Adult (8–10 wk of age) female mice exposed to room air or hyperoxia at birth were anesthetized by intraperitoneal injection with avertin (2,2,2-tribromoethanol; Sigma-Aldrich, Milwaukee, WI) and inoculated intranasally with 120 hemagglutinating units (25 μl) of influenza A virus (strain HKx31; H3N2) (32, 41). Mice were weighed before infection and every 2 days following infection, as well as on the day of death. Mice were housed in microisolator cages in a specified pathogen-free environment according to a protocol approved by the University Committee on Animal Resources at the University of Rochester and were provided food and water ad libitum.
Quantification of MCP-1 protein.
Levels of MCP-1 in BAL fluid were determined using an MCP-1-specific sandwich enzyme-linked immunoabsorbent assay, according to the manufacturer's instructions (R & D Systems, Minneapolis, MN). The lower limit of detection for this assay was 3.9 pg/ml.
Collection and quantification of immune cells.
Airway-derived immune cells were collected by BAL as previously described (5). Lung-derived immune cells were collected by collagenase digestion of lung tissues as previously described (41). The total number of airway- and lung-derived immune cells collected from each mouse was determined using a TC10 automated cell counter (Bio-Rad, Hercules, CA). Airway- and lung-derived immune cells collected from individual mice were transferred to separate microscope slides using a cytological centrifuge and were stained with Diff-Quik. The percentage and number of macrophages, neutrophils, and lymphocytes was then determined by differential leukocyte counts of at least 200 cells on coded slides by two independent investigators.
Lung histology and immunohistochemistry.
Lungs from mice were inflation fixed with 10% neutral-buffered formalin, embedded in paraffin, and sectioned as previously described (50, 51). Tissue sections were stained with hematoxylin and eosin to evaluate changes in lung structure. Collagen deposition in tissue sections was detected with Gomori's Trichrome stain according to the manufacturer's instructions (Richard-Allan Scientific, Kalamazoo, MI). Separate tissue sections were rehydrated and incubated overnight at 4°C with anti-α-smooth muscle actin (α-SMA) primary antibody (33). Immune complexes were detected with fluorescently labeled secondary antibodies before tissue sections were counterstained with 4′,6-diamidino-2-phenylindole. Stained tissue sections were visualized with a Nikon E-800 fluorescence microscope (Nikon, Melville, NY), and images were captured with a SPOT-RT digital camera (Diagnostic Instruments, Sterling Heights, MI).
Collagen assay.
Total lung collagen was determined using the Sircol Collagen Assay kit (Biocolor, Belfast, UK) according to the manufacturer's instructions and as previously described (5).
Statistical analysis.
StatView statistical software (SAS Institute, Cary, NC) was used for all data analysis. Group means were compared by one-way analysis of variance, followed by a Fisher's least-significant difference post hoc test. Single comparisons of values were analyzed by an unpaired Student's t-test. Survival was analyzed by a Mantel-Cox test. All values are expressed as means ± SE with P < 0.05 being considered significant.
RESULTS
EC-SOD preserves alveolar development and the health of infected mice exposed to neonatal hyperoxia.
Adult WT and EC-SOD transgenic (EC-SOD Tg) mice were exposed to room air or hyperoxia between PND 0 and 4, and then the oxygen-exposed mice were recovered in room air. At 8 wk of age, alveolar simplification was evident in WT mice exposed to hyperoxia (Fig. 1). Alveolar simplification was markedly reduced in EC-SOD Tg mice exposed to hyperoxia. Adult WT and EC-SOD Tg mice exposed to room air or hyperoxia as neonates were infected with a sublethal dose of influenza A virus (strain HKx31, H3N2). As previously reported (33), neonatal hyperoxia significantly reduced the mean body weight of infected WT mice on postinfection days 6 through 14 (Fig. 2A). Overexpression of EC-SOD blunted this response such that body weight loss following infection in EC-SOD Tg mice was comparable to the body weight loss observed in infected mice exposed to room air as neonates. Additionally, overexpression of EC-SOD significantly reduced mortality of infected mice exposed to hyperoxia as neonates (Fig. 2B).
Fig. 1.

Extracellular (EC)-superoxide dismutase (SOD) preserves alveolar development in adult mice exposed to hyperoxia at birth. Representative hematoxylin-eosin-stained lung tissues from naïve adult wild-type (WT) and EC-SOD transgenic (EC-SOD Tg) mice exposed to room air (RA) or 100% oxygen (O2) at birth. Each image is representative of 5 mice examined/group. Scale bar = 100 μm.
Fig. 2.

EC-SOD reduces morbidity and enhances survival of infected adult mice exposed to hyperoxia at birth. Adult female WT and EC-SOD Tg mice exposed to RA or O2 at birth were intranasally infected with a sublethal dose of influenza A virus. Body weight (A) and survival (B) were monitored for 14 days following influenza A virus infection, and results are shown as %body weight and %survival, respectively (n = 8–12 mice/group). Analysis of differences between mean body weights over time was first determined by a one-way analysis of variance (P < 0.05), followed by an unpaired t-test (*P < 0.05 when compared with RA WT). Analysis of differences in survival over time was determined by a Mantel-Cox test (P < 0.05).
EC-SOD prevents fibrotic lung disease following infection in adult mice exposed to hyperoxia at birth.
As previously observed (5, 33), fibrotic lung disease was readily evident by postinfection day 14 in infected WT mice that had been exposed to hyperoxia, but not room air, as neonates. Increased collagen deposition was observed by Trichrome staining and by quantifying total soluble collagen levels in lung homogenates of infected mice exposed to hyperoxia (Fig. 3). Fibrotic regions also stained positive for α-SMA, a marker of activated myofibroblasts. However, fibrotic lung disease was not observed in infected adult EC-SOD Tg mice exposed to hyperoxia as neonates.
Fig. 3.
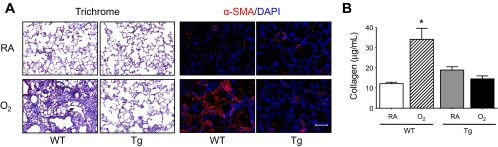
EC-SOD prevents fibrotic lung disease following infection in adult mice exposed to hyperoxia at birth. Adult female WT and EC-SOD Tg mice exposed to RA or O2 at birth were intranasally infected with a sublethal dose of influenza A virus. A: lung tissues were harvested on postinfection day 14 and were stained with Gomori's Trichrome stain to visualize collagen deposition and antibodies against α-smooth muscle actin (α-sma, red), followed by counterstaining with 4′,6-diamidino-2-phenyl-indole (DAPI, blue) to visualize activated myofibroblasts. Each image is representative of 5 mice examined/group. B: total collagen protein in the lungs of infected adult mice exposed to RA or O2 at birth was measured by the Sircol Collagen Assay on postinfection day 14 (n = 4–9 mice/group, *P < 0.05 compared with RA WT). Scale bar in A = 100 μm.
EC-SOD does not ameliorate the exacerbated inflammatory response to infection in mice exposed to hyperoxia at birth.
Exposure to neonatal hyperoxia promotes excessive leukocyte recruitment to the lungs of mice on postinfection days 5–9 (33). On postinfection day 7, significantly more leukocytes were observed in BAL fluid of WT and EC-SOD Tg mice exposed to hyperoxia at birth compared with infected siblings birthed into room air (Fig. 4A). However, in the room air-exposed groups, the mean number of airway leukocytes was lower in EC-SOD Tg mice compared with WT mice. Therefore, we considered whether the significant difference in mean number of leukocytes in WT and EC-SOD Tg mice exposed to hyperoxia at birth was reflective of this difference. When we compared the means as fold change from room air-exposed mice of the same genotype, it is clear that EC-SOD Tg mice are not protected against the hyperoxia-mediated increase in leukocyte recruitment (Fig. 4B). Moreover, in both genotypes, the increased mean leukocyte number observed in mice that had been exposed to hyperoxia at birth corresponded with an enhancement in neutrophils, monocytes/macrophage, and lymphocytes, that is, although the overall number of leukocytes was higher, there were no differences in the percentage of these leukocyte phenotypes in infected mice that had been exposed to room air or hyperoxia at birth (Table 1). This finding suggests a general increase in the number of leukocytes recruited to the lung as opposed to a change in the frequency of a particular cell type.
Fig. 4.

EC-SOD does not ameliorate the exacerbated inflammatory response to infection observed in mice exposed to hyperoxia at birth. Adult female WT and EC-SOD Tg mice exposed to RA or O2 at birth were intranasally infected with a sublethal dose of influenza A virus. Total leukocyte number (A), fold change in total leukocyte number (B), levels of monocyte chemoattractant protein (MCP)-1 (C), and fold change in MCP-1 present in bronchoalveolar lavage fluid (D) were determined on postinfection day 7 (n = 5–7 mice/group, *P < 0.05 compared with RA WT, #P < 0.05 compared with RA Tg).
Table 1.
Differential leukocyte counts on postinfection day 7
|
Day 7 |
||||
|---|---|---|---|---|
| WT |
Tg |
|||
| BAL Fluid | RA | O2 | RA | O2 |
| Neutrophils, % | 21.1 ± 3.0 | 18.1 ± 2.0 | 13.7 ± 1.3 | 17.5 ± 2.1 |
| Macrophages, % | 62.5 ± 3.1 | 64.7 ± 2.2 | 69.1 ± 1.6 | 66.9 ± 1.9 |
| Lymphocytes, % | 16.4 ± 3.2 | 17.2 ± 1.9 | 17.1 ± 1.4 | 15.5 ± 2.5 |
Values are means ± SE; n = 5 mice/group. Adult female wild-type (WT) and extracellular superoxide dismutase transgenic (Tg) mice exposed to room air (RA) or 100% oxygen (O2) at birth were intranasally infected with a sublethal dose of influenza A virus. Leukocytes in bronchoalveolar lavage (BAL) fluid were collected on postinfection day 7, and percentages of neutrophils, macrophages, and lymphocytes were determined.
Neonatal hyperoxia selectively increases levels of MCP-1, but not IFN-γ, IL-1β, IL-6, TNF-α, keratinocyte chemokine, granulocyte-macrophage colony-stimulating factor, or macrophage inflammatory protein-1α following infection (33). Because MCP-1 is a regulator of inflammation, its abundance in BAL fluid was determined. Levels of MCP-1 were significantly increased in WT mice exposed to hyperoxia as neonates compared with siblings birthed into room air (Fig. 4C). Levels of MCP-1 were modestly increased in EC-SOD Tg mice exposed to hyperoxia at birth compared with siblings birthed into room air, although the fold change observed was not significantly different (Fig. 4D). Nevertheless, while previously investigating the contribution of MCP-1 to the altered host response to infection (5), we generated findings suggesting that the exacerbated inflammatory response observed in mice exposed to hyperoxia as neonates may be attenuated in the absence of MCP-1 gene expression. Because MCP-1 represents a putative pathway contributing to the altered host response to infection in mice exposed to hyperoxia at birth, we decided to further investigate these findings.
Neonatal hyperoxia enhances leukocyte recruitment to infected lungs through MCP-1 signaling.
MCP-1 can contribute to significant lung inflammation and fibrosis when aberrantly expressed (3, 19, 39, 48). Lung inflammation and fibrosis were assessed following infection in Mcp-1 WT and Mcp-1 KO mice exposed to room air or hyperoxia as neonates. When lung structure was assessed before infection, alveolar simplification was readily observed in both Mcp-1 WT and KO mice exposed to hyperoxia as neonates (Fig. 5). Mcp-1 WT and KO mice exposed to room air or hyperoxia as neonates were infected with influenza A virus. Levels of MCP-1 in BAL fluid were always greater following infection in Mcp-1 WT mice exposed to hyperoxia at birth compared with siblings birthed into room air, with statistical significance on postinfection days 5 and 7 (Fig. 6). As expected, levels of MCP-1 were undetectable at all time points postinfection in BAL fluid collected from Mcp-1 KO mice.
Fig. 5.

Neonatal hyperoxia promotes alveolar simplification independent of MCP-1. Representative hematoxylin-eosin-stained lung tissues from adult Mcp-1 WT and Mcp-1 knockout (KO) mice exposed to RA or O2 at birth. Each image is representative of 6 mice examined/group. Scale bar = 100 μm.
Fig. 6.
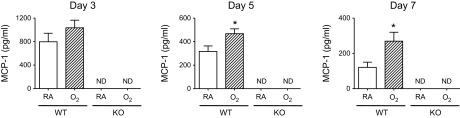
Neonatal hyperoxia enhances MCP-1 expression in mice infected with influenza A virus. Adult Mcp-1 WT and KO mice exposed to RA or O2 at birth were intranasally infected with a sublethal dose of influenza A virus. Levels of MCP-1 in bronchoalveolar lavage fluid were determined on postinfection days 3, 5, and 7 (ND = not detected, n = 4–6 mice/group, *P < 0.05).
The total number of leukocytes in BAL fluid and lung interstitium was measured on postinfection days 3, 5, and 7 in WT and Mcp-1 KO mice exposed to room air or hyperoxia at birth (Fig. 7). The mean total number of leukocytes in BAL fluid collected from Mcp-1 WT mice exposed to hyperoxia at birth was significantly increased compared with siblings birthed into room air on postinfection days 3 and 7. In contrast, no differences in the mean total number of leukocytes were observed between Mcp-1 KO mice exposed to room air or hyperoxia as neonates at any of the time points examined postinfection. To determine if the lack of excessive leukocyte recruitment to the airways observed in Mcp-1 KO mice exposed to hyperoxia at birth was attributed to cells being trapped within the lung itself, total leukocyte number was measured within lung interstitium. Similar to findings observed in the airways, the mean total number of leukocytes was increased in Mcp-1 WT mice exposed to hyperoxia at birth on postinfection day 3, whereas no differences in total leukocyte number were observed in Mcp-1 KO mice. Notable differences in the mean percentage of the various leukocyte phenotypes examined in BAL fluid primarily included a significant increase in neutrophils and a significant decrease in macrophages in Mcp-1 KO mice exposed to room air or hyperoxia at birth compared with their WT counterparts on postinfection days 3 and 5 (Table 2). No remarkable differences in the mean percentage of neutrophils, macrophages, and lymphocytes were observed in the lung interstitium of Mcp-1 KO mice exposed to room air or hyperoxia at birth (data not shown).
Fig. 7.

Excessive recruitment of leukocytes to lungs of infected mice exposed to neonatal hyperoxia is dependent upon MCP-1. Adult Mcp-1 WT and KO mice exposed to RA or O2 at birth were intranasally infected with a sublethal dose of influenza A virus. Total leukocyte number in bronchoalveolar lavage (BAL) fluid and postlavaged interstitium (Lung) were determined on postinfection days 3, 5, and 7 (n = 5–7 mice/group, *P < 0.05 compared with RA WT).
Table 2.
Differential leukocyte counts on postinfection days 3, 5, and 7
|
Day 3 |
Day 5 |
Day 7 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT |
KO |
WT |
KO |
WT |
KO |
|||||||
| BAL Fluid | RA | O2 | RA | O2 | RA | O2 | RA | O2 | RA | O2 | RA | O2 |
| Neutrophils, % | 43.1 ± 1.8 | 53.0 ± 2.2* | 60.6 ± 4.0Δ | 65.4 ± 1.9† | 34.7 ± 1.6 | 33.1 ± 2.0 | 52.7 ± 3.1Δ | 44.3 ± 2.6 | 12.1 ± 2.4 | 10.7 ± 2.1 | 14.5 ± 2.4 | 17.0 ± 2.9 |
| Macrophages, % | 54.4 ± 2.0 | 43.9 ± 1.8* | 38.8 ± 3.8Δ | 32.1 ± 2.0† | 62.1 ± 1.6 | 61.5 ± 2.0 | 43.6 ± 2.7Δ | 51.5 ± 2.7† | 65.5 ± 2.1 | 71.4 ± 1.4* | 62.6 ± 2.9 | 66.6 ± 2.0 |
| Lymphocytes, % | 3.1 ± 0.6 | 3.1 ± 0.6 | 3.8 ± 1.3 | 2.5 ± 0.4 | 3.2 ± 0.6 | 5.4 ± 0.8 | 4.0 ± 0.8 | 4.2 ± 0.7 | 22.3 ± 3.1 | 17.9 ± 1.3 | 23.0 ± 3.2 | 16.4 ± 1.9 |
Values are means ± SE; n = 5–7 mice/group. Adult monocyte chemoattractant protein-1 WT and knockout (KO) mice exposed to RA or O2 at birth were intranasally infected with a nonlethal dose of influenza A virus. Leukocytes in BAL fluid were collected on postinfection days 3, 5, and 7, and percentages of neutrophils, macrophages, and lymphocytes were determined.
P < 0.05 compared with RA WT,
P < 0.05 compared with RA WT, and
P < 0.05 compared with O2 WT.
Neonatal hyperoxia promotes lung fibrosis in infected mice independent of MCP-1.
Lung fibrosis was assessed on postinfection day 14 by staining lung tissues with Trichrome and antibodies against α-SMA (Fig. 8A). Interstitial thickening of alveolar septae and extensive collagen deposition, as evidenced by Trichrome staining, were readily apparent in lung tissues harvested from infected mice exposed to hyperoxia at birth. These fibrotic regions of lung tissue stained positive for α-SMA, a marker of activated myofibroblasts. Mean levels of total soluble collagen in lung tissues harvested from infected Mcp-1 WT and Mcp-1 KO mice exposed to hyperoxia at birth were comparable and significantly greater compared with levels observed in infected siblings birthed into room air (Fig. 8B).
Fig. 8.
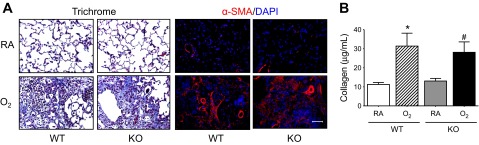
Fibrotic lung disease observed in infected mice exposed to hyperoxia as neonates is not dependent upon MCP-1. Adult Mcp-1 WT and KO mice exposed to RA or O2 at birth were intranasally infected with a sublethal dose of influenza A virus. A: lung tissues were harvested on postinfection day 14 and were stained with Gomori's Trichrome stain to visualize collagen deposition and antibodies against α-α-sma (red), followed by counterstaining with DAPI (blue) to visualize activated myofibroblasts. Each image is representative of 5 mice examined/group. B: total collagen protein in the lungs of infected adult mice exposed to RA or O2 at birth was measured by the Sircol Collagen Assay on postinfection day 14 (n = 7–8 mice/group, *P < 0.05 compared with RA WT, #P < 0.05 compared with RA KO). Scale bar in A = 100 μm.
DISCUSSION
Increasing evidence supports the concept that early life exposure to environmental factors, such as hyperoxia, can interfere with the developmental programming of the lung, resulting in altered lung structure and increased risk for respiratory disease in susceptible individuals later in life (14, 36, 43, 46). If we consider that oxygen will continue to be used therapeutically to treat preterm infants, then it is important to understand how it can adversely change respiratory health later in life. Here, using an established mouse model of persistent pulmonary disease, we provide evidence that exposure to neonatal hyperoxia increases leukocyte recruitment and fibrosis in adult mice infected with influenza virus through different pathways (Fig. 9). While transgenic EC-SOD was involved in a pathway that prevented hyperoxia-mediated lung fibrosis following infection, it did not protect against a pathway that promoted excessive recruitment of leukocytes or the production of MCP-1. In contrast to EC-SOD, MCP-1 was required for the pathway promoting excessive recruitment of leukocytes to the lungs, but not the pathway leading to the development of fibrosis, suggesting an uncoupling of inflammation from interstitial lung disease. Hence, our findings suggest neonatal hyperoxia disrupts at least two distinct pathways that separately control inflammation and alveolar repair following influenza A virus infection.
Fig. 9.
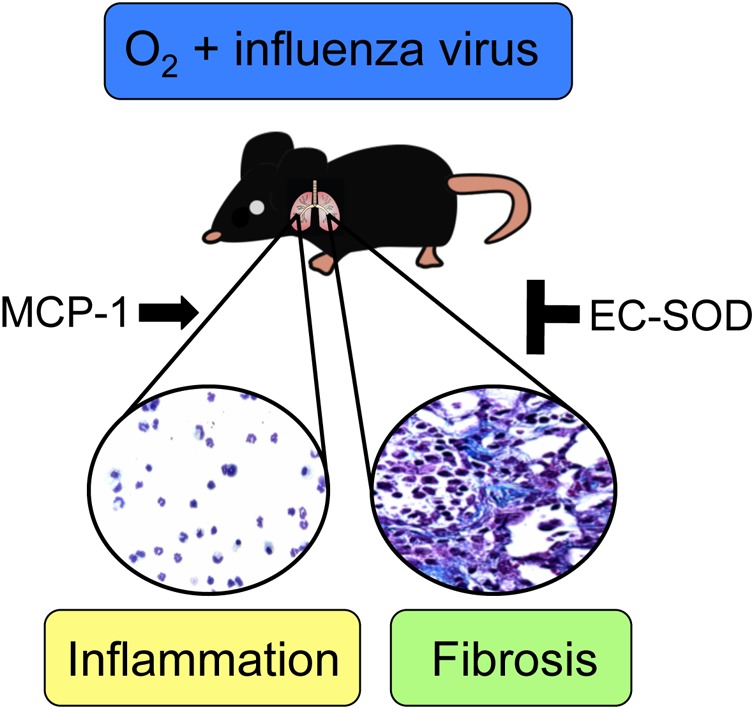
Cartoon model depicting how neonatal hyperoxia reprograms the host response to influenza A virus through different pathways. MCP-1 is involved in a pathway that promotes excessive leukocyte recruitment to the lungs following influenza virus infection in adult mice that had been exposed to hyperoxia as neonates, whereas EC-SOD is involved in a pathway that prevents the development of fibrotic lung disease.
It is generally accepted that oxygen-induced injury in the lung is mediated through ROS (45, 47). While ROS are known to be important mediators in pathways contributing to normal cell growth and differentiation during lung development, they can cause cellular and tissue injury when their production exceeds the antioxidant capacity of the cell (45). Thus, any interference with the developmental programming of the lung, such as that mediated by increased production of ROS, during a critical period of growth and development may increase the risk for respiratory disease later in life. In fact, mice exposed to neonatal hyperoxia demonstrate fewer alveolar type II cells as adults and are more sensitive to infection with influenza A virus, as defined by increased inflammation and the development of fibrotic lung disease (5, 33, 51). Because alveolar type II cells are considered progenitor cells within the lung (15), their depletion as a result of exposure to hyperoxia at birth may be contributing to the impaired ability of these mice to effectively respond to and repair viral-mediated injury. Interestingly, it has been shown that transgenic overexpression of human EC-SOD preserves lung development in newborn mice exposed to hyperoxia (1). This finding was subsequently attributed, at least in part, to preservation of alveolar type II cell proliferation, which was associated with a reduction in oxidative DNA damage to the alveolar epithelium (2). Consistent with preservation of lung development, overexpression of EC-SOD limited hyperoxia-induced alveolar simplification, as well as weight loss, mortality, and fibrotic lung disease following infection with influenza A virus. Surprisingly, overexpression of EC-SOD did not blunt the excessive recruitment of leukocytes or production of MCP-1. It is difficult to directly compare our findings with children who were born prematurely and treated with SOD for one month because transgenic EC-SOD was constitutively expressed in type II cells throughout the life of the mouse. Nevertheless, our findings show that EC-SOD is involved in a pathway through which neonatal hyperoxia reprograms normal alveolar repair, but not excessive leukocyte recruitment, following infection. Therefore, EC-SOD may be involved in a pathway that preserves alveolar epithelial balance, thereby promoting normal repair in response to injury.
Although the inflammatory infiltrate following a respiratory viral infection is helpful in initially controlling an infection, it can often be injurious to the host when excessive, resulting in severe lung pathology (10, 25). Therefore, identifying factors that contribute to the excessive recruitment of proinflammatory leukocytes following infection in mice exposed to hyperoxia at birth may help explain the augmented lung pathology observed in these mice. One such factor is MCP-1, which, to date, is the only leukocyte chemoattractant we have identified that is selectively different in BAL fluid following infection in adult mice exposed to hyperoxia at birth (33). Because levels of MCP-1 in WT and EC-SOD Tg mice exposed to hyperoxia at birth were elevated following infection compared with siblings birthed into room air, it may be contributing to the excessive leukocyte recruitment also observed in these mice. Neutrophil and monocyte recruitment to the lungs is altered in Mcp-1- or Mcp-1 receptor (CCR2)-deficient mice, and the mice are unable to effectively clear pathogen, suggesting the importance of MCP-1 in mediating an adequate immune response (9, 11, 44). We report here that the absence of MCP-1 gene expression in infected adult mice exposed to hyperoxia at birth leads to a significant reduction in the total number of leukocytes, primarily macrophages, on postinfection days 3 and 7 in both BAL fluid and lung interstitium. Our finding that total leukocyte numbers were not different between any of the experimental test groups on postinfection day 5 may be attributable to variability in the kinetics of the innate immune response often observed in this model. Regardless, our findings reveal the existence of a pathway, involving MCP-1 gene expression, through which exposure to neonatal hyperoxia promotes excessive recruitment of leukocytes to lungs of infected mice.
In addition to its role in mediating an adequate immune response to influenza virus infection, MCP-1 has also been suggested to contribute to lung fibrosis when aberrantly expressed. For example, clinical studies and in vivo studies in mice have shown an association between elevated levels of MCP-1 and the development of bronchiolitis obliterans, a nonreversible obstructive lung disease (3, 48). Increased levels of MCP-1 have also been detected in serum or BAL fluid obtained from children and adults with interstitial lung disease (19, 39). Moreover, overexpression of mutant MCP-1 or knockout of the MCP-1 receptor (CCR2) confers protection against bleomycin- or FITC-induced lung fibrosis in mice (21, 30, 35). In contrast to those findings, we report here that, even in the absence of MCP-1 gene expression, fibrosis clearly remains evident following infection in adult mice exposed to hyperoxia at birth, as defined by increased collagen staining, increased staining of α-sma-expressing myofibroblasts, and increased total collagen protein in the lungs. Similarly, in a mouse model of experimental allergic asthma, airway fibrosis was demonstrated in the absence of MCP-1 or its receptor (26). Given the potential for chemokine redundancy in our model, it is possible that other monocyte chemoattractants could be compensating for the lack of MCP-1 gene expression and contributing to the observed fibrosis in infected mice exposed to hyperoxia at birth. For example, increased levels of the monocyte chemoattractant protein MIP-1α have been observed in patients with cystic fibrosis and in a mouse model of bleomycin-induced pulmonary fibrosis (31, 37). However, we have previously shown that levels of MIP-1α in BAL fluid collected from infected adult mice exposed to hyperoxia at birth were not different from levels in infected siblings birthed into room air during any of the postinfection time points examined in the study (33). Nevertheless, additional studies are needed to determine if proinflammatory chemokines or cytokines other than MCP-1 are able to contribute to the observed lung fibrosis in our model.
On the other hand, lung fibrosis is currently considered to be principally due to recurrent epithelial injury and abnormal repair (20). Our finding that excessive leukocyte recruitment to the lungs following influenza virus infection in adult mice exposed to hyperoxia at birth is uncoupled from lung fibrosis is consistent with this concept. In fact, studies examining bleomycin-induced lung fibrosis in Smad3-deficient mice or mice treated with anti-ICAM-1 antibody have shown that inflammation is not solely responsible for the fibrotic phenotype (28, 52). Inflammation may therefore play a role in the progression, but not initiation, of fibrotic disease (6). Interestingly, the enhanced inflammation associated with certain viral infections has recently been recognized as a potential exacerbating agent in the development of lung fibrosis (42). Moreover, we have recently shown that neonatal hyperoxia increases the sensitivity of adult mice to bleomycin-induced lung fibrosis, which was primarily associated with a disproportionate increase in neutrophils, which is not observed in mice infected with influenza A virus (49). Depletion of neutrophils in these mice with anti-Gr-1 antibody reduced the early activation of transforming growth factor-β1 and attenuated the hyperoxia-enhanced fibrosis. Hence, identifying pathways that contribute to the progression of lung fibrosis is important and may be useful for the identification of individuals born prematurely who are at increased risk for interstitial lung disease later in life.
The primary objective of this study was to identify possible pathways through which neonatal hyperoxia alters the sensitivity to viral infection later in life. There are, of course, limitations that make it difficult to extrapolate our experimental findings to humans born preterm and prematurely exposed to high levels of oxygen at birth. To successfully carry out this work, we deliberately infected immune competent adult mice with a sublethal viral inoculum to investigate how early life exposure to oxygen compromises host responses to infection later in life. This experimental approach was chosen since it is difficult to manipulate neonatal mice and achieve sublethal doses of influenza virus. In addition, the work reported herein used only a single strain of influenza A virus. We have previously shown that host resistance of adult mice to several different strains of influenza virus, including HKx31 (H3N2), A/CA/04/09 (H1N1), and A/PR/8/34 (H1N1), was similarly reduced by exposure to neonatal hyperoxia (18). Therefore, while our findings do not address the role of MCP-1 or EC-SOD during infection with other strains of influenza virus (or in other strains of mice), identification of these pathways within this system improves our understanding of why individuals born preterm are at increased risk for respiratory disease later in life.
In summary, we have shown that EC-SOD is involved in a pathway that prevents neonatal hyperoxia-mediated lung fibrosis following influenza virus infection, and that MCP-1 gene expression is involved in a pathway that contributes to the excessive recruitment of leukocytes in these mice. These data suggest that neonatal hyperoxia differentially reprograms lung development and likely disrupts multiple pathways contributing to the pulmonary response to viral infection later in life. Ultimately, our findings suggest that multiple therapeutic strategies may be needed to provide complete protection against diseases attributed to prematurity and early life exposure to oxygen.
GRANTS
This work was funded in part by March of Dimes Grant 06-FY08-264 (M. A. O'Reilly) and National Institutes of Health (NIH) Grants HL-091968 (M. A. O'Reilly), HL-097141 (M. A. O'Reilly and B. P. Lawrence), and ES-017250 (B. P. Lawrence). NIH Training Grants ES-07026 and HL-66988 supported B. W. Buczynski. NIH Center Grant ES-01247 supports the animal inhalation facility and the tissue-processing core.
DISCLOSURES
The authors declare no conflicts of interest, financial or otherwise.
AUTHOR CONTRIBUTIONS
Author Contributions: B.W.B. and M.A.O. conceptualized and designed experiments; B.W.B., M.Y., and K.C.M. performed animal procedures and data collection; B.P.L. provided the influenza A virus; B.W.B. and M.A.O. performed data analysis; B.W.B prepared figures and drafted manuscript; B.W.B., M.Y., K.C.M., B.P.L., and M.A.O. critiqued and approved the final version of the manuscript.
ACKNOWLEDGMENTS
We thank Gabrielle Crandall for technical assistance with the collagen digestion of lung tissues and differential leukocyte counts.
REFERENCES
- 1. Ahmed MN, Suliman HB, Folz RJ, Nozik-Grayck E, Golson ML, Mason SN, Auten RL. Extracellular superoxide dismutase protects lung development in hyperoxia-exposed newborn mice. Am J Respir Crit Care Med 167: 400–405, 2003 [DOI] [PubMed] [Google Scholar]
- 2. Auten RL, O'Reilly MA, Oury TD, Nozik-Grayck E, Whorton MH. Transgenic extracellular superoxide dismutase protects postnatal alveolar epithelial proliferation and development during hyperoxia. Am J Physiol Lung Cell Mol Physiol 290: L32–L40, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Belperio JA, Keane MP, Burdick MD, Lynch JP, 3rd, Xue YY, Berlin A, Ross DJ, Kunkel SL, Charo IF, Strieter RM. Critical role for the chemokine MCP-1/CCR2 in the pathogenesis of bronchiolitis obliterans syndrome. J Clin Invest 108: 547–556, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bouvier NM, Lowen AC. Animal models for influenza virus pathogenesis and transmission. Viruses 2: 1530–1563, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Buczynski BW, Yee M, Paige Lawrence B, O'Reilly MA. Lung development and the host response to influenza A virus are altered by different doses of neonatal oxygen in mice. Am J Physiol Lung Cell Mol Physiol 302: L1078–L1087, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Coward WR, Saini G, Jenkins G. The pathogenesis of idiopathic pulmonary fibrosis. Ther Adv Respir Dis 4: 367–388, 2010 [DOI] [PubMed] [Google Scholar]
- 7. Dauger S, Ferkdadji L, Saumon G, Vardon G, Peuchmaur M, Gaultier C, Gallego J. Neonatal exposure to 65% oxygen durably impairs lung architecture and breathing pattern in adult mice. Chest 123: 530–538, 2003 [DOI] [PubMed] [Google Scholar]
- 8. Davis JM, Parad RB, Michele T, Allred E, Price A, Rosenfeld W. Pulmonary outcome at 1 year corrected age in premature infants treated at birth with recombinant human CuZn superoxide dismutase. Pediatrics 111: 469–476, 2003 [DOI] [PubMed] [Google Scholar]
- 9. Dawson TC, Beck MA, Kuziel WA, Henderson F, Maeda N. Contrasting effects of CCR5 and CCR2 deficiency in the pulmonary inflammatory response to influenza A virus. Am J Pathol 156: 1951–1959, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, Hoang DM, Chau NV, Khanh TH, Dong VC, Qui PT, Cam BV, Ha do Q, Guan Y, Peiris JS, Chinh NT, Hien TT, Farrar J. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med 12: 1203–1207, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dessing MC, van der Sluijs KF, Florquin S, van der Poll T. Monocyte chemoattractant protein 1 contributes to an adequate immune response in influenza pneumonia. Clin Immunol 125: 328–336, 2007 [DOI] [PubMed] [Google Scholar]
- 12. Doyle LW. Respiratory function at age 8–9 years in extremely low birthweight/very preterm children born in Victoria in 1991–1992. Pediatr Pulmonol 41: 570–576, 2006 [DOI] [PubMed] [Google Scholar]
- 13. Doyle LW, Anderson PJ. Adult outcome of extremely preterm infants. Pediatrics 126: 342–351, 2010 [DOI] [PubMed] [Google Scholar]
- 14. Doyle LW, Faber B, Callanan C, Freezer N, Ford GW, Davis NM. Bronchopulmonary dysplasia in very low birth weight subjects and lung function in late adolescence. Pediatrics 118: 108–113, 2006 [DOI] [PubMed] [Google Scholar]
- 15. Fehrenbach H. Alveolar epithelial type II cell: defender of the alveolus revisited. Respir Res 2: 33–46, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Folz RJ, Abushamaa AM, Suliman HB. Extracellular superoxide dismutase in the airways of transgenic mice reduces inflammation and attenuates lung toxicity following hyperoxia. J Clin Invest 103: 1055–1066, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Frank L, Bucher JR, Roberts RJ. Oxygen toxicity in neonatal and adult animals of various species. J Appl Physiol 45: 699–704, 1978 [DOI] [PubMed] [Google Scholar]
- 18. Giannandrea M, Yee M, O'Reilly MA, Lawrence BP. Memory CD8+ T cells are sufficient to alleviate impaired host resistance to influenza A virus infection caused by neonatal oxygen supplementation. Clin Vaccine Immunol 19: 1432–1441, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hartl D, Griese M, Nicolai T, Zissel G, Prell C, Reinhardt D, Schendel DJ, Krauss-Etschmann S. A role for MCP-1/CCR2 in interstitial lung disease in children (Abstract). Respir Res 6: 93, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Homer RJ, Elias JA, Lee CG, Herzog E. Modern concepts on the role of inflammation in pulmonary fibrosis. Arch Pathol Lab Med 135: 780–788, 2011 [DOI] [PubMed] [Google Scholar]
- 21. Inoshima I, Kuwano K, Hamada N, Hagimoto N, Yoshimi M, Maeyama T, Takeshita A, Kitamoto S, Egashira K, Hara N. Anti-monocyte chemoattractant protein-1 gene therapy attenuates pulmonary fibrosis in mice. Am J Physiol Lung Cell Mol Physiol 286: L1038–L1044, 2004 [DOI] [PubMed] [Google Scholar]
- 22. Jobe AH, Bancalari E. Bronchopulmonary dysplasia. Am J Respir Crit Care Med 163: 1723–1729, 2001 [DOI] [PubMed] [Google Scholar]
- 23. Jobe AH, Kallapur SG. Long term consequences of oxygen therapy in the neonatal period. Semin Fetal Neonatal Med 15: 230–235, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kinsella JP, Parker TA, Davis JM, Abman SH. Superoxide dismutase improves gas exchange and pulmonary hemodynamics in premature lambs. Am J Respir Crit Care Med 172: 745–749, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kobasa D, Jones SM, Shinya K, Kash JC, Copps J, Ebihara H, Hatta Y, Kim JH, Halfmann P, Hatta M, Feldmann F, Alimonti JB, Fernando L, Li Y, Katze MG, Feldmann H, Kawaoka Y. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 445: 319–323, 2007 [DOI] [PubMed] [Google Scholar]
- 26. Koth LL, Rodriguez MW, Bernstein XL, Chan S, Huang X, Charo IF, Rollins BJ, Erle DJ. Aspergillus antigen induces robust Th2 cytokine production, inflammation, airway hyperreactivity and fibrosis in the absence of MCP-1 or CCR2 (Abstract). Respir Res 5: 12, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lakshminrusimha S, Russell JA, Wedgwood S, Gugino SF, Kazzaz JA, Davis JM, Steinhorn RH. Superoxide dismutase improves oxygenation and reduces oxidation in neonatal pulmonary hypertension. Am J Respir Crit Care Med 174: 1370–1377, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matsuse T, Teramoto S, Katayama H, Sudo E, Ekimoto H, Mitsuhashi H, Uejima Y, Fukuchi Y, Ouchi Y. ICAM-1 mediates lung leukocyte recruitment but not pulmonary fibrosis in a murine model of bleomycin-induced lung injury. Eur Respir J 13: 71–77, 1999 [DOI] [PubMed] [Google Scholar]
- 29. McGrath-Morrow SA, Cho C, Soutiere S, Mitzner W, Tuder R. The effect of neonatal hyperoxia on the lung of p21Waf1/Cip1/Sdi1-deficient mice. Am J Respir Cell Mol Biol 30: 635–640, 2004 [DOI] [PubMed] [Google Scholar]
- 30. Moore BB, Paine R, 3rd, Christensen PJ, Moore TA, Sitterding S, Ngan R, Wilke CA, Kuziel WA, Toews GB. Protection from pulmonary fibrosis in the absence of CCR2 signaling. J Immunol 167: 4368–4377, 2001 [DOI] [PubMed] [Google Scholar]
- 31. Mrugacz M, Zelazowska B, Bakunowicz-Lazarczyk A, Kaczmarski M, Wysocka J. Elevated tear fluid levels of MIP-1alpha in patients with cystic fibrosis. J Interferon Cytokine Res 27: 491–495, 2007 [DOI] [PubMed] [Google Scholar]
- 32. Neff-LaFord HD, Vorderstrasse BA, Lawrence BP. Fewer CTL, not enhanced NK cells, are sufficient for viral clearance from the lungs of immunocompromised mice. Cell Immunol 226: 54–64, 2003 [DOI] [PubMed] [Google Scholar]
- 33. O'Reilly MA, Marr SH, Yee M, McGrath-Morrow SA, Lawrence BP. Neonatal hyperoxia enhances the inflammatory response in adult mice infected with influenza A virus. Am J Respir Crit Care Med 177: 1103–1110, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. O'Reilly MA, Yee M, Buczynski BW, Vitiello PF, Keng PC, Welle SL, Finkelstein JN, Dean DA, Lawrence BP. Neonatal oxygen increases sensitivity to influenza A virus infection in adult mice by suppressing epithelial expression of Ear1. Am J Pathol 42: 258–266, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Okuma T, Terasaki Y, Kaikita K, Kobayashi H, Kuziel WA, Kawasuji M, Takeya M. C-C chemokine receptor 2 (CCR2) deficiency improves bleomycin-induced pulmonary fibrosis by attenuation of both macrophage infiltration and production of macrophage-derived matrix metalloproteinases. J Pathol 204: 594–604, 2004 [DOI] [PubMed] [Google Scholar]
- 36. Robin B, Kim YJ, Huth J, Klocksieben J, Torres M, Tepper RS, Castile RG, Solway J, Hershenson MB, Goldstein-Filbrun A. Pulmonary function in bronchopulmonary dysplasia. Pediatr Pulmonol 37: 236–242, 2004 [DOI] [PubMed] [Google Scholar]
- 37. Smith RE, Strieter RM, Phan SH, Lukacs N, Kunkel SL. TNF and IL-6 mediate MIP-1alpha expression in bleomycin-induced lung injury. J Leukoc Biol 64: 528–536, 1998 [PubMed] [Google Scholar]
- 38. Smith VC, Zupancic JA, McCormick MC, Croen LA, Greene J, Escobar GJ, Richardson DK. Rehospitalization in the first year of life among infants with bronchopulmonary dysplasia. J Pediatr 144: 799–803, 2004 [DOI] [PubMed] [Google Scholar]
- 39. Suga M, Iyonaga K, Ichiyasu H, Saita N, Yamasaki H, Ando M. Clinical significance of MCP-1 levels in BALF and serum in patients with interstitial lung diseases. Eur Respir J 14: 376–382, 1999 [DOI] [PubMed] [Google Scholar]
- 40. Suliman HB, Ryan LK, Bishop L, Folz RJ. Prevention of influenza-induced lung injury in mice overexpressing extracellular superoxide dismutase. Am J Physiol Lung Cell Mol Physiol 280: L69–L78, 2001 [DOI] [PubMed] [Google Scholar]
- 41. Teske S, Bohn AA, Regal JF, Neumiller JJ, Lawrence BP. Activation of the aryl hydrocarbon receptor increases pulmonary neutrophilia and diminishes host resistance to influenza A virus. Am J Physiol Lung Cell Mol Physiol 289: L111–L124, 2005 [DOI] [PubMed] [Google Scholar]
- 42. Vannella KM, Moore BB. Viruses as co-factors for the initiation or exacerbation of lung fibrosis (Abstract). Fibrogenesis Tissue Repair 1: 2, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Walter EC, Ehlenbach WJ, Hotchkin DL, Chien JW, Koepsell TD. Low birth weight and respiratory disease in adulthood: a population-based case-control study. Am J Respir Crit Care Med 180: 176–180, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wareing MD, Lyon A, Inglis C, Giannoni F, Charo I, Sarawar SR. Chemokine regulation of the inflammatory response to a low-dose influenza infection in CCR2−/− mice. J Leukoc Biol 81: 793–801, 2007 [DOI] [PubMed] [Google Scholar]
- 45. Weinberger B, Laskin DL, Heck DE, Laskin JD. Oxygen toxicity in premature infants. Toxicol Appl Pharmacol 181: 60–67, 2002 [DOI] [PubMed] [Google Scholar]
- 46. Weisman LE. Populations at risk for developing respiratory syncytial virus and risk factors for respiratory syncytial virus severity: infants with predisposing conditions. Pediatr Infect Dis J 22: S33–S39, 2003 [DOI] [PubMed] [Google Scholar]
- 47. Wilborn AM, Evers LB, Canada AT. Oxygen toxicity to the developing lung of the mouse: role of reactive oxygen species. Pediatr Res 40: 225–232, 1996 [DOI] [PubMed] [Google Scholar]
- 48. Winter C, Taut K, Srivastava M, Langer F, Mack M, Briles DE, Paton JC, Maus R, Welte T, Gunn MD, Maus UA. Lung-specific overexpression of CC chemokine ligand (CCL) 2 enhances the host defense to Streptococcus pneumoniae infection in mice: role of the CCL2-CCR2 axis. J Immunol 178: 5828–5838, 2007 [DOI] [PubMed] [Google Scholar]
- 49. Yee M, Buczynski BW, Lawrence BP, O'Reilly MA. Neonatal hyperoxia increases sensitivity of adult mice to bleomycin-induced lung fibrosis. Am J Respir Cell Mol Biol 48: 258–266, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yee M, Chess PR, McGrath-Morrow SA, Wang Z, Gelein R, Zhou R, Dean DA, Notter RH, O'Reilly MA. Neonatal oxygen adversely affects lung function in adult mice without altering surfactant composition or activity. Am J Physiol Lung Cell Mol Physiol 297: L641–L649, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yee M, Vitiello PF, Roper JM, Staversky RJ, Wright TW, McGrath-Morrow SA, Maniscalco WM, Finkelstein JN, O'Reilly MA. Type II epithelial cells are critical target for hyperoxia-mediated impairment of postnatal lung development. Am J Physiol Lung Cell Mol Physiol 291: L1101–L1111, 2006 [DOI] [PubMed] [Google Scholar]
- 52. Zhao J, Shi W, Wang YL, Chen H, Bringas P, Jr, Datto MB, Frederick JP, Wang XF, Warburton D. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am J Physiol Lung Cell Mol Physiol 282: L585–L593, 2002 [DOI] [PubMed] [Google Scholar]