

Abstract
The effects of substituents on the aryl ring were studied by the preparation and testing of several PD173955 analogs. Inserting a single carbon atom into the C-N bond in the aniline subunit (PDC) reduced the kinase inhibition by a factor of 200. Despite its decreased affinity for Abl compared with PD173955, PDC exhibits a Ki very similar to that reported for Imatinib. Increased water solubility is also gained by replacing the thiomethyl group with an amino or glycyl moiety. For both PD173955 and PDC, the analogs with amino groups in place of the methylthio group are 10 times more inhibitory than the parent molecules. Two molecules were identified with Kis about three orders of magnitude lower than reported for Imatinib.
1. Introduction
Because tyrosine protein kinases regulate cell proliferation, drugs that control these kinases provide important treatments for proliferative diseases such as cancer and atherosclerosis [1]. Chronic myelogenous leukemia (CML) is a hematopoietic stem cell disease that accounts for 15% of all adult leukemias and is characterized by the clonal expansion of cells carrying the Philadelphia (Ph) chromosome [2]. The Ph chromosome, resulting from the translocation of genes from chromosomes 9 and 22 encodes the chimeric protein Bcr-Abl [2, 3]. It is now well recognized that the Bcr-Abl protein is both the initial cause and major driver of CML. The rearranged Bcr-Abl gene encodes a constitutively active Abl kinase that confers the property of excessive and uncontrolled proliferation to the myelogenous cells in which it is expressed [4]. Thus, this enzyme makes it an attractive target for therapeutic intervention [5].
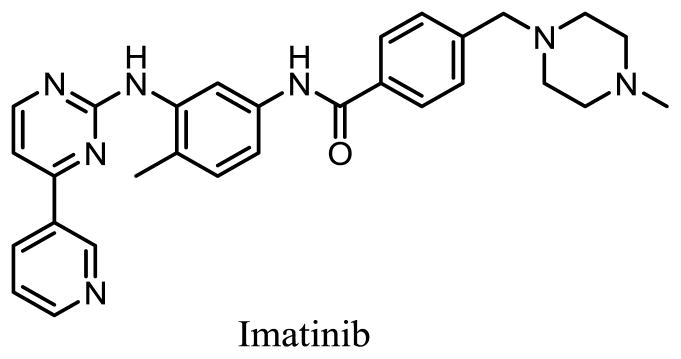
Imatinib, a phenylaminopyrimidine-derived inhibitor of the Abelson tyrosine kinase (c-Abl) shown in Figure 1, has been a very successful treatment for CML and other cancers. However, resistance has developed in many cancer patients [6]. Thus, new drugs that inhibit Imatinib-resistant kinases are needed.
Figure 1.
Substituted pyrido[2,3-d]pyrimidines are potent inhibitors that vary in their specificities for different tyrosine kinases [7]. PD173955 (1), shown in Figure 2, inhibits the Abelson kinase (Abl) and the Src and Yes tyrosine kinases that are also often up-regulated in cancer [8]. PD173955 inhibits the proliferation of many cancer cell types, but two limitations prevent it from being applied in a clinical setting. These are its ability to inhibit kinases other than the Abl kinase, which results in toxicity for proliferating normal cells [9] and its low solubility in water. In its favor, its affinity is much lower for most kinases other than those of the Src family of which Abl is a member, and PD173955 inhibits both the active and inactive forms of Abl. By contrast, Imatinib only inhibits the active form of the enzyme. In addition, the Ki for inhibition of Abl by PD173955 is very low, making it a more potent inhibitor of Abl and a more effective inhibitor of cancer cell proliferation than Imatinib [10, 12, 15]. Thus we speculated that, with further modification, PD173955 may be developed as an effective inhibitor of c-Abl to treat patients with CML and other cancers, including those that have developed Imatinib-resistance.
Figure 2.
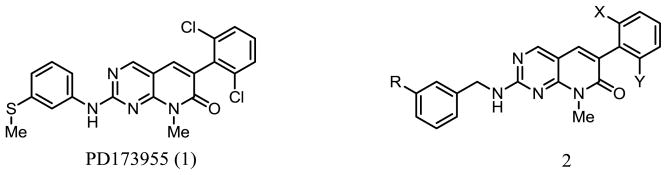
Clarkson and Duyster showed that 1 (Fig. 2) and related compounds inhibit Bcr-Abl kinase activity with greater potency than Imatinib. Moreover, many of these compounds also inhibit kinase domain mutants of Bcr-Abl that are resistant to Imatinib [11]. With the emerging need for kinase inhibitors that will kill cells that express Imatinib-resistant Bcr-Abl kinase domain mutants, PD173955 is a prime candidate for further development.
To investigate the structural elements of PD173955 that make it a good inhibitor of the Abl kinase, we planned to synthesize analogs of 1. The bicyclic ring and the halogen containing ring subunit of PD173955 interact with several amino acid residues in the crystal structure of the kinase complex [12]. As discussed below, many analogs of 2 were also synthesized. These were expected to also be good inhibitors of the Abl kinase because the crystal structure of Abl with PD173955 shows the methylthiophenyl ring extending into the solution space and not interacting extensively with the protein [12].
2. Chemistry
Our synthetic route to analogs of PD173955 began from the known aldehyde 3, readily available from the commercially available ethyl 4-chloro-2-methylthio pyrimidine-5-carboxylate by amination, reduction and manganese dioxide oxidation [13]. The reaction of aldehyde 3 with substituted phenylacetic acid ester 4 was hampered by problems related to solubility and low yields. After several experiments, condensation of aldehyde 3 with various substituted phenyl acetic acid esters was successfully performed following the method developed by Blass et al. using KF/Al2O3 and dimethyl acetamide at room temperature to give functionalized pyrido[2,3-d]pyrimidinones 5a–e in moderate to excellent yields (Scheme 1) [14]. Oxidation of 5a–e with 2 equiv. of m-chloroperbenzoic acid in dichloromethane gave corresponding sulfones 6a–e in excellent yields.
Scheme 1.

Initially, the conversion of sulfone 6 into analogs of PD173955 using various anilines in boiling aprotic solvents failed. Interestingly, the reaction of sulfone 6 with 3-aminobenzylamine, a more reactive nucleophile, in boiling DMF generated an adduct in good yield (Scheme 2).
Scheme 2.

This adduct (which we term PDC because of the extra carbon atom between the phenyl group and the secondary amino group) was more soluble than PD173955 in aqueous solutions, particularly when R = −NH2 or -NHGly. Our interest in understanding the effect of changes in the PD structure on its ability to inhibit Abl kinase led us to synthesize various PD and PDC analogs 7a–7e, 8a–8d, 9a–9b and 1. The reason to develop the analogs 8c and 9b was to circumvent the problem of low chemical reactivity of aromatic amine moiety of analogs 7a–7e. The synthetic route towards the synthesis of 8c and 9b consist of standard two step sequence where 7a and 9a were reacted with bromoacetyl bromide under basic conditions followed by the treatment with ammonia to get 8c and 9b in good yields (Scheme 3).
Scheme 3.

3. Results and discussion
The results of inhibition of Abl kinase (Ki) are shown below in Figures 3 – 5. The number in parentheses after the Ki is the number of independent experiments performed to obtain the Ki. The asterisks represent the p values determined from the Pearson product moment correlation analysis (***, p<0.0005, **, p<0.005, *, p<0.03)
Figure 3.
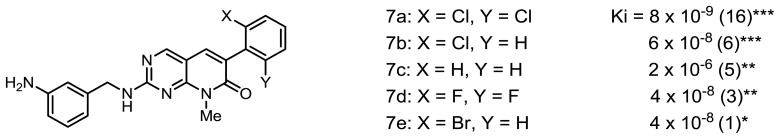
Figure 5.
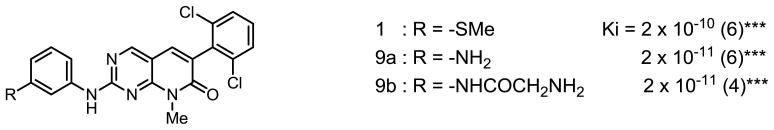
The 2,6-dichlorophenyl substituent 7a clearly exhibited the strongest inhibition. Analog 8a was prepared by the reaction of m-methylthioaniline with the corresponding sulfone (Scheme 2). The analogs 8b–d were synthesized by modifying 7a (Scheme 3).
PD173955 was ultimately synthesized using boiling diglyme in sealed tube, conditions much harsher than those for the synthesis of the PDC and its analogs (Scheme 2). The activity of 1 and adducts 9a and 9b was tested using the standard protocol and the results are shown below.
Compounds 7a–e were synthesized to probe the steric and electronic influences on the aryl ring. Molecular models clearly show that the 2,6-dichlorophenyl group in 1 is perpendicular to the pyrido[2,3-d]pyrimidine subunit as a result of nonbonded interactions of the chlorine atoms [12]. The effectiveness with which c-Abl was inhibited by the synthesized analogs was determined from the Ki for each compound, which increases with decreasing inhibitory effectiveness. As shown above, compared with the 2-chlorophenyl substituent, the monochlorophenyl and phenyl substituents have increasingly higher Ki values. The 2,6-difluorophenyl substituent (7d), which is sterically the same size as the phenyl group, has about the same inhibitory efficiency as PDC with the 2-chloro (7b) and 2-bromo (7e) substituents. Structural analysis of the interaction of PD173955 with the Abl kinase identified the chloro substituents of the aryl ring as being embedded in the enzyme’s ATP-binding pocket and held with multiple van der Waals interactions, which contribute to the affinity of the molecule for the Abl kinase [12]. The chlorine atoms limit the rotation of the aryl ring to a rotational angle that is the same as for the central phenyl group of Imatinib. These phenyl groups of Imatinib and PD173955 sit in the same position, similarly rotated, in the protein. The removal of one or two chlorine atoms from 7a or their replacement with fluoro constituents would allow rotational flexibility of the compound when not bound to the enzyme and might increase the Ki compared with 7a by increasing koff in the equilibrium between the Abl and PD compound and the Abl-PD complex due to increased entropy of the unbound 7a.
The much higher Ki of 7c compared with 7b, 7d, or 7e suggests that electronic effects also play an important role in the interaction of PDC with the enzyme. It is likely that the relevant interacting amino residue might be lys271 that lies close enough to the inhibitor in the enzyme pocket to interact with PD173955 (and presumably also PDC) through van der Waals forces as described for the crystal structure, but which might also be close enough to interact electrostatically with the aryl ring constituents through its epsilon amino group.
Although most van der Waals interactions reported for PD173955 with Abl are located around the bicyclic constituent and the dichlorophenyl group, two of the ten interactions are with amino acids that contact the methylthiophenyl segment of PD173955. Alteration of the molecular structure in this region by insertion of an additional carbon between the secondary amino group and the aryl groups results in a increase in Ki of about 200-fold, which is consistent with the loss of 2 of the 10 van der Waals interactions that hold the inhibitor in place in the protein’s ATP-binding pocket. The 10-fold decrease in the Ki with the replacement of the methylthio group by an amino group on either PD or PDC probably reflects the gain in van der Waals or an electrostatic interaction with the enzyme by way of the amino group.
4. Conclusion
In summary, the effects of substituents on the aryl ring were studied by the preparation and testing of several PD173955 analogs. The results are consistent with the observation from the crystal structure that electronic and van der Waals forces are likely to be involved in the interaction of inhibitor and enzyme. They also show that inserting a single carbon atom into the C-N bond in the aniline subunit (PDC) reduced the kinase inhibition by a factor of 200, consistent with the loss of 20% of the van der Waals interactions between inhibitor and protein. Despite its decreased affinity for Abl compared with PD, PDC (7a) exhibits a Ki very similar to that reported for Imatinib [4 × 10−8] [15] and is significantly more water soluble compared with PD173955. Furthermore, replacement of the thiomethyl group on PD173955 with either an amino or a glycyl group resulted in a decrease in the Ki of 10-fold, which is 1000-fold lower than the Ki reported for Imatinib.
5. Experimental
5.1 Chemistry
5.1.1 Materials and Instrumentation
THF was distilled from sodium benzophenone ketyl. Methylene chloride, DMA and DMF were purchased from Aldrich. All experiments were performed under an argon atmosphere. Organic extracts were dried over anhydrous MgSO4. NMR experiments were performed with either a Varian 300 MHz or a Bruker 400 MHz instrument. HRMS were recorded on a Kratos model MS-50 spectrometer and LRMS were performed with a Finnegan 4023 mass spectrometer. Standard grade silica gel (60 Å) was used for flash column chromatography.
5.1.2 Representative procedure for the preparation of 5a–e
To a stirred solution of 0.400 g (2.19 mmol) of 4-amino-2-(methylthio) pyrimidine-5-carbaldehyde 3 and 0.638 g (2.74 mmol) of ethyl 2-(2,6-dichlorophenyl)acetate 4a in 10.0 mL of dry DMA, 2.536 g of KF/Al2O3 (40 wt %, KF, Sigma–Aldrich catalog #316385) was added in batches of 0.5 g every 30 min. The reaction mixture was then stirred at RT for 24 h under argon after which it was filtered through celite, the residual solid was washed with methylene chloride and the combined filtrates were concentrated. Due to the poor solubility of dichloropyridopyrimidinones derivative 5a in most solvents, it was purified by recrystallization from DMF to obtain 0.480 g (62%). Other pyridopyrimidinones were purified by column chromatography over silica gel using 1:1 hexanes:EtOAc to give pure products.
5.1.2.1 6-(2,6-dichlorophenyl)-8-methyl-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (5a)
Yield 62%; 1H-NMR (400 MHz, CDCl3): δ 8.66 (s, 1H), 7.60 (s, 1H), 7.41 (d, J = 8.0 Hz, 2H), 7.29 (d, J = 8.0 Hz, 1H), 3.83 (s, 3H), 2.67 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ 173.9, 161.1, 156.7, 154.6, 136.1, 135.6, 133.9, 130.4, 129.7, 128.3, 109.4, 28.7, 14.8; LRMS: 352.0, 200.0, 154.1; HRMS calculated M+ for C15H11Cl2N3OS: 352.0073; found: 352.0074.
5.1.2.2 6-(2-chlorophenyl)-8-methyl-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (5b)
Yield 73%; 1H-NMR (400 MHz, CDCl3): δ 8.63 (s, 1H), 7.64 (s, 1H), 7.42 – 7.49 (m, 1H), 7.27 – 7.37 (m, 3H), 3.80 (s, 3H), 2.65 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ 173.5, 161.8, 156.5, 154.4, 135.0, 134.8, 133.8, 131.7, 131.5, 130.0, 129.9, 126.9, 109.5, 28.6, 14.7; LRMS: 318.0; HRMS calculated M+ for C15H12ClN3OS: 317.0390; found: 317.0396
5.1.2.3 8-methyl-2-(methylthio)-6-phenylpyrido[2,3-d]pyrimidin-7(8H)-one (5c)
Yield 89%; 1H-NMR (400 MHz, CDCl3): δ 8.61 (s, 1H), 7.67 (s, 1H), 7.61 – 7.66 (m, 2H), 7.34 – 7.45 (m, 3H), 3.79 (s, 3H), 2.63 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ 172.8, 162.4, 156.3, 153.9, 135.6, 132.8, 132.6, 128.9, 128.7, 128.4, 109.9, 28.5, 14.6; LRMS: 284.1, 176.1, 158.1; HRMS calculated M+ for C15H13N3OS: 283.0779; found: 283.0778
5.1.2.4 6-(2-bromophenyl)-8-methyl-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (5d)
Yield 83%; 1H-NMR (400 MHz, CDCl3): δ 8.63 (s, 1H), 7.65 (d, J = 8.0 Hz, 1H), 7.61 (s, 1H), 7.29 – 7.40 (m, 2H), 7.21 – 7.28 (m, 1H), 3.80 (s, 3H), 2.64 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ 173.5, 161.8, 156.6, 154.5, 136.9, 135.0, 133.3, 133.1, 131.6, 130.2, 127.6, 123.9, 109.5, 28.7, 14.8; LRMS: 364.0, 362.0; HRMS calculated M+ for C15H12BrN3OS: 360.9884; found: 360.9876.
5.1.2.5 6-(2,6-difluorophenyl)-8-methyl-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (5e)
Yield 92%; 1H-NMR (400 MHz, CDCl3): δ 8.64 (s, 1H), 7.72 (s, 1H), 7.30 – 7.40 (m, 1H), 6.93 – 7.04 (m, 2H), 3.81 (s, 3H), 2.64 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ 173.9, 162.0 (d, J = 28.0 Hz), 161.2, 159.5 (d, J = 28.0 Hz), 156.7, 154.5, 136.9, 130.6 (t, J = 40.0 Hz), 122.4, 111.7 (d, J = 100.0 Hz), 109.3, 28.7, 14.7; LRMS: 320.1; HRMS calculated M+ for C15H11F2N3OS: 319.0591; found: 319.0591.
5.1.3 Representative procedure for the preparation of 6a–e
To a solution of 0.500 g (1.42 mmol) of 5a in 40 mL of chloroform, was added 0.800 g (3.12 mmol) of 75% m-chloroperoxybenzoic acid. The solution was stirred at RT for 6 h. After the completion of reaction, 2 mL of DMSO was added to the reaction mixture to neutralize unreacted m-chloroperoxybenzoic acid and it was further stirred for 15 minutes after which the reaction mixture was diluted with methylene chloride and washed with saturated NaHCO3, water and brine. The organic phase was dried over anhydrous MgSO4 and concentrated. The crude was used for the next step without further purification.
5.1.3.1 6-(2,6-dichlorophenyl)-8-methyl-2-(methylsulfonyl)pyrido[2,3-d]pyrimidin-7(8H)-one (6a)
Yield 87%; 1H-NMR (300 MHz, CDCl3): δ 9.03 (s, 1H), 7.78 (s, 1H), 7.42 – 7.48 (m, 2H), 7.31 – 7.38 (m, 1H), 3.91 (s, 3H), 3.44 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ 165.0, 160.5, 157.5, 155.5, 135.2, 135.0, 134.7, 132.9, 131.0, 128.4, 115.0, 39.5, 29.5; LRMS: 352.0, 200.0, 154.1; HRMS calculated M+ for C15H11Cl2N3O3S: 383.9971; found: 383.9970
5.1.3.2 6-(2-chlorophenyl)-8-methyl-2-(methylsulfonyl)pyrido[2,3-d]pyrimidin-7(8H)-one (6b)
Yield 85%; 1H-NMR (400 MHz, CDCl3): δ 9.00 (s, 1H), 7.82 (s, 1H), 7.47 – 7.53 (m, 1H), 7.33 – 7.43 (m, 3H), 3.89 (s, 3H), 3.43 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ 164.6, 161.2, 157.3, 155.2, 136.6, 133.8, 133.7, 133.5, 131.3, 130.7, 130.1, 127.1, 115.1, 39.4, 29.4; LRMS: 452.3, 411.1, 391.3, 350.0, 331.1, 302.1, 268.1, 249.1, 215.1; HRMS calculated M+ for C15H12ClN3O3S: 349.0310; found: 349.0299.
5.1.3.3 8-methyl-2-(methylsulfonyl)-6-phenylpyrido[2,3-d]pyrimidin-7(8H)-one (6c)
Yield 65%; 1H-NMR (400 MHz, CDCl3): δ 8.99 (s, 1H), 7.85 (s, 1H), 7.64 – 7.71 (m, 2H), 7.42 – 7.49 (m, 3H), 3.88 (s, 3H), 3.41 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ 164.2, 162.0, 157.0, 154.7, 137.7, 134.7, 131.2, 129.8, 129.1, 128.7, 115.7, 39.5, 29.4; LRMS: 318.0; HRMS calculated M+ for C15H13N3O3S: 317.0390; found: 317.0396.
5.1.3.4 6-(2-bromophenyl)-8-methyl-2-(methylsulfonyl)pyrido[2,3-d]pyrimidin-7(8H)-one (6d)
Yield 84%; 1H-NMR (400 MHz, CDCl3): δ 9.00 (s, 1H), 7.79 (s, 1H), 7.70 (d, J = 8.0 Hz, 1H), 7.42 (t, J = 8 Hz, 1H), 7.29 – 7.36 (m, 2H), 3.90 (s, 3H), 3.43 (s, 1H); 13C-NMR (100 MHz, CDCl3): δ 164.7, 161.2, 157.3, 155.3, 138.3, 135.8, 133.7, 133.3, 131.2, 130.9, 127.8, 123.4, 115.2, 39.5, 29.5; LRMS: 457.0, 396.0, 348.0, 254.1, 215.1; HRMS calculated M+ for C15H12BrN3O3S: 392.9783; found: 392.9792.
5.1.3.5 6-(2,6-difluorophenyl)-8-methyl-2-(methylsulfonyl)pyrido[2,3-d]pyrimidin-7(8H)-one (6e)
Yield 82%; 1H-NMR (400 MHz, CDCl3): δ 9.02 (s, 1H), 7.90 (s, 1H), 7.37 – 7.47 (m, 1H), 7.03 (t, J = 8.0 Hz, 2H), 3.89 (s, 3H), 3.42 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ 164.9, 161.9 (d, J = 28.0 Hz), 160.6, 159.4 (d, J = 28.0 Hz), 157.5, 155.4, 135.7, 131.4 (t, J = 40.0 Hz), 127.8, 114.9, 111.9 (d, J = 100.0 Hz), 39.5, 29.5; LRMS: 413.1, 352.0, 333.1, 304.1, 251.1, 209.2, 121.0; HRMS calculated M+ for C15H11F2N3O3S: 351.0489; found: 351.0491.
5.1.4 Representative procedure for the preparation of 7a–e and 8a
A stirred mixture of 0.135 g (0.352 mmol) of sulfone 6a and 0.086 g (0.704 mmol) of 3-aminobenzylamine in DMF (5 mL) was refluxed overnight. The resultant reaction mixture was cooled to RT and diluted with water. This aqueous solution was extracted with ethyl acetate (3x 15 mL). The ethyl acetate layer was subjected to water wash (2×10 mL) and brine wash followed by drying over anhydrous MgSO4 and concentrated in vacuo to give the crude product which was chromatographed over silica gel using 2% MeOH in CH2Cl2 to give 0.091 g (61%) of pure 7a.
5.1.4.1 2-(3-aminobenzylamino)-6-(2,6-dichlorophenyl)-8-methylpyrido[2,3-d]pyrimidin-7(8H)-one (7a)
Yield 61%; 1H-NMR (400 MHz, CDCl3): δ 8.27 (bs, 1H), 7.44 (s, IH), 7.40 (d, J = 8.0 Hz, 2H), 7.25 (t, J = 8.0 Hz, 1H), 7.14 (t, J = 8.0 Hz, 1H), 6.61 – 6.78 (m, 3H), 4.64 (d, J = 5.2 Hz, 2H), 3.72 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ 161.67, 161.19, 158.67, 156.22, 147.00, 139.75, 136.71, 135.94, 134.55, 129.87, 129.79, 128.12, 120.27, 119.82, 118.58, 118.09, 114.42, 114.32, 113.83, 46.05, 28.33; LRMS: 427.1, 425.1, 392.1, 390.1, 287.1, 285.1, 106.1; HRMS calculated M+ for C21H17Cl2N5O: 452.0810; found: 425.0813.
5.1.4.2 2-(3-aminobenzylamino)-6-(2-chlorophenyl)-8-methylpyrido[2,3-d]pyrimidin-7(8H)-one (7b)
Yield 77%; 1H-NMR (400 MHz, CDCl3): δ 8.32 (bs, 1H), 7.52 (s, IH), 7.47 (t, J = 4.0 Hz, 1H), 7.30 – 7.36 (m, 3H), 7.14 (t, J = 8.0 Hz, 1H), 6.77 (d, J = 7.6 Hz, 1H), 6.70 (s, 1H), 6.62 (d, J = 7.6 Hz, 1H), 4.64 (d, J = 5.2 Hz, 2H), 3.71 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ 162.5, 161.6, 158.8, 156.1, 147.0, 140.0, 135.8, 135.5, 134.0, 131.9, 129.9, 129.6, 129.5, 126.8, 118.0, 114.4, 114.2, 46.0, 28.4; LRMS: 391.1, 356.2, 251.1, 106.1, 69.0; HRMS calculated M+ for C21H18ClN5O: 391.1200; found: 391.1204.
5.1.4.3 2-(3-aminobenzylamino)-8-methyl-6-phenylpyrido[2,3-d]pyrimidin-7(8H)-one (7c)
Yield 53%; 1H-NMR (400 MHz, CDCl3): δ 8.20 (bs, 1H), 7.64 (d, J = 7.6 Hz, 2H), 7.55 (s, 1H), 7.43 – 7.32 (m, 3H), 7.13 (t, J = 7.6 Hz, 1H), 6.77 – 6.59 (m, 3H), 4.62 (d, J = 5.2 Hz, 2H), 3.71 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ 163.1, 161.3, 158.6, 155.6, 147.0, 139.9, 136.5, 133.7, 129.8, 129.6, 128.9, 128.4, 128.0, 120.4, 118.6, 118.0, 114.3, 46.0, 28.3; LRMS: 358.2, 357.2, 356.2, 252.1, 121.1, 106.1, 77.0; HRMS calculated M+ for C21H19N5O: 357.1590; found: 357.1600.
5.1.4.4 2-(3-aminobenzylamino)-6-(2-bromophenyl)-8-methylpyrido[2,3-d]pyrimidin-7(8H)-one (7e)
Yield 48%; 1H-NMR (400 MHz, CDCl3): δ 8.18 (bs, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.46 (bs, 1H), 7.37 – 7.32 (m, 2H), 7.26 – 7.20 (m, 2H), 7.13 (t, J = 7.6 Hz, 1H), 6.78 – 6.60 (m, 3H), 4.62 (d, J = 5.2 Hz, 2H), 3.71 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ 162.5, 161.6, 161.3, 158.8, 156.1, 147.1, 139.9, 137.6, 135.8, 133.1, 131.9, 129.9, 129.7, 127.5, 124.3, 120.4, 118.7, 118.1, 114.4, 46.1, 28.4; LRMS: 435, 358, 356, 252, 237, 179, 106, 77; HRMS calculated M+ for C21H18BrN5O: 435.06947; found: 435.07033.
5.1.4.5 2-(3-aminobenzylamino)-6-(2,6-difluorophenyl)-8-methylpyrido[2,3-d]pyrimidin-7(8H)-one (7d)
Yield 67%; 1H-NMR (400 MHz, CDCl3): δ 8.37 (bs, 1H), 7.59 (s, IH), 7.32 (dt, J = 7.4 Hz, J = 2.0 Hz, 1H), 7.14 (t, J = 7.6 Hz, 1H), 6.97 (t, J = 8.0 Hz, 2H), 6.76 (d, J = 7.2 Hz, 1H), 6.69 (s, 1H), 6.61 (dd, J = 8.0 Hz, J = 2.0 Hz, 1H), 4.64 (d, J = 5.2 Hz, 2H), 3.71 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ 162.3, 162.0, 161.7, 161.2, 159.8, 159.0, 156.3, 147.0, 139.8, 137.6, 129.9, 120.5, 119.9, 118.7, 118.0, 114.4, 111.7, 111.5, 105.6, 46.1, 28.5; LRMS: 394.1, 393.1, 392.1, 288.1, 269.1, 121.1, 106.1, 77.0, 69.0; HRMS calculated M+ for C21H17F2N5O: 393.1401; found: 393.1404.
5.1.4.6 6-(2,6-dichlorophenyl)-8-methyl-2-(3-(methylthio)benzylamino)pyrido[2,3-d]pyrimidin-7(8H)-one (8a)
Yield 64%; 1H-NMR (400 MHz, CDCl3): δ 8.19 (bs, 1H), 7.42 – 7.38 (m, 3H), 7.27 – 7.22 (m, 3H), 7.18 – 7.16 (m, 2H), 6.84 (bs, 1H), 4.68 (d, J = 4.4 Hz, 2H), 3.71 (s, 3H), 2.46 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ 161.6, 158.9, 156.3, 139.4, 139.3, 136.7, 136.6, 136.0, 134.6, 129.9, 129.4, 128.2, 126.0, 125.7, 124.6, 45.9, 28.4, 16.0; LRMS: 457, 422, 295, 188; HRMS calculated M+ for C22H18Cl2N4OS: 456.05783; found: 456.05881.
5.1.5 Representative procedure for the preparation of 1 and 9a
To a solution of sulfone 6a (0.187 g, 0.5 mmol) in freshly distilled diglyme (5 ml), 1,3-phenylenediamine (0.120 g, 1.1 mmol) was added and resulting reaction mixture was refluxed for 12 h. After the completion of reaction, most of the solvent was evaporated invacuo and residue was purified by preparative TLC to get amine 9a (0.110 g, 55%).
5.1.4.7 6-(2,6-dichlorophenyl)-8-methyl-2-(3-(methylthio)phenylamino)pyrido[2,3-d]pyrimidin-7(8H)-one (1)
Yield 57%; 1H-NMR (400 MHz, CDCl3): δ 8.60 (s, 1H), 7.75 (bs, 1H), 7.54 – 7.52 (m, 2H), 7.42 – 7.38 (m, 3H), 7.33 – 7.25 (m, 2H), 7.01 (d, J = 8.0 Hz, 1H), 3.82 (s, 3H), 2.53 (s, 3H); LRMS: 442, 407, 362, 313, 269, 203, 196, 180; HRMS calculated M+ for C21H16Cl2N4OS: 442.04219; found: 442.04315.
5.1.4.8 2-(3-aminophenylamino)-6-(2,6-dichlorophenyl)-8-methylpyrido[2,3-d]pyrimidin-7(8H)-one (9a)
Yield 55%; 1H-NMR (400 MHz, CDCl3): δ 8.58 (s, 1H), 7.53 (s, 1H), 7.46 – 7.40 (m, 3H), 7.28 – 7.25 (m, 1H), 7.23 – 7.11 (m, 2H), 7.07 – 6.98 (bs, 1H), 6.48 (d, J = 8.0 Hz, 1H), 3.81 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ 161.6, 159.3, 159.2, 158.6, 156.1, 147.4, 139.8, 136.5, 136.5, 136.4, 136.4, 135.9, 134.4, 130.1, 128.2, 126.3, 110.7, 110.3, 106.8, 106.5, 28.8; LRMS: 412, 377, 376, 284, 242; HRMS calculated M+ for C20H15Cl2N5O: 411.06536; found: 411.06606.
5.2 Pharmocology
5.2.1 Kinase Assays and Analysis
The recombinant v-Abl tyrosine kinase was from EMD Calbiochem, Cat#102555 (San Diego, CA), its abltide substrate was from Upstate Biochemicals (Lake Placid, NY), the adenosine 5′-triphosphate (ATP) disodium salt was from Sigma, and the 32P-ATP was from MP Biomedicals (Solon, OH). The tyrosine kinase activity was determined from the rate of incorporation of 32P into abltide from γ 32P-ATP. The reaction mixtures, containing 0.4 pg/ml v-Abl and various concentrations of PD173955, PDC or their analogs in 50 mM Tris-HCl, 0.8 mM MOPS, 10 mM MgCl2, 120 μM EDTA, 0.015 % BRIJ35, 1 mM DTT, 50 μM Abltide, 10 % DMSO, (pH 7.5). All reaction conditions were tested in triplicate. The reactions were started by the addition of γ32P-ATP (1 μCi/10−6 mmol) to a final concentration of 100 μM. They were incubated for 30 min at 30 °C and stopped by spotting on P30 filtermat glass fiber filters (cat#1450–523, Wallac-Perkin Elmer, Waltham, MA). The free γ32P-ATP and 32P was separated from the phosphorylated peptide by ascending thin layer chromatography on P30 Wallac Filtermat with a mobile phase of 0.75 % phosphoric acid. The filters were cut to isolate the phosphorylated peptide and the incorporated cpm was determined by measuring Cerenkov radiation using a scintillation spectrometer. Triplicate values were averaged to obtain the value for kinase activity in the presence of each tested concentration of PD173955, PDC or their analogs.
The Kis were determined using all the data from multiple experiments for each compound by non-linear least squares fits using the Hill plot to determine IC50 and the formula: Ki =10IC50/(1+([ATP]/Kd) in which the Kd for ATP was taken as ATP 3.8 × 10−6 M. The data was analyzed for statistical significance with the Pearson product moment correlation coefficient (PMCC) (calculated by the Costat program CoHort Software, Monterey, CA). From this analysis all titrations were fit with PMCCs of 0.9 and above (with p values of 0.0014 and lower) except for the data for compound 6e for which the PMCC was 0.87 (p value of 0.023). The relative p values are represented by asterisks in figures 3 through 5.
Supplementary Material
Figure 4.
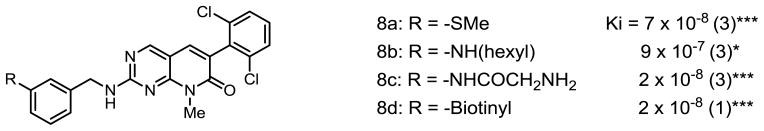
Acknowledgments
We thank Howard A. Levine for early guidance in data analysis. This work was funded by NIH grant R43 CA-110222, STTR to Molecular Express Inc., and a subcontract to Iowa State University.
Footnotes
Supplementary data associated with this article can be found in the online version at
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Klein S, Levitzki A. Targeted cancer therapy: promise and reality. Advances in Cancer Research. 2007;97:295–319. doi: 10.1016/S0065-230X(06)97013-4. [DOI] [PubMed] [Google Scholar]
- 2.National Comprehensive Cancer Network. Clinical Practice Guidelines in Oncology: Chronic Myelogenous Leukemia. 2009;1 doi: 10.6004/jnccn.2009.0065. [DOI] [PubMed] [Google Scholar]
- 3.Schiffer CA. BCR-ABL tyrosine kinase inhibitors for chronic myelogenous leukemia. N Engl J Med. 2007;357(3):258–265. doi: 10.1056/NEJMct071828. [DOI] [PubMed] [Google Scholar]
- 4.Wolf D, Tilg H, Rumpold H, Gastl G, Wolf AM. The Kinase Inhibitor Imatinib - An Immunosuppressive Drug? Current Cancer Drug Targets. 2007;7:251–258. doi: 10.2174/156800907780618293. [DOI] [PubMed] [Google Scholar]
- 5.Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105(7):2640–2653. doi: 10.1182/blood-2004-08-3097. [DOI] [PubMed] [Google Scholar]
- 6.Ramirez P, DiPersio JF. Therapy options in imatinib failures. Oncologist. 2008;13(4):424–434. doi: 10.1634/theoncologist.2007-0170. [DOI] [PubMed] [Google Scholar]
- 7.Hamby JM, Connolly CJ, Schroeder MC, Winters RTo, Showalter HD, Panek RL, Major TC, Olsewski B, Ryan MJ, Dahring T, Lu GH, Keiser J, Amar A, Shen C, Kraker AJ, Slintak V, Nelson JM, Fry DW, Bradford L, Hallak H, Doherty AM. Structure-activity relationships for a novel series of pyrido[2,3-d]pyrimidine tyrosine kinase inhibitors. J Med Chem. 1997;40:2296–2303. doi: 10.1021/jm970367n. [DOI] [PubMed] [Google Scholar]
- 8.Moasser MM, Srethapakdi M, Sachar KS, Kraker AJ, Rosen N. Inhibition of Src kinases by a selective tyrosine kinase inhibitor causes mitotic arrest. Cancer Res. 1999;59:6145–6152. [PubMed] [Google Scholar]
- 9.Wissing J, Godl K, Brehmer D, Blencke S, Weber M, Habenberger P, Stein-Gerlach M, Missio A, Cotton M, Muller S, Daub H. Chemical proteomic analysis reveals alternative modes of action for pyrido[2,3-d]pyrimidine kinase inhibitors. Cell Proteomics. 2004;3:1181–1193. doi: 10.1074/mcp.M400124-MCP200. [DOI] [PubMed] [Google Scholar]
- 10.Wisniewski D, Lambek CL, Liu C, Strife A, Veach DR, Nagar B, Young MA, Schindler T, Bornmann WG, Bertino JR, Kuriyan J, Clarkson B. Characterization of Potent Inhibitors of the Bcr-Abl and the c-Kit Receptor Tyrosine Kinases. Cancer Res. 2002;62:4244–4255. [PubMed] [Google Scholar]
- 11.von Bubnoff Nikolas, Veach Darren R, Miller W Todd, Wanqing Li, Jana Saenger, Christian Peschel, Bornmann William G, Bayard Clarkson, Duyster Justus. Inhibition of Wild-Type Mutant Bcr-Abl by Pyrido-Pyrimidine-Type Small Molecule Kinase Inhibitors. Cancer Res. 2003;63:6395–6404. [PubMed] [Google Scholar]
- 12.Nagar B, Bornmann WG, Pellicena P, Schindler T, Veach DR, Miller WT, Clarkson B, Kuriyan J. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571) Cancer Res. 2002;62:4236–4243. [PubMed] [Google Scholar]
- 13.Klutchko SR, Hamby JM, Boschelli DH, Wu Z, Kraker AJ, Amar AM, Hartl BG, Shen C, Klohs WD, Steinkampf RW, Driscoll DL, Nelson JM, Elliott WL, Roberts BJ, Stoner CL, Vincent PW, Dykes DJ, Panek RL, Lu GH, Major TC, Dahring TK, Hallak H, Bradford LA, Showalter HDH, Doherty AM. 2-Substituted aminopyrido[2,3-d]pyrimidin-7(8H)-ones. Structure-Activity Relationships against selected Tyrosine Kinases and in Vitro and in Vivo Anticancer Activity. J Med Chem. 1998;41:3276. doi: 10.1021/jm9802259. [DOI] [PubMed] [Google Scholar]
- 14.Blass BE, Coburn K, Fairweather N, Sabat M, West L. A facile, KF/Al2O3 mediated method for the preparation of functionalized pyrido[2,3-d]pyrimidin-7(8H)-ones. Tet Lett. 2006;47(18):3177–3180. [Google Scholar]
- 15.Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural Mechanism for STI-571 Inhibition of Abelson Tyrosine Kinase. Science. 2000;289:1938–1942. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.