

Abstract
Background: Apoptosis, reactive oxygen species (ROS) and inflammatory cytokines have all been implicated in the development of Alzheimer’s disease (AD).
Objectives: The present study identifies the apoptotic factor that was responsible for the fourfold increase in apoptotic rates that we previously noted when pig proximal tubule, LLC-PK1, cells were exposed to AD plasma as compared to plasma from normal controls and multi-infarct dementia.
Patients and Methods: The apoptotic factor was isolated from AD urine and identified as lipocalin-type prostaglandin D2 synthase (L-PGDS). L-PGDS was found to be the major apoptotic factor in AD plasma as determined by inhibition of apoptosis approximating control levels by the cyclo-oxygenase (COX) 2 inhibitor, NS398, and the antibody to L-PGDS. Blood levels of L-PGDS, however, were not elevated in AD. We now demonstrate a receptor-mediated uptake of L-PGDS in PC12 neuronal cells that was time, dose and temperature-dependent and was saturable by competition with cold L-PGDS and albumin. Further proof of this endocytosis was provided by an electron microscopic study of gold labeled L-PGDS and immunofluorescence with Alexa-labeled L-PGDS.
Results: The recombinant L-PGDS and wild type (WT) L-PGDS increased ROS but only the WTL-PGDS increased IL6 and TNFα, suggesting that differences in glycosylation of L-PGDS in AD was responsible for this discrepancy.
Conclusions: These data collectively suggest that L-PGDS might play an important role in the development of dementia in patients on dialysis and of AD.
Keywords: Dialysis Dementia, Alzheimer’s disease, Apoptosis, Reactive oxygen species, Inflammatory cytokines, Receptor-mediated endocytosis, Lipocalin-type prostaglandin D2 synthase
1. Background
Lipocalin-type prostaglandin (PG) D2 synthase (L-PGDS) is a member of a superfamily of lipocalins that have diverse physiological functions, including binding and transporting lipophilic compounds, induction of sleep, regulation of body temperature, inhibition of platelet aggregation, vasodilatation, modulation of pain, convulsions, hormone release, immunomodulation, inflammation and apoptosis (1-5). L-PGDS, also known as β trace protein, makes up ~3% of proteins in the cerebrospinal fluid (CSF) and is the second most common protein in the CSF (6,7). It is produced by the leptomeninges, choroid plexus and parenchymal oligodendrites in brain and has been localized in various neuronal cells by immunohistochemical and mRNA determinations, including lysosomes in the cytosol, suggesting receptor-mediated endocytosis (8-10). While there is ample evidence for characterization of cell-surface receptor binding for some lipocalins, the specific receptor for all lipocalins has not been identified and as a result, there is limited delineation of the precise mechanisms for receptor binding, post receptor mediation of ligand endocytosis and biological translation (11). Part of this difficulty in the characterization of lipocalin receptors lies in the identification of only a limited number of specific receptors for lipocalins (11).
The enzymatic role of L-PGDS in the biosynthetic pathway of prostanoid metabolism has been clearly established. In this pathway, cyclo-oxygenase (COX) 1 and 2 convert arachidonic acid to PGH2, which, in the presence of L-PGDS, converts to PGD2, the most common prostanoid in brain (8). PGD2 in turn converts to PGJ2 and then to 15 deoxy PGΔ12,14 J2 (15dPG J2), the primary ligand for peroxisome proliferator-activator receptor-gamma (PPARγ)(12). Fifteen dPGJ2 has been reported to induce apoptosis in human astrocytes and cortical neurons (13,14). We previously demonstrated the induction of apoptosis by L-PGDS in PC12, LLC-PK1 and rat mesangial cells that was inhibited by L-PGDS inhibitors, growth factors such as insulin growth factor, insulin and erythropoietin in addition to PGE1, PGE2, and COX 2 and caspase inhibition (1,3,4). We also reported a fourfold increase in apoptotic activity in the plasma of patients with AD as compared to plasma from patients with multi-infarct dementia and normal age and gender-matched controls (15).
2. Objectives
In the present report, we present data on the isolation and identification of L-PGDS as a dominant inducer of apoptosis in AD plasma. We provide evidence for a receptor-mediated endocytosis of L-PGDS, which induces apoptosis and generates reactive oxygen species (ROS) and inflammatory cytokines (IC) in PC12 neuronal cells.
3. Materials and Methods
Purification Procedure
Urine Processing
We reasoned that the apoptotic factor we described in AD plasma was a small protein that would be filtered and excreted in urine. Detection of the apoptotic factor in urine would provide a virtual unlimited supply of the factor after being immeasurably purified by the kidneys and thus facilitate identification. Urine was, therefore, collected separately from two patients with AD whose diagnoses were established by previously described criteria (15). Protein from 3-5 L of urine was precipitated with ammonium sulfate, 80% (w/v). The precipitate was pelleted at 13,000 x g for 35 min at 4°C. Protein in the pelleted fraction was resuspended in 1/35 volume of 25 mM Tris-HCl buffer [pH 7.4] and dialyzed in a 10 kDa M.W. cutoff dialysis membrane (Spectrum, Los Angeles, CA) against 3 x 4 L of the same buffer for 24 h at 4oC. Insoluble material was removed by centrifugation as above and the supernatant filtered through a #1 Whatman filter paper. The filtered dialysate was positive for apoptotic activity in cultured LLC-PK1 cells as determined by terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end-labeling (TUNEL) assay using the ApopDetek Cell Death Assay Kit (Enzo, Farmingdale, NY) as previously described (15,16).
Ion Exchange Chromatography
A 57 x 2.5 cm column was packed with Q Sepharose Fast Flow media (Pharmacia, Uppsala, Sweden) and equilibrated with 3 column volumes of 25 mM Tris-HCl buffer [pH 7.4] at 4o C. The filtered dialysate was loaded onto the column and washed with column buffer until absorbance returned to near background. The column was then developed, using step elution with an increasing salt concentration of 100, 200, 300, 400, 500 mM and 2.5 M NaCl gradient in Tris-HCl buffer, pH 7.4, at 8 ml/min until absorbance returned to near background each time. Apoptotic activity was detected primarily in the 100 mM NaCl eluate. Some activity was also noted in the 400 mM eluate. The 100 mM NaCl eluate was concentrated forty-fold in a 1 kDa M.W. cutoff dialysis membrane against solid PEG 8000 (Sigma, St. Louis, MO). The concentrate was then transferred to a 10 kDa M.W. cutoff dialysis membrane and dialyzed overnight at 4o C against 4 L of 10 mM sodium phosphate buffer [pH 7.1].
High Performance Liquid Chromatography (HPLC) and SDS polyacrylamide gel electrophoresis-Identification of apoptotic factor
Three to 6 ml of the concentrated 100 mM NaCl eluate was subjected to HPLC chromatography on a 4.6 cm x 250 mm Rainin anion exchange column (Hydropore 5-SAX; Woburn, MA). Bound material was eluted by use of a linear gradient from 0 to 200 mM NaCl in 10 mM sodium phosphate buffer, pH 7.1, over 60 min at a flow rate of 0.5 ml/min, collecting 1 ml fractions. The protein eluted as a single broad peak in 4 fractions, each of which had apoptotic activity by the TUNEL assay. An aliquot of the peak HPLC fraction at 22 min was subjected to electrophoresis on a 15% SDS polyacrylamide gel, transferred to a PVDF membrane, and to N-terminal amino acid sequence analysis at the Laboratory for Macromolecular Analysis, Albert Einstein College of Medicine, Bronx, NY on a Perkin Elmer-ABI analyzer (Model Procise). A subsequent Blast P search produced a 12/12 match for beta trace protein-glutathione independent L-PGDS with a score of 28.2 bits and expect value of 1.9 (17).
Contribution of L-PGDS to Apoptosis in AD Plasma
In order to determine the contribution of L-PGDS to the apoptotic activity that we described previously in AD plasma, we determined apoptotic activity in the plasma of 18 AD subjects with a clinical diagnosis of AD by methods previously described in LLC-PK1 cells, kindly supplied by Dr. Julia Lever, Houston, TX (2). LLC-PK1 cells were selected because we had previously reported the apoptotic activity in AD plasma in this cell line. We incubated LLC-PK1 cells with 60 ul AD plasma or 50 ug/ml recombinant (r)L-PGDS as previously described (3,15) for 2 h with and without 10 mg/ml of the COX2-specific inhibitor, NS-398 (RBI, Natick, MA), dissolved to 10µM in DMSO (18). In other experiments, 40 ug of affinity purified antibody to L-PGDS, kindly provided by D.C. Chen (Protein Chemistry Core Laboratory, Rockefeller University, New York, NY USA) was incubated for 2 h with and without 60 ul AD plasma or 50 ugrL-PGDS and apoptosis determined by the TUNEL assay (15). L-PGDS levels in plasma were determined by an antibody sandwich ELISA as described by Oda et al with modifications (19). In these assays, the secondary polyclonal antibody was raised in chicken and the horseradish peroxidase-labeled goat anti-chicken IgG antibody was obtained from Aves Labs, Tigard, OR.
Cell Culture
PC12 cells, obtained from ATCC (Manassas, VA), were grown in RPMI 1640 medium (Life Technologies, Gaithersburg, MD) that was supplemented with 10 % horse serum, 5% fetal calf serum, 25 U/ml penicillin, and 25 ug/ml streptomycin in a 5% CO2 atmosphere at 37°C. Cells were fed three times a week by replacing two thirds of the medium each time. The cells were plated onto Permanox plastic chamber plates that had been previously coated with 5ug/cm2 for 1 h rat tail collagen in 0.02N acetic acid to promote neuritic outgrowth. Cells were fed three times a week by replacing two thirds of the medium each time. A unicellular suspension was created by passing the cells through a 21g needle and cells plated at 105 cells/well. The following morning, RPMI that was supplemented with 1% horse serum and 50 ng/ml mouse nerve growth factor (Boehringer Mannheim Biochemicals) was substituted for the medium. The cells were used 4-5 days after plating when there was neuritic outgrowth.
Uptake of 125I rL-PGDS
Time and Temperature-Dependent Uptake Studies
All uptake studies utilized purified rL-PGDS that was labeled with 125I (Amersham, Piscataway, NJ). One hundred ug of purified rL-PGDS was dissolved in 200 ul PBS, Ph 7.4, and iodinated with 1 mCi125I by the Iodo bead method, using 3.175 mm diameter size beads (Pierce Chemical, Rockford, IL). After 15 min incubation, the reaction mixture was separated from the beads and dialyzed overnight by using a Sephadex G-25 column to separate the iodinated rL-PGDS from free 125I. This iodination procedure yielded ~30 Ci/mmolrL-PGDS. All timed uptake studies were performed 4-5 days after plating PC12 cells in six-chamber plates, by adding 0.5 ml RPMI medium containing sufficient 125I L-PGDS and 2.5 ug cold L-PGDS at room temperature. The reaction was stopped by adding 1 ml ice cold saline at 3, 5, 10, 15, 30 and 60 seconds and at 3, 5 10, 15, 30, 60 and 120 minutes. Because of the tendency of the cells to detach from the plate surface, the cells were detached from the plate surface to create a cell suspension, transferred to microcentrifuge tubes and centrifuged at 1500 RPM for 4 min in a microcentrifuge. The supernatant was then decanted and the cells gently resuspended by pipeting repeatedly in 1 ml ice cold saline. After a total of three saline washes, the cells were suspended in 1 ml of 1N NaOH and incubated for 30 min at 37°C. The NaOH was transferred to counting vials and 10 ul removed for protein analysis by bicinchoninic acid (20). One ml NaOH was added to rinse the microcentrifuge tube and added to the counting vial. To test the effect of temperature on uptake of L-PGDS similar studies were performed with 15 min incubations at 4°C, room temperature and 37°C. Radioactivity was measured in a LKB Wallace 1282 compugamma CS universal counter.
Dose-Dependent Uptake Studies
Under the same conditions as the time-dependent studies, the RPMI medium containing 125I-labeled recombinant L-PGDS (rL-PGDS) alone and with progressively increasing concentrations of cold rL-PGDS, ranging from 100, 500, 1000 to 2000 uM, were incubated for 15 min at 4°C. The cells were washed 3 times with ice cold saline and then 1N NaOH as above.
Determination of Membrane-Bound and Cytosolic L-PGDS
In order to separate membrane-bound from cytosolic rL-PGDS, PC12 cells were incubated for 15 min with the same 125I rL-PGDS and 2.5 ug unlabeled rL-PGDS in RPMI medium at 37°C and washed 3 times with ice cold saline as above. The cells were then suspended in 1 ml of an acid wash solution (50 mM glycine and 100 mM NaCl, pH 5.0) for 5 minutes to remove the membrane-bound fraction of 125I rL-PGDS. After gentle mixing, the cells were centrifuged and the supernatant transferred to a counting vial. This process was repeated and the supernatant was added to the same counting vial for determination of membrane-bound rL-PGDS. The cells were then treated with 1N NaOH for determination of protein and cytosolic rL-PGDS as above.
Competition Studies
Competition of uptake of rL-PGDS was performed by incubating 125I rL-PGDS alone and with increasing concentrations of unlabeled albumin or unlabeled rL-PGDS at concentrations of 100, 250, 500, 1000 and 2000 uM or 100, 500, 1000 and 2000 uM, respectively, for 15 minutes at 4°C. The cells were then washed with ice cold saline followed by1N NaOH for determination of protein and radioactivity as above.
Electron microscopy of gold-labeled rL-PGDS
Tissue was fixed in 2% paraformaldehyde, 0.1% glutaraldehyde, 0.15 M sodium cacodylate buffer, pH 7.4, dehydrated in a graded series of ethanol, infiltrated in LR-White resin (London Resin Co. Ltd., Hampshire, England), and polymerized with UV irradiation at 282 nm. Thin sections were picked up on nickel formvar coated grids, blocked in 0.01M glycine, 1% nonfat dry milk, 0.5% gelatin in 0.1M phosphate buffer, pH 7.4, exposed to a 1:100 dilution of a rabbit polyclonal antibody against L-PGD2S, and finally exposed to 10nm gold labeled anti-rabbit antibody( EMS, Ft. Washington, PA) diluted 1:20 in phosphate buffered saline, pH 7.4. Sections were then fixed with 2% glutaraldehyde, stained with 2% uranyl acetate, and scoped on a Zeiss EM10 transmission electron microscope.
Fluorescent labeling of L-PGD2S for determination of endocytosis
An Alexa Fluor 488 protein labeling kit, A-10235 (Molecular Probes, Eugene, OR) was used to label rL-PGDS for immunohistochemistry studies as per instructions by the manufacturer. PC12 cells were plated on glass slides and cultured as described above, fixed with cold methanol and incubated with 50 ug/mL L-PGDS for 2 h. The slide was rinsed with PBS, blocked with 10% goat serum, and incubated with purified primary L-PGDS antibody (polyclonal rabbit, 5 ug/ml) at room temperature for 60 min. The slide was then washed with PBS and re-incubated with secondary antibody (anti rabbit IgG-FITC, 10 ug/ml), (Santa Cruz Biotechnology, Santa Cruz, CA). The cells were mounted with water-soluble mounting media and examined by fluorescent microscopy, using a FITC wavelength filter. (Nikon Eclipes E400, Columbia, MD, USA).
Analysis for Reactive Oxygen Species
PC12 cells were suspended in HANK’s buffer and loaded with 5 uM DCF-DA (5(and-6)-chloromethyl 12’7’ dichlorodihydrofluorescein) (Molecular Probes, Eugene, OR) for 15 min at 37°C. The cells were then washed 3 times with HANK’s buffer, resuspended at 106 cells/ml, incubated with 50 ugrL-PGDS or WTL-PGDS, and 10 mg/ml albumin for 2h at 37°C and read on a PTI fluorometer (Photon Technology International, Canada) at 498 nm excitation and 522 nm emission while the cells were maintained in suspension with a stirring pellet in the cuvette.
Analysis for Inflammatory Cytokines
Panomics Cytokine Array 1.0 (Panomics, Redwood City, CA) was used for these experiments. Briefly, 2 ml of conditioned medium was added to the membrane arrays after a 1h protein blocking step. The array was then washed and a biotin-conjugated anti-cytokine mix added for 2 h. The membrane was washed again and reacted with Streptavidin-HRP for 1h and the signal detected with ECL. The membrane was then exposed to Xray film. Densitometry on the spots was determined with SigmaGel. IL-6, IL-8, MCP-1, TGF-β1, TNF-α and IL-1 were measured in the supernatants using commercial human or porcine ELISA kits (Quantikine: R&D Systems, MN, USA) according to the manufacturer’s protocol. The sensitivity of the ELISA is less than 0.7 pg/ml, 4.4 pg/ml and 1 pg/ml for IL-6, IL-8, TNF-α and IL-1 assays, respectively. Cytokine concentration in the unknown samples were determined by interpolation into a standard curve developed with known amounts of recombinant human cytokines., Experiments were conducted in triplicate using 96-well micro plates and results were read in a micro plate reader. Cells were trypsinized and counted to express the amount of cytokine as pg/ml medium or 106 cells.
Statistical analysis
All data are expressed as the mean ± SEM of at least 3 experiments performed in duplicates. Statistical analysis of the data was done by unpaired Student’s t test except for IL-6 and TNFα, which were analyzed by the GraphPad Prism, version 4 for Macintosh, using the Newman-Keuls multiple comparison test. Statistical significance was defined as p<0.05.
4. Results
Isolation and identification of apoptotic factor
The demonstration of apoptotic activity in ammonium precipitated proteins from the Alzheimer urine greatly simplified identification of an apoptotic factor in Alzheimer plasma. An aliquot of the four fractions from the broad HPLC peak with apoptotic activity demonstrated a single broad band at ~29 kD when subjected to electrophoresis on a 15% SDS-PAGE gel, figures 1 and 2. As noted above, the single band was identified as beta trace protein-lipocalin-type L-PGDS with a score of 28.2 bits and expected value of 1.9. (17) The protein was over-expressed and purified from Escherichia coli and a western analysis of the WTL-PGDS and the rL-PGDS revealed the WT L-PGDS to be more heavily glycosylated by migrating to ~29 kD as compared to ~20 kDa for the rL-PGDS, figure 2. (3,4) These results are identical to the results previously reported from this laboratory (4).
Figure 2.
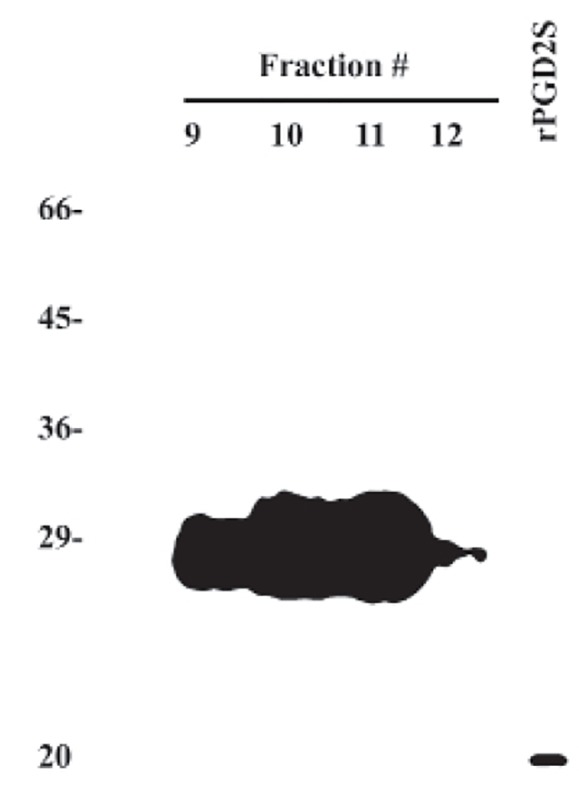
Figure 1.

Estimation of L-PGDS contribution to Apoptotic Activity in AD Plasma
As noted in figure 3a , incubation of plasma from 18 subjects with AD had a mean apoptotic rate of 35.1 ± 3.1% which was significantly higher than the control of 9.4 ± 0.43 %, p<0.001 and reduced to means of 14.3 ± 0.30%, p< 0.001, by NS 398, and 12.1 ± 0.27%, p< 0.001, by affinity-purified antibodies to L-PGDS as compared to control and AD plasma. The failure of both inhibitors to reduce mean apoptotic rates to control values, p<0.001, suggests the presence of an apoptotic factor(s) other than L-PGDS in AD plasma. On the other hand, while rL-PGDS increased mean apoptosis from a control mean of 7.2 ± 0.22% to 22.9 ± 0.35%, p <0.001, the incubation of NS-398 and antibody to L-PGDS with rL-PGDS inhibited apoptosis to mean apoptotic rates of 5.5 ± 0.1% and 9.5 ± 1.3%, respectively. Apoptosis after inhibition by NS398 was lower than control, p<0.002, whereas inhibition by the antibody was higher than control, p<0.001, figure 3b . Despite these differences in apoptotic activity between control and AD plasma, however, the mean plasma levels of L-PGDS levels of 190.2 ± 67.9ng/mL in 18 controls subjects was not statistically different from the mean of 185.6 ± 45.4 ng/mL in a total of 31 AD subjects, suggesting that the larger, glycosylated isoforms of L-PGDS in AD plasma was apoptotically more active than the rL-PGDS are present in AD plasma. Non-aqueous solvents used as vehicles did not inhibit or exhibit apoptotic activity.
Figure 3a.

Figure 3b.

Time and Dose-Dependent Uptake of L-PGDS
As noted in figure 4, there was a rapid uptake of rL-PGDS, reaching its peak in 3 seconds and remaining at this level for 120 minutes. The same peak levels were noted at 18 h after exposure, data not shown. There was also a dose-dependent uptake of rL-PGDS, uptake increasing progressively as rL-PGDS dose increased from 100 to 2000uM with saturation occurring at 1000 uM at 4°C, figure 5. Kinetic analyses suggest auMKd that is consistent with a low affinity, high capacity receptor.
Figure 4.

Figure 5.

Uptake of L-PGDS at 37°C for 15 min was largely in the cytosol as compared to being membrane bound, figure 6a , and was temperature-dependent as uptake progressively increased as incubation temperatures were increased from 4° to 22° to 37°C, figure 6b.
figure 6a.

figure 6b.

Competition Studies
Competition studies performed with increasing concentrations of unlabeled albumin and rL-PGDS demonstrated a progressive decrease in 125I rL-PGDS uptake from 100 uM to a nadir at 1000 uM for unlabeled L-PGDS and 2000uM for unlabeled albumin, figure 7.
figure 7.
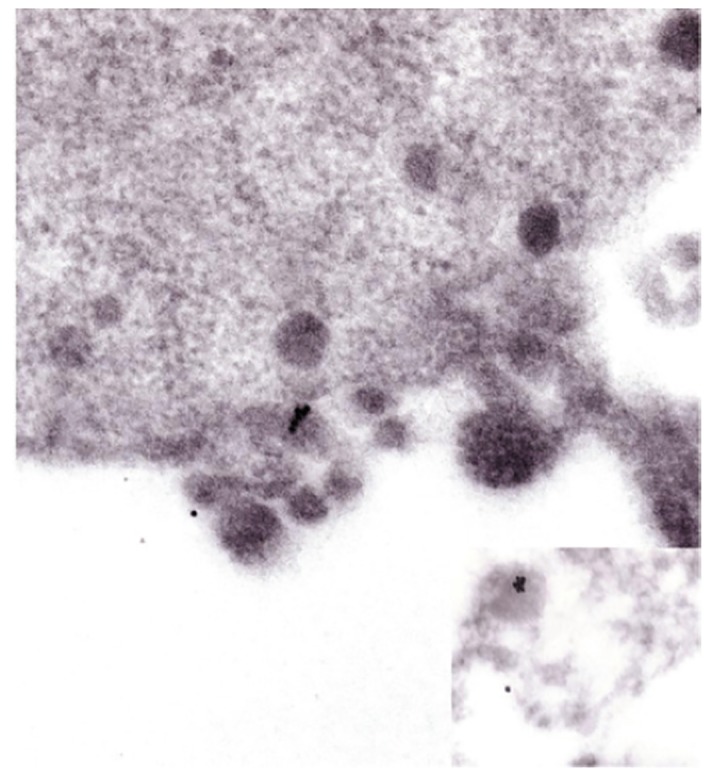
Endocytosis of PGDS by electronmicroscopy and immunofluorescence.
In order to demonstrate further the endocytosis of PGDS, we performed electron microscopic studies of gold-labeled PGDS and uptake of fluorescent-labeled PGDS. On electronmicroscopy rL-PGDS was localized on the membrane surface and within lysosomes 7a and 7b of PC12 cells, figure 7c and 7d . Endocytosis of PGDS was further demonstrated by fluorescent-labeling of rPGDS, figures.
Induction of reactive oxygen species by L-PGDS
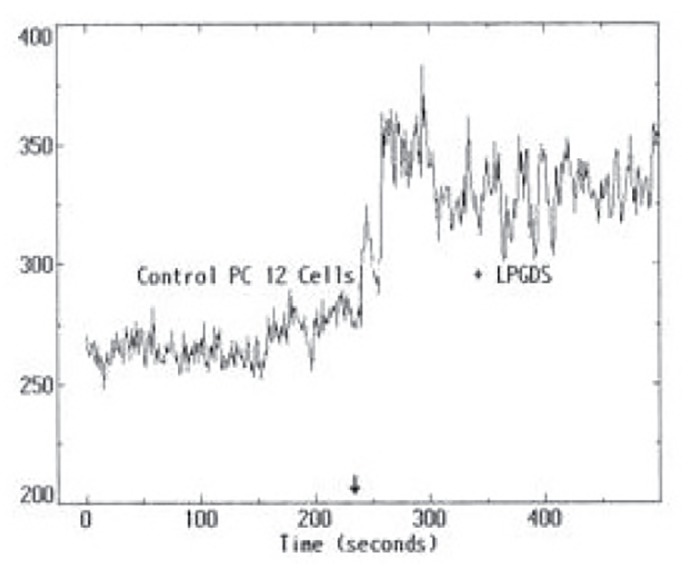
The induction of ROS by 50 ugrL-PGDS or WTL-PGDS and 10 mg/mL albumin was evaluated after a 2 h exposure to either form of L-PGDS or albumin using DCF-DA fluorescent probe for ROS production. Both therL-PGDS, figure 9, WTL-PGDS and albumin increased ROS production after 2 h, data not shown.
figure 9.
Induction of inflammatory cytokines by L-PGDS
The generation of IC by proteins such as albumin and light chains prompted the present investigations into the generation of IC by rL-PGDS (1,3,21). After an overnight exposure of PC12 cells to 50 ugrL-PGDS or WT L-PGDS, we demonstrated an increase in IL6 and TNFα by the WT L-PGDS, p<0.01, but not rL-PGDS, p>0.05, figures 10a and 10b. Albumin increased IL-6, p<0.05, but not TNFα production, p>0.05. IL8 and MCP1 did not increase over baseline by either source of L-PGDS.
figure 10a.

figure 10b.

5. Discussion
The present studies provide data on the isolation and identification of L-PGDS as the predominant apoptotic factor in AD plasma, which we reported to be fourfold greater in AD plasma than in plasma from normal age and gender-matched controls and from multi-infarct dementia (15). This conclusion is supported by the significant inhibition of the apoptotic activity noted in AD plasma by COX-2 inhibition and the antibody to L-PGDS. Since the COX-2 inhibitor and antibody to L-PGDS did not reduce apoptosis to control levels when incubated with AD plasma, however, apoptotic factor(s) other than L-PGDS appears to exist in AD plasma.
We previously demonstrated induction of apoptosis by L-PGDS in PC12 neuronal cells, LLC-PK1 and rat glomerular mesangial cells that appears to involve the prostaglandin pathway, including arachidonic acid conversion to PGH2 by COX-1 and 2, the conversion of PGH2 to PGD2 by L-PGDS, eventual conversion of PGD2 to 15dPGJ2, a major ligand for the transcription factor, PPARγ, and apoptosis. We have shown that apoptosis induced by L-PGDS can be inhibited by COX-1 and/or 2 inhibition, inhibition of L-PGDS by selenium or its specific antibody, and by erythropoietin, PGE1, PGE2, and PGF2α, and by growth factors such as insulin, insulin-like growth factor and platelet derived growth factor (1,3,4). Moreover, our recent studies suggest that phorbol ester-induced apoptosis is mediated by PGDS phosphorylation and activation of calcium-dependent protein kinase C, and is accompanied by an inhibition of phosphatidylinositol 3-kinase/protein kinase B signaling pathways (22). The role of apoptosis in the pathogenesis of AD remains to be resolved. Several studies have supported an apoptotic or apoptotic-like mechanism in AD and other neurodegenerative diseases, although others do not support such a mechanism (23-25). The general consensus, however, does not seem to view apoptosis as a major contributor to the development of AD.
In the present study, we demonstrate internalization of L-PGDS in PC12 neuronal cells by a pathway consistent with receptor-mediated endocytosis. The uptake of L-PGDS peaks rapidly within 3 seconds after exposure of PC12 cells to the ligand and remains at this level after 18 hours. The uptake is dose-dependent, is reduced at 4°C and is predominantly within the cytosol as compared to the membrane at 37°C. Competition of cellular uptake between radiolabeled L-PGDS and unlabeled L-PGDS or albumin reveals saturability of receptor sites that is consistent with other lipocalin receptors and demonstrates low affinity, high capacity characteristics (11). Immunofluorescence studies of Alexa-labeled L-PGDS further confirm endocytosis of the ligand and electron microscopic analysis of gold-labeled L-PGDS demonstrates the ligand to be membrane-bound and within lysosomes, suggesting a receptor-mediated endocytosis of the ligand. The present studies, however, do not identify the receptor to L-PGDS in PC12 cells. Specific receptors have been identified for lipocalins such as for retinol binding protein by megalin, α-1-microglobulin, β-lactoglobulin, odorant binding protein and lipocalin-1 where binding characteristics have been defined more precisely (11,26,27). Receptors can be characterized as being general for a number of ligands or specific to one particular lipocalin but even of the known lipocalin receptors, there is still discrepancy over molecular weights, binding characteristics or binding affinities (11). There is, moreover, awareness that the functional diversity of the lipocalins and failure to identify and characterize the molecular basis of the receptor(s) responsible for binding and endocytosis create an exciting conundrum for future investigations to resolve.
The induction of ROS and IC by overloading of proteins such as albumin or light chains has been the subject of great interest (28-30). Their contribution to the progression of organ failure, including AD, has gained increasing support and interest (31,32). In the present study, we demonstrate the induction of ROS by immunofluorescence studies that reflect a generalized increase in ROS but with an emphasis on H2O2 (33). H2O2 induces a concentration-dependent increase in phosphorylation of extracellular signal-regulated kinases 1 and 2 (ERK1/2) and the kinase Akt/PKB or c-jun N-terminal kinase (JNK), which were partially or totally dependent on extracellular calcium, respectively, and on Phosphatidylinositol 3-kinase PI3-kinase (34). The induction of apoptosis by L-PGDS appears to occur through an induction of ROS and not via Peroxisome Proliferator-Activated Receptorγ, PPARγ, the downstream product of prostaglandin D2 pathway. H2O2 could also lead to the generation of hydroxyl radicals via the Fenton reaction, which may have important consequences that involve DNA disruption, the release of intracellular calcium, induction of apoptosis and cell death. It can also initiate lipid peroxidation of arachidonic acid with the release of AD-related isoprostanes. (35,36) The potential role of L-PGDS in the generation of isoprostanes through lipid peroxidation of arachidonic acid via an ROS mechanism needs to be tested. On the other hand, nitric oxide might lead to the generation of the highly toxic ONOO- in the setting of a reduction in the ONOO- scavenger, uric acid, in AD where hypouricemia due to a defect in renal tubular transport of uric acid has been reported from our laboratory (37). Because of the instability of ROS in autopsied tissues, measurements of ROS have been limited in autopsied brains of AD subjects. In brain homogenates from autopsied samples, however, there has been de novo generation of ROS that was further increased by the addition of ferrous sulfate (38). The net result of ROS damage leading to neuronal cell death, however, must be balanced by an antioxidant system that protects against these toxic reactive species, specifically catalase, superoxide dismutase and glutathione peroxidase. Although there is some inconsistency in the levels of these antioxidants reported in AD brain, reduction in the levels of all three antioxidants has been reported in key areas of AD brain (21). It would appear that the increased generation of ROS and reduction in endogenous antioxidants provide an environment that favors neuronal damage.
An extension of the induction of ROS by L-PGDS is the activation of transcription factors that lead to IC. ROS such as H2O2 and NO are known activators of the transcription factor, NF-κB, and production of IC (39). The increase in IL6 and TNFα noted in the present study after exposure to L-PGDS is consistent with this pathway for IC production. An unexpected outcome was the failure of the rL-PGDS to induce these IC as compared to WTL-PGDS. This difference in activity might result from the formation of glycosylated isoforms of L-PGDS that was distinguishable from the rL-PGDS on SDS PAGE and western analysis, figure 2. There was a fourfold increase in apoptotic rates and induction of IC by the WTL-PGDS as compared to equal doses of rL-PGDS. It would appear that the glycosylated isoforms of L-PGDS are more toxic than the recombinant form of L-PGDS.
L-PGDS is a secreted, membrane-associated protein that is the second most abundant protein (~3%) in CSF (6,7). It is primarily synthesized in epithelial cells of the choroid plexus, oligodendrocytes in white matter, leptomeninges and male gonads (9,10). L-PGDS levels increase progressively as renal function decreases and have been shown to be a better marker of glomerular filtration rate than creatinine (40). Blood levels of L-PGDS are also increased 35 to 150 fold over normal values in dialysis patients, increase geometrically like creatinine with progressive kidney disease and are elevated in blood and urine of hypertensive patients (41,42). CSF and blood levels of L-PGDS are normal in neurological diseases such as multiple sclerosis (6), while low CSF levels have been associated with brain tumor (43). L-PGDS has been implicated in various functions, including the induction of sleep, regulation of body temperature, convulsions, inhibition of platelet aggregation, vasodilatation, modulation of pain, hormone release and allergic and inflammatory response (2,5). Because of elevated blood levels in renal failure and dialysis patients, L-PGDS has been implicated as a contributor to progression of renal failure and dialysis dementia (44).
The upregulation of COX-2 in AD brain and the observation that PGD2 is the most common prostaglandin in brain suggest that L-PGDS diverts the prostaglandin pathway to PGD2 rather than to other metabolites such as PGI2, PGE2, thromboxane A2 and PGF2α (8,45,46). The expected generation of downstream products of PGD2 such as the activation of PPARγ by 15dPGJ2, production of ROS and IC by as yet unknown mechanisms and the role of receptor-mediated endocytosis in rendering L-PGDS cytotoxic require further investigations.
Dialysis dementia is a condition that is characterized by progressive dementia, psychoses, speech disturbances, multifocal seizures and death within 6 months after onset of symptoms in patients who had been on hemodialysis for more than 2 years (47). This form of dementia was attributed to contamination of dialysate with aluminum in the water supply or by intestinal absorption of aluminum by aluminum-containing phosphate binders that were used to control the putative effects of elevated plasma phosphate in these patients (47,48). Support for such a proposal was the dramatic reduction in this form of dementia after eliminating aluminum from the water supply and as phosphate binders (48). Interest in dialysis dementia waned after elimination of aluminum exposure of dialysis patients but there has not been a systematic study to determine the prevalence and extent of dialysis dementia. Although, dialysis dementia probably represents the final pathway for multiple factors that contribute to this condition, it is tempting to invoke the role of L-PGDS in this unresolved problem. Other potentially reversible causes such as drugs, electrolyte disturbances, infections, trace elements, vitamin deficiency, or intracranial diseases such as infections, bleeding or tumor should be considered.
Blood levels of L-PGDS have been reported to be 35 to 150 fold higher in patients on dialysis as compared to normal controls (42). With a molecular weight approximating 29 kDa, L-PGDS can be considered a middle molecule that would not readily pass through the dialysis membrane and thus explain higher blood levels noted in dialysis patients. The wild type L-PGDS has been shown to have greater apoptotic activity than recombinant L-PDS with a molecular weight of about 20 kDa, suggesting that glycosylation of L-PGDS provides not only greater apoptotic activity but greater induction of IC as demonstrated in this study.
It would be interesting to note that interest in dialysis dementia might have waned because dementia may have decreased significantly after the introduction of erythropoietin into the therapy of anemia for the majority of patients on dialysis. It has been well documented that there is a rapid improvement in cognitive function after administration of erythropoietin, suggesting an improvement in neuronal endplate function (49) . Other data suggest that cellular protective effects of erythropoietin that may have attenuated or eliminated neuronal cell death in these patients. Erythropoietin has been shown to reduce brain shrinkage when administered prior to brain damage by blunt trauma or ischemia (50,51). Darbepoetin was also shown to inhibit apoptosis induced by L-PGDS, anoxia and camptothecin in LLC-PK1, pig proximal tubule, and mouse glomerular mesangial cells, but did not inhibit apoptosis induced by H2O2 (1). The present studies suggest that the apoptosis induced by L-PGDS is largely due to generation of H2O2 and not via the PPARγ pathway. The inhibition of apoptosis induced by erythropoietin, therefore, appears to be an upstream effect of L-PGDS, before production of H2O2.
6. Conclusion
Based on these data, it would appear that dialysis dementia might have decreased significantly after the advent of erythropoietin. Erythropoietin inhibited the toxic effects of markedly increased levels of L-PGDS in dialysis plasma, inhibiting apoptosis by reducing H2O2 production, possibly attenuating the putative effects of IC and entering into other protective cellular mechanisms.
Authors’ contributions
JKM, BS and TP prepared the primary draft. LR, VB, NM,SS,SY and MES provided extensive intellectual contribution. JKM prepared the final manuscript.
Acknowledgment
This study was in part supported by the Winthrop-University Hospital intramural fund for biomedical research.
Conflict of Interest
There are no conflicts of interest to report for any of the authors.
Implication for health policy/practice/research/medical education:
Lipocalin-type prostaglandin D2 synthase might play an important role in the development of dementia in patients on dialysis and of Alzheimer’s disease.
Please cite this paper as:Maesaka JK, Sodam B, Palaia T, Ragolia L, Batuman V, Miyawaki N, Shastry S, Youmans S, El-Saban M. Prostaglandin D2 synthase: Apoptotic factor in alzheimer plasma, inducer of reactive oxygen species, inflammatory cytokines and dialysis dementia. J Nephropathology. 2013; 2(3): 166-180. DOI: 10.5812/nephropathol.10732
References
- 1.Fishbane S, Ragolia L, Palaia T, Johnson B, Elzein H, Maesaka JK. Cytoprotection by Darbepoetin Alfa in Renal Tubular and Mesangial Cells. Kidney Int. 2004;65:452–458. doi: 10.1111/j.1523-1755.2004.00400.x. [DOI] [PubMed] [Google Scholar]
- 2.Hayaishi O. Prostaglandin D synthase beta-trace and sleep. Adv Exp Med Biol. 1997;433:347–350. doi: 10.1007/978-1-4899-1810-9_74. [DOI] [PubMed] [Google Scholar]
- 3.Maesaka JK, Palaia T, Frese L, Fishbane S, Ragolia L. Prostaglandin D2 synthase induces apoptosis in pig kidney LLC-PK1 cells. Kidney Int. 2001;60:1692–1698. doi: 10.1046/j.1523-1755.2001.00989.x. [DOI] [PubMed] [Google Scholar]
- 4.Ragolia L, Palaia T, Frese L, Fishbane S, Maesaka JK. Prostaglandin D2 synthase induces apoptosis in PC12 neuronal cells. Neuro Report. 2001;12:2623–2628. doi: 10.1097/00001756-200108280-00008. [DOI] [PubMed] [Google Scholar]
- 5.Urade Urade, Y Y, Hayaishi O. Prostaglandin D synthase: Structure and function. Vit Horm. 2000;58:89–120. doi: 10.1016/s0083-6729(00)58022-4. [DOI] [PubMed] [Google Scholar]
- 6.Melegos DN, Freedman MS, Diamandis EP. Prostaglandin D synthase concentration in cerebrospinal fluid and serum of patients with neurological disorders. Prostaglandins. 1997;54:463–474. doi: 10.1016/s0090-6980(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 7.Zahn M, Mader M, Schmidt B, Bollensen E, Feigenhauer K. Purification and N-terminal sequence of beta-trace, a protein abundant in human cerebrospinal fluid. Neurosci Lett. 1993;154:93–95. doi: 10.1016/0304-3940(93)90179-o. [DOI] [PubMed] [Google Scholar]
- 8.Beuckmann CT, Lazarus M, Gerashchenko D, Mizoguchi A, Nomura S, Mohri I, Uesugi A, Kaneko T, Mizuno N, Hayashi O, Urade Y. Cellular localization of lipocalin-type prostaglandin D synthase (β trace) in the central nervous system of the adult rat. J Comp Neurol. 2000;428:62–78. doi: 10.1002/1096-9861(20001204)428:1<62::aid-cne6>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 9.Blodorn G, Mader M, Urade Y, Hayaishi O, Felgenhauer K, Bruck W. Choroid plexus, the major site of mRNA expression for the beta-trace protein (prostaglandin synthase) in human brain. Neurosci Lett. 1996;209:117–120. doi: 10.1016/0304-3940(96)12614-8. [DOI] [PubMed] [Google Scholar]
- 10.Urade Y, Kitahama K, Ohishi H, Kaneko T, Mizuno N, Hayaishi O. Dominant expression of mRNA for prostaglandin D synthase in leptomeninges, choroid plexus and oligodendrocytes of the adult rat brain. Proc Natl Acad Sci U.S.A. 1993;90:9070–9074. doi: 10.1073/pnas.90.19.9070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fowler DR. Beyond the superfamily: the lipocalin receptors. Biochim Biophys Acta. 2000;1472:327–336. doi: 10.1016/s0167-4838(00)00169-2. [DOI] [PubMed] [Google Scholar]
- 12.Fitzpatrick FA, Wynalda MA. Albumin-catalyzed metabolism of prostaglandin D2Identification of products formed in vitro. J Biol Chem. 1983;258:11713–11718. [PubMed] [Google Scholar]
- 13.Chattopadhay N, Singh DP, Heese O, Godbole MM, Sinohara T, Black PM, Brown EM. Expression of peroxisome proliferator-activated receptors (PPARS) in human astrocytes. J Neurosci Res. 2000;61:67–74. doi: 10.1002/1097-4547(20000701)61:1<67::AID-JNR8>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 14.Rohn TT, Wong SM, Cotman CW, Cribbs DH. 15-deoxy-delsta 12,14-prostaglandin J2, a specific ligand for peroxisome proliferators-activated receptor-gamma, induces neuronal apoptosis. Neuro Report. 2001;16:839–843. doi: 10.1097/00001756-200103260-00043. [DOI] [PubMed] [Google Scholar]
- 15.Maesaka JK, Palaia T, Chowdhury SA, Shimamura T, Fishbane S, Reichman W, Coyne A, O’Rear JJ, El-Sabban ME. Partial characterization of apoptotic plasma factor in Alzheimer’s disease. Am J Physiol. 1999;276:F521–F527. doi: 10.1152/ajprenal.1999.276.4.F521. [DOI] [PubMed] [Google Scholar]
- 16.Gavrieli Y, Sherman YY, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Bio. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST, a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Futaki N, Takahashi S, Yokoyama M, Arai I, Higuchi S, Otomo S. NS-398, a new anti-inflammatory agent, selectively inhibits prostaglandin G/H synthase/cyclooxygenase (COX-2) activity in vitro. Prostaglandins. 1994;47:55–59. doi: 10.1016/0090-6980(94)90074-4. [DOI] [PubMed] [Google Scholar]
- 19.Oda H, Shiina Y, Seiki K, Sato N, Eguchi N, Urade Y. Development and evaluation of a practical ELISA for human urinary lipocalin-type prostaglandin D synthase. Clin Chem. 2002;48:1375–1376. [PubMed] [Google Scholar]
- 20.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 21.Marcus DL, Thomas C, Rodriguez C, Simberkoff K, Tsai JS, Strafaci JA, Freedman ML. Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer’s disease. Exp Neurol. 1998;150:40–44. doi: 10.1006/exnr.1997.6750. [DOI] [PubMed] [Google Scholar]
- 22.Ragolia L, Palaia T, Paric E, Maesaka JK. Elevated prostaglandin D2 synthase activity contributes to PMA-induced apoptosis in LLC-PK1 cells concomitant with down-regulation of PI13-K. Am J Physiol Cell Physiol. 2003;284:C119–C126. doi: 10.1152/ajpcell.00247.2002. [DOI] [PubMed] [Google Scholar]
- 23.Dragunow M, MacGibbon GA, Lawlor P, Butterworth N, Connor B, Henderson C, Walton M, Woodgate A, Hughes P, Faull RL. Apoptosis, neurotrophic factors and neurodegeneration. Rev Neurosc. 1997;8(3-4):223–265. doi: 10.1515/revneuro.1997.8.3-4.223. [DOI] [PubMed] [Google Scholar]
- 24.Martin LJ, Al-Abdulla NA, Brambrink AM, Kirsch JR, Sieber FE, Portera-Cailliau C. Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation, A perspective on the contributions of apoptosis and necrosis. Brain Res Bull. 1998;46(4):281–309. doi: 10.1016/s0361-9230(98)00024-0. [DOI] [PubMed] [Google Scholar]
- 25.Nishimoto I, Okamoto T, Giambarella U, Iwatsubo T. Apoptosis in neurodegenerative diseases. Adv Pharmacol. 1997;41:337–68. doi: 10.1016/s1054-3589(08)61064-9. [DOI] [PubMed] [Google Scholar]
- 26.Christiansen EI, Moskaug JO, Vorum H, Gunderson TE, Nykjaer A, Blomhoff R, Willnow TE, Moestrup SK. Evidence for an essential role of megalin in transepithelial transport of retinol. J Am Soc Nephrol. 1999;10:685–695. doi: 10.1681/ASN.V104685. [DOI] [PubMed] [Google Scholar]
- 27.Wojnar P, Lechner M, Merschak P, Redl B. Molecular cloning of a novel lipocalin-1 interacting human cell membrane receptor using phage display. J Biol Chem. 2001;276:20206–20212. doi: 10.1074/jbc.M101762200. [DOI] [PubMed] [Google Scholar]
- 28.Abbate M, Zoja C, Corna D, Capitanio M, Bertani T, Remuzzi G. In progressive nephropathies, overload of tubular cells with filtered proteins translates glomerular permeability dysfunction into cellular signals of interstitial inflammation. J Am Soc Nephrol. 1998;9:1213–1224. doi: 10.1681/ASN.V971213. [DOI] [PubMed] [Google Scholar]
- 29.Batuman V, Guan V. Receptor-mediated endocytosis of immunoglobulin light chains by renal proximal tubule cells. Am J Physiol. 1997;272(Renal Physiol 41):F521–F530. doi: 10.1152/ajprenal.1997.272.4.F521. [DOI] [PubMed] [Google Scholar]
- 30.Tang S, Leung JCK, Abe K, Chan KW, Chan LYY, Chan TM, Lai KN. Albumin stimulates interleukin-8 expression in proximal tubular epithelial cells in vitro and in vivo. J Clin Invest. 2003;111:515–527. doi: 10.1172/JCI16079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Halliwell B, Gutteridge J. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J. 1984;219:1–14. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGeer PL, McGeer EG. Inflammation and the degenerative diseases of aging. Ann N Y AcadSci. 2004;1035:104–115. doi: 10.1196/annals.1332.007. [DOI] [PubMed] [Google Scholar]
- 33.Tarpey MM, Wink DA, Grisham MB. Methods for detection of reactive metabolites of oxygen and nitrogen: in vitro and in vivo considerations. Am J Physiol Regul Integr Comp Physiol. 2004;286:R431–R444. doi: 10.1152/ajpregu.00361.2003. [DOI] [PubMed] [Google Scholar]
- 34.Crossthwaite AJ, Sumaera H, Williams RJ. Hydrogen peroxide-mediated phosphorylation of ERK1/2, Akt/PKB and JNK in cortical neurons: dependence on Ca2+ and PI3-kinase. J Neurochem. 2002;80:24–35. doi: 10.1046/j.0022-3042.2001.00637.x. [DOI] [PubMed] [Google Scholar]
- 35.Pratico D, Zhukareva V, Yao Y, Uryu K, Fund CK, Lawson JA, Trojanowski JQ, Lee VMY. 12/15-Lipoxygenase is increased in Alzheimer’s disease. Am J Path. 2004;164:1655–1662. doi: 10.1016/S0002-9440(10)63724-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Volicer M. Free radicals in the development of Alzheimer’s diseaseNeurobiol. Aging. 1990;11:567–571. doi: 10.1016/0197-4580(90)90119-k. [DOI] [PubMed] [Google Scholar]
- 37.Maesaka JK, Wolf-Klein G, Piccione JM, Ma CM. Hypouricemia, abnormal renal tubular urate transport, and plasma natriuretic factor (s) in patients with Alzheimer’s disease . J Am Geriatri Soc. 1993;41:501–50. doi: 10.1111/j.1532-5415.1993.tb01885.x. [DOI] [PubMed] [Google Scholar]
- 38.Zhou Y, Richardson JS, Mombourquette MJ, Weill JA. Free radical formation in autopsy samples of Alzheimer and control cortex. Neurosci Letters. 1995;195:89–92. doi: 10.1016/0304-3940(94)11787-j. [DOI] [PubMed] [Google Scholar]
- 39.Rahman A, Kefer J, Bando M, Niles WD, Malik AB. E-selectin expression in human endothelial cells by TNF-alpha-induced oxidant generation and NF-κB activation. Am J Physiol. 1998;275:L533–L544. doi: 10.1152/ajplung.1998.275.3.L533. [DOI] [PubMed] [Google Scholar]
- 40.Priem F, Althaus H, Mirnbaum M, Sinha P, Conradt HS, Jung K. Beta-trace protein in serum: A new marker of glomerular filtration in the creatinine-blind range. Clin Chem. 1999;45:567–568. [PubMed] [Google Scholar]
- 41.Hirawa N, Uehara Y, Yamakado M, Toya Y, Gomi T, Ikeda T, Eguchi Y, Takagi M, Oda H, Seiki K, Urade Y, Umemura S. Lipocalin-type prostaglandin D synthase in essential hypertension. Hypertension. 2002;39:449–454. doi: 10.1161/hy0202.102835. [DOI] [PubMed] [Google Scholar]
- 42.Melegos DN, Grass L, Pierratos A, Diamandis EP. Highly elevated levels of prostaglandin D synthase in serum of patients with renal failure. Urology. 1999;53:32–37. doi: 10.1016/s0090-4295(98)00453-1. [DOI] [PubMed] [Google Scholar]
- 43.Saso L, M.G. Leone C, Sorrentino S, Giacomelli B, Silvestrini J, Grima J.C, Li E, Samy D. Quantification of prostaglandin D synthetase in cerebrospinal fluid, a potential marker for brain tumor. Biochem. Mol. Biol. Int. 1998;46:643–656. doi: 10.1080/15216549800204172. [DOI] [PubMed] [Google Scholar]
- 44.Maesaka JK, Palaia T, Fishbane S, Ragolia L. Contribution of prostaglandin D2 synthase to progression of renal failure and dialysis dementia. Sem Nephrol. 2002;22:407–414. doi: 10.1053/snep.2002.34726. [DOI] [PubMed] [Google Scholar]
- 45.Ho L, Pieronie C, Winger D, Purohit DP, Aisen PS, Pasinetti GM. Regional distribution of cyclooxygenase-2 in the hippocampal formation in Alzheimer’s disease. J Neurosci Res. 1999;57:295–303. doi: 10.1002/(SICI)1097-4547(19990801)57:3<295::AID-JNR1>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 46.Qin W, Ho L, Pampl PN, Peng Y, Zhao Z, Xiang Z, Robakis NK, Shioi J, Suh J, Pasinetti GM. Cyclooxygenase (COX)-2 and COX-1 potentiate beta-amyloid peptide generation through mechanisms that involve gamma secretase activity. J BiolChem. 2003;278(51):50970–50977. doi: 10.1074/jbc.M307699200. [DOI] [PubMed] [Google Scholar]
- 47. Fraser CL, Arieff AI: Metabolic encephalopathy as a complications of renal failure: Mechanisms and mediators. New Horizons 2:518-526, 1994. [PubMed]
- 48. Dunea G: Dialysis dementia: An epidemic that came and went. ASAIO J 47:192-194, 2001. [DOI] [PubMed]
- 49. Pickett JL, Theberge DC, Brown WS, et al: Normalizing hematocrit in dialysis patients improves brain function. Am J Kidney Dis 33:1122-1130, 1999. [DOI] [PubMed]
- 50. Brines ML, Ghezzi P, Keenal, et al: Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc Nat AcadSci 97:10526-10531, 2000. [DOI] [PMC free article] [PubMed]