

Abstract
High levels of chenodeoxycholic acid (CDCA) and deoxycholic acid stimulate Cl− secretion in mammalian colonic epithelia. While different second messengers have been implicated in this action, the specific signaling pathway has not been fully delineated. Using human colon carcinoma T84 cells, we elucidated this cascade assessing Cl− transport by measuring I− efflux and short-circuit current (Isc). CDCA (500 μM) rapidly increases I− efflux, and we confirmed by Isc that it elicits a larger response when added to the basolateral vs. apical surface. However, preincubation with cytokines increases the monolayer responsiveness to apical addition by 55%. Nystatin permeabilization studies demonstrate that CDCA stimulates an eletrogenic apical Cl− but not a basolateral K+ current. Furthermore, CDCA-induced Isc was inhibited (≥67%) by bumetanide, BaCl2, and the cystic fibrosis transmembrane conductance regulator (CFTR) inhibitor CFTRinh-172. CDCA-stimulated Isc was decreased 43% by the adenylate cyclase inhibitor MDL12330A and CDCA increases intracellular cAMP concentration. The protein kinase A inhibitor H89 and the microtubule disrupting agent nocodazole, respectively, cause 94 and 47% reductions in CDCA-stimulated Isc. Immunoprecipitation with CFTR antibodies, followed by sequential immunoblotting with Pan-phospho and CFTR antibodies, shows that CDCA increases CFTR phosphorylation by approximately twofold. The rapidity and side specificity of the response to CDCA imply a membrane-mediated process. While CDCA effects are not blocked by the muscarinic receptor antagonist atropine, T84 cells possess transcript and protein for the bile acid G protein-coupled receptor TGR5. These results demonstrate for the first time that CDCA activates CFTR via a cAMP-PKA pathway involving microtubules and imply that this occurs via a basolateral membrane receptor.
Keywords: CDCA, CFTR, T84 cells, cAMP signaling
in normal physiology, >95% of the bile acids secreted into the duodenum are efficiently reabsorbed in the terminal ileum. Less than 5% enters the colon where they are deconjugated and then dehydroxylated by bacteria and either reabsorbed or excreted (15). However, in the pathologies of diseases associated with ileal bile acid malabsorption, such as Crohn's disease, radiation ileitis, and ileal resection, there is an increase in colonic levels of bile acids. The resultant excess of primary and secondary bile acids in the colonic lumen leads to an increase in net water and electrolyte secretion causing diarrhea (28).
In a variety of colonic epithelia, such as mouse (14), rabbit (36), and human (29), including the human colon carcinoma cell line T84 (11, 20), we and others have shown that the endogenous primary and secondary dihydroxy bile acids chenodeoxycholic acid (CDCA), deoxycholic acid (DCA), respectively, and their taurine conjugates (TCDC and TDC) stimulate Cl− secretion. Depending on the species, both a direct action of the bile acids on epithelial cells and an indirect action involving other epithelial and subepithelial elements (12, 14, 18, 36, 37) have been demonstrated. It is well recognized that in colonocytes Cl− secretion occurs by the concerted effort of apical membrane Cl− channels, chiefly the cystic fibrosis transmembrane conductance regulator (CFTR) and basolateral transporters Na+-K+-ATPase, Na+-K+-2Cl− cotransporter (NKCC1), and K+ channels (3, 28, 35). However, the specific transport proteins and molecular mechanism(s) mediating the effects of bile acids on Cl− secretion are not fully understood. Thus, while the mechanism for bile acid-induced Cl− secretion has been linked to intracellular messengers such as cAMP and calcium (12), it has not been directly determined if bile acids indeed activate CFTR in the colon.
The stimulation of Cl− secretion in the colon by bile acids was shown to occur mainly by exposure to the serosal side of the colonic epithelia, suggesting the involvement of receptor(s) expressed on the basolateral membrane of colonocytes. However, the receptor underlying the secretagogue action of bile acids has not been implicated. Bile acids can act either via nuclear receptors, the most prominent being the farnesoid receptor FXR, and/or via membrane localized, heptahelical G protein-coupled receptors (GPCRs). In terms of GPCRs, bile acids have been shown to activate the muscarinic M3 receptor, which in turn transactivates the epidermal growth factor receptor to stimulate reversible activation of the p44/42 MAP kinase signaling cascade and proliferation of colon carcinoma cells (8). More recently, the GPCR TGR5, also known as GPBAR-1, M-BAR, BG37, GPCR19, or GPR131, has been reported to be widely distributed in human tissues, including most segments of the digestive tract with the exception of the esophagus and rectum (19, 24). In terms of mechanism, binding of bile acids to TGR5 has been shown to increase cAMP production leading to activation of protein kinase A; for example, in rabbit alveolar macrophages, bile acids increase cAMP production and suppress LPS-stimulated cytokine production (19).
The human colon carcinoma cell line, T84, has been a useful model in which to study the cellular regulation of intestinal Cl− secretion. Therefore, in this study, we used T84 cells to determine if bile acids act through CFTR and to delineate the underlying signaling cascade. Our data represent the first direct demonstration of the involvement of CFTR, activated by the cAMP cascade, in bile acid action in colonic epithelial cells.
MATERIALS AND METHODS
Materials.
Tissue culture media, Ham's F-12 nutrient mixture, bovine calf serum, fluo-3 AM, TRIzol reagent, superscript II reverse transcriptase, and rabbit polyclonal anti-phosphorylated proteins (Pan-phospho) were obtained from Invitrogen (Carlsbad, CA). CDCA, TCDC, H89, CFTRinh-172, nocodazole, interferon-γ, interleukin (IL)-1β, forskolin, carbachol, 8-bromo (Br)-cAMP, isobutyl methyl xanthine (IBMX), A23187, bumetanide, H89, CdCl2, nystatin, MDL12330A, RIPA buffer, Amplification Grade DNase I, protease inhibitor cocktail, and phosphatase inhibitor cocktail 2 were purchased from Sigma-Aldrich (St. Louis, MO). Tumor necrosis factor (TNF)-α was from Promega (Madison, WI). Monoclonal anti-human CFTR COOH-terminal antibody was from R&D Systems (Minnneapolis, MN). Rabbit polyclonal anti-human CFTR NH2-terminal antibody and protein A/G plus-agarose immunoprecipitation reagent were from Santa Cruz Biotechnology (Santa Cruz, CA). All other reagents were of analytical grade and were purchased from either Sigma-Aldrich or Fisher Scientific (Hanover Park, IL) except if stated otherwise.
Cell culture.
The T84 cell line was purchased from American Type Culture Collection (ATCC; Manassas, VA). Cells were grown in DMEM/F-12 medium containing bovine calf serum (GIBCO-BRL), penicillin (100 U/ml), streptomycin (100 μg/ml), and ampicillin (8 μg/ml). The cells were incubated in a humidified atmosphere of 5% CO2 at 37 °C.
Iodide effluxes.
Iodide efflux studies were performed as we have described earlier (5) and are based on the original method of Venglarik et al. (34) modified by Chappe et al. (7). Briefly, T84 cells were grown in six-well plates. One million cells were seeded per well, and it took 4–5 days for the cells to reach 90% confluence at which time they were incubated with iodide loading buffer [containing in mM: 136 NaI, 3 KNO3, 2 Ca(NO3)2, 11 glucose, and 20 HEPES pH 7.4] for 1 h at room temperature in the dark. The cells were then rinsed three times with iodide-free efflux buffer (same as the iodide loading buffer except NaNO3 replaced NaI). Individual wells were exposed to DMSO, CDCA (500 μM), TCDC (500 μM), or a cAMP cocktail composed of 100 μM 8-Br-cAMP + 10 μM forskolin + 100 μM IBMX. Efflux buffer (1 ml) was then added to the dish; after 2 min, the buffer was removed and saved and another 1 ml fresh efflux buffer was added into each well. Samples were thus collected at 2-min intervals for the duration of the experiment. The iodide concentration of the collected samples was determined using an iodide-sensitive electrode (Orion 96–53; Fisher Scientific) and a pH/mV meter. The iodide concentration of samples were calculated based on a standard curve as previously described (5) and depicted as the iodide efflux rate (nmol/min) at every 2-min intervals.
Electrophysiological measurements.
For Ussing chamber measurements, T84 cells were seeded at a density of ∼250,000 cells per insert into 24-well, 0.4-μm pore size Transwell tissue culture inserts (Corning 3413; Corning Life Sciences, Lowell, MA), coated with mouse tail collagen. The transepithelial resistance (TER) was measured using EVOM2 voltohmmeter and a STX2 electrode (World Precision Instruments, Sarasota, FL). When TER reached values of ≥950 Ω·cm2 (∼9–14 days), the inserts were mounted in Ussing chambers (area: 0.1 cm2; Physiologic Instruments, San Diego, CA) and bathed with oxygenated (95% O2-5% CO2) buffer A (5 ml/reservoir) of the following composition (in mM): 115.4 NaCl, 5.4 KCl, 1.2 CaCl2, 1.2 MgCl2, 21.0 NaHCO3, 0.6 NaH2PO4, 2.4 Na2HPO4 pH 7.4, and 10 d-glucose at 37°C. Transmural short-circuit current (Isc; μA/cm2) was monitored throughout the experiment using an automatic voltage-clamp apparatus (VCC-MC6; Physiologic Instruments, San Diego, CA) as described earlier (37). TER was obtained by introduction of a 5-mV bipolar pulse at 10-s intervals and TER calculated by Ohm's law. For nystatin experiments, cells were exposed to 200 μg/ml nystatin for 10 to 20 min either on the apical or on the basolateral surface. Following this equilibration period, bile acid was added to the basolateral surface. When basolateral membrane was permeablized, a Cl− gradient was established by replacing NaCl in basolateral reservoir with sodium gluconate and the concentration of CaCl2 was increased from 1.2 to 2 mM to account for chelation of Ca2+ by gluconate (23). When the apical membrane was permeablized, a K+ gradient was created by substituting NaCl with KCl in apical reservoir (13). At the end of each experimental run, forskolin (10 μM) and carbachol (100 μM) were sequentially added to the basolateral solution to assess tissue viability.
Intracellular cAMP measurement.
Intracellular cAMP was measured using an Amersham cAMP Biotrak Enzymeimmunoassay (EIA) System from GE Healthcare Bio-Sciences (Piscataway, NJ). T84 cells were seeded at a density of 105 cells per well in 96-well plates to reach confluence, which was generally achieved the next day. Fresh media with different secretagogues or solvent (DMSO) were added to the wells, and the plates were placed in the cell culture incubator at 37°C during the stimulation period. Cell lysis and preparation for enzymeimmunoassay were according to EIA kit instructions. The EIA microplate wells were sequentially incubated with antiserum (4°C, 2 h) and cAMP-peroxidase (4°C, 1 h) and then washed and incubated with enzyme substrate for 30 min at room temperature. The reaction was stopped with 1.0 M sulfuric acid, and the optical density was read at 450 nm in a microplate autoreader (Bio-Tek Instruments, Winooski, Vermont). The cAMP concentrations of samples were calculated from the standard curve generated as per manufacturer's instructions.
RT-PCR.
RT-PCR was performed as we have previously described (37) with one important modification. We harvested T84 cells grown to confluence both in six-well plates and in Transwells to account for variation in expression, if any, due to the different growth conditions. Results for both are shown. Briefly, total RNA was isolated using TRIzol reagent and was treated with DNase to eliminate potential contaminating DNA. Total RNA was quantitated photometrically at 260 nm. One microgram of total RNA was used for the RT assays employing superscript II reverse transcriptase. Using the strategy of Hov et al. (16), we employed two sets of primers. The products of primer TGR5-IE span part of the intron between the two exons and part of exon 2 (product size 653 bp), while the products of primer TGR5-E2 are entirely within exon 2 (product size 321 bp). The primer pairs used for TGR5 are TGR5-IE (forward primer: AGCATCTTCCTTCCTCTCAGC and reverse primer: TTGTGTATCCCTGCCTCCAC) and TGR5-E2 (forward primer: GCTGCTTCTTCCTRAGCCTA and reverse primer: TGGGAGCTGCAGTTGGCA). The PCR was performed at annealing temperature of 64°C (TGR5-IE) or 55°C (TGR5-E2), and PCR was run for 36 cycles. The primer pairs used for FXR are as follows: forward primer: ATTTTGACGGAAATGGCAAC and reverse primer: AGCTAGACCCCTCCCCTGTA. The PCR was performed at annealing temperature of 55°C, and PCR was run for 36 cycles.
Immunoprecipitation and immunoblot analysis.
Immunoprecipitation and immunoblot analysis were conducted according to Sakesena et al. (30). The cells were subjected to a growth and treatment regimen similar to that used for the Ussing chamber experiments. Briefly, T84 cells were seeded at a density of 1.5 × 106 cells per insert in six-well Transwell tissue culture inserts. When TER reached values of ≥950 Ω·cm2 (∼9–14 days) as determined by EVOM2 voltohmmeter and STX2 electrode, the cells were treated basolaterally with DMSO (0.1%), CDCA (500 μM), or forskolin (10 μM) for 20 min. Cells were washed three times with PBS, and the membrane was cut and immersed in lysis buffer (20 mM Tris·HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM EGTA, protease inhibitor cocktail, and phosphatase inhibitor cocktail 2). Cells from four wells were pooled together as one sample. The cells were sonicated on ice (25 s, Branson Sonifier Cell Disruptor Model 350). The homogenate was centrifuged (1,000 g for 10 min at 4°C) to pellet out the nuclei and unbroken cells. The supernatant containing 5 mg protein was incubated with 3 μg monoclonal anti-human CFTR COOH-terminal antibody overnight at 4°C on a shaker. After incubation, immune complexes were precipitated using the protein A/G plus-agarose immunoprecipitation reagent. Pellets were washed four times with RIPA buffer, and after the final wash, pellets were resuspended in 50 μl of SDS-containing Laemmli buffer. Proteins were separated by electrophoresis on 7.5% SDS-polyacrylamide gels and transferred to PVDF membrane (Millipore, Bedford, MA). The PVDF membranes were blocked with 3% BSA for 1 h at room temperature and incubated with 1 μg/ml of rabbit polyclonal anti-phosphorylated proteins (Pan-phospho) in 1% BSA overnight at 4°C on a shaker. The membranes were washed three times using TBS containing 0.1% Tween 20 (TBS-T) and were incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit antibody (1:10,000 dilution) for 1 h at room temperature. Finally, the membranes were washed three times with TBS-T and visualized with Pierce SuperSignal West Pico Chemiluminescent Substrate kit (Thermo Scientific, Rockford, IL). The membranes were then stripped by agitating for 30 min at 50°C in stripping buffer (100 mM β-mercaptoethanol, 2% SDS, and 62.5 mM Tris·HCl pH 6.7) and reprobed with rabbit polyclonal anti-human CFTR NH2-terminal antibody (1:1,000 dilution in 1% milk, overnight at 4°C). The secondary antibody used was HRP-conjugated goat anti-rabbit antibody (1:10,000 dilution), and Pierce SuperSignal West Pico Chemiluminescent Substrate kit was again used to visualize the reaction product. Immunoblot bands were quantified by ImageQuant software (GE Healthcare) after scanning densitometry. Phosphorylated CFTR protein was normalized to total CFTR protein, and the values for DMSO treated samples were set at 1.
To prepare membrane fractions of T84 cells for TGR5 immunoblots, cells were homogenized in a buffer containing the following (in mM): 1 EDTA, 2 MgCl2, 5 β-mercaptoethanol, 1 DTT, 25 Tris·HCl pH 7.4, and protease inhibitor cocktail as described previously (2). The homogenate was centrifuged at 1,000 g for 10 min at 4°C to pellet out the nuclei and unbroken cells. The resultant supernatant was then centrifuged at 100,000 g for 30 min at 4°C (2). The final membrane pellet was resuspended in lysis buffer. Rabbit polyclonal antibody to TGR5 (1:1,000 dilution) from Abcam (Cambridge, MA) was used to probe for the presence of the protein and visualized with HRP-conjugated goat anti-rabbit secondary antibody as described above for the immunoblotting procedure.
Preparation of detergent-soluble and insoluble microtubules.
Detergent-soluble and -insoluble tubulin was prepared according to Yu et al. (38). In brief, T84 cells grown in six-well plates were equilibrated at 4°C for 30 min. Nocodazole (33 μM) was then added to both apical and basolateral sides, and the cells were kept on ice for an additional 30 min. Next, the cells were rinsed once with 37°C PBS and once with extraction buffer (0.1 M PIPES, 1 mM MgSO4, 2 mM EGTA, 0.1 mM EDTA, and 2 M glycerol pH 6.75). Cells were subsequently extracted twice for 8 min each with 250 μl of extraction buffer containing 0.1% Triton X-100 and protease inhibitors and the fractions collected to yield the detergent-soluble fraction. After excess extraction buffer was drained from each well, 250 μl of lysis buffer (25 mM Na2HPO4, 0.4 M NaCl, and 0.5% SDS pH 7.2) were added to each well. The cytoskeletal lysate was boiled for 3 min and then centrifuged for l0 min (2,000 g) to get rid of the DNA-containing pellet, and the supernatant was collected to yield the detergent-insoluble fraction. Equal amounts of detergent-soluble (5 μg) and -insoluble (10 μg) proteins from each treatment group were loaded onto SDS-polyacrylamide gels, and the tubulin content was assessed by immunoblot using α-tubulin antibody from Sigma (1:2,500 dilution).
Statistical analysis.
Data from at least three individual experiments were analyzed and presented as means ± SE. Statistical significance was determined using one-way ANOVA or Student's t-test, and values of P ≤ 0.05 were considered statistically significant.
RESULTS
Effect of CDCA on chloride transport in T84 cells.
The iodide efflux assay is a convenient method to assess Cl− transport via channels (2). Iodide effluxes from T84 cells treated with 0.2% DMSO (control), a cAMP cocktail (100 μM 8-Br-cAMP + 10 μM forskolin + 100 μM IBMX), CDCA (500 μM), and TCDC (500 μM) were compared. As shown in Fig. 1A, and as previously reported by us (2), the cAMP cocktail caused a significant increase in iodide efflux rate (nmol/min) at 2, 4, and 6 min compared with DMSO control. As shown in Fig. 1B, treatment with CDCA caused significant increase in iodide efflux rate at 2, 4, and 6 min, while treatment with TCDC caused significant increase at 6 min compared with DMSO control. These data indicate that CDCA and TCDC activate Cl− channel(s) in T84 cells.
Fig. 1.

Effect of chenodeoxycholic acid (CDCA) on iodide efflux in T84 cells. T84 cells grown in 6-well plates were treated with 0.2% DMSO, cAMP cocktail [100 μM 8-bromo (Br)-cAMP + 10 μM forskolin + 100 μM IBMX; A], CDCA (500 μM), or taurine conjugate TCDC (500 μM; B). Iodide efflux assay was performed as described in materials and methods. Bar graphs show iodide efflux rate (nmol/min), which was calculated over a 2-min interval. Data are expressed as the means ± SE of 3 independent experiments. *P < 0.05, compared with 0.2% DMSO.
Effect of CDCA on Isc in T84 cells.
We confirmed the findings of I− efflux studies by examining the effects of CDCA on transepithelial Isc, a measure of Cl− secretion across T84 cells. T84 cells grown on 24-well Transwell tissue culture inserts were mounted in Ussing chambers. Representative tracing of Isc are shown in Fig. 2, A and B. As has been reported by others (20), we also observe that basolateral addition of CDCA caused more than a 15-min sustained increase in Isc (ΔIsc: 24.3 ± 6.6 μA/cm2; n = 4), while apical addition of CDCA caused a much smaller response (ΔIsc: 6.6 ± 1.6 μA/cm2; n = 7; Fig. 2A). In addition, the rate of response to basolateral addition (slope: 0.38 ± 0.06; n = 4) is much higher than in response to apical addition (slope: 0.11 ± 0.02; n = 7). The TER remained steady for 15 min and did not drop below 95% after apical or basolateral addition of CDCA.
Fig. 2.

Effect of CDCA on short-circuit current (Isc) in T84 monolayers in the absence and presence of cytokines. T84 cell monolayers were incubated with a cytokine cocktail containing TNFα (10 ng/ml), interferon-γ (30 ng/ml), and IL-1β (10 ng/ml) for 24 h before being mounted in Ussing chambers. CDCA (500 μM) was first added to the apical chamber and then added to the basolateral chamber. Representative tracing of Isc shows the effect of CDCA in the control (A; n = 3) and cytokines-treated (B; n = 4) T84 monolayers. AM, exposure of apical membrane to CDCA; BLM, exposure of basolateral membrane to CDCA.
Proinflammatory cytokines are known to alter transepithelial permeability (26). To examine if the sidedness of bile acid action is altered in the presence of inflammatory mediators, we preincubated T84 cells with a cytokine cocktail containing TNFα (10 ng/ml), interferon-γ (30 ng/ml), and IL-1β (10 ng/ml) for 24 h in serum free medium. As shown in Fig. 2B, pretreatment with cytokines potentiated the effects of apical addition of CDCA on Cl− secretion by 55% (ΔIsc: 13.5 ± 2.1, vs. control ΔIsc: 8.7 ± 3.2; n = 3). As anticipated, exposure to cytokines caused a significant decrease in TER (control: 1,068 ± 41 Ω·cm2; n = 3; cytokine treated: 569 ± 20 Ω·cm2; n = 4). These results suggest that the sidedness of bile acid action on Isc depends on the epithelial integrity.
Effect of transporter inhibitors and permeabilization on CDCA-induced changes in Isc.
Transepithelial Cl− secretion is governed by the concerted activation of apical Cl− channels and of basolaterally located NKCC1 cotransporter, K+ channels, and Na+-K+-ATPase. To confirm that the induction of Isc by bile acids reflects Cl− secretion, we first blocked the route of Cl− entry, using the NKCC inhibitor, bumetanide. As shown in Fig. 3A, the action of CDCA was inhibited 100% by bumetanide (100 μM; n = 3). In addition, the K+ channel blocker BaCl2 (5 mM) completely abolished the effect of CDCA on Isc (n = 3).
Fig. 3.
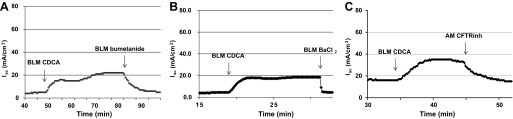
Effect of inhibitors of transepithelial Cl− secretion on CDCA-induced changes in Isc. T84 cells grown in Transwells were mounted in Ussing chambers. CDCA (500 μM) was added to the basolateral solution. Bumetanide (100 μM BLM; A), BaCl2 (5 mM BLM; B), or CFTRinh-172 (10 μM AM; C) was added after Isc reached plateau and stayed steady for ∼10 min. Data depicted are representative tracings of n ≥ 3 experiments.
The results of iodide efflux and Isc suggested CDCA may possibly act via a Cl− channel, and in this cell type, CFTR is the most likely candidate. Therefore, we examined the effects of the specific CFTR inhibitor, CFTRinh-172 (10 μM), on the CDCA elicited Isc responses in T84 cells. As shown in Fig. 3C, CFTRinh-172, added to the apical chamber, greatly attenuated (97%, n = 4) CDCA-stimulated Isc and therefore Cl− secretion. We also studied the effect of CDCA after T84 cells were pretreated with each of these inhibitors for 20 min. Addition of 100 μM bumetanide to the basolateral solution did not alter the basal Isc, and it inhibited the CDCA response by 72 ± 6% (n = 3). Addition of 10 μM CFTRinh-172 to the apical solution caused a small transient, insignificant decrease (∼1 μA/cm2) in the basal Isc. Pretreatment with CFTRinh-172 inhibited the action of CDCA by 69 ± 7% (n = 6). In this treatment regimen, addition of 5 mM BaCl2 (basolateral) before CDCA did not change the basal Isc, and it inhibited the action of CDCA by 67 ± 4% (n = 4).
To further delineate the effect(s) of CDCA on apical Cl− or basolateral K+ conductances, we electrically isolated these membranes by permeabilizing the contralateral membrane with nystatin (200 μg/ml) (23). As shown in Fig. 4A, permeabilizing the basolateral membrane, followed by exposure to CDCA (500 μM) resulted in an increase in Isc across the apical membrane (ΔIsc: 72.3 ± 12.2 μA/cm2; n = 4). During this 15-min period, TER dropped to 76% after CDCA was added. Not surprisingly, the rate of response was faster in the permeabilized monolayers (slope of Isc: 1.18 ± 0.37; n = 4) compared with that in nonpermeabilized monolayers (0.38 ± 0.06; n = 4) mentioned above. Further addition of forskolin caused a small increase in Isc which was greatly inhibited by the addition of 10 μM CFTRinh-172 (80% inhibition; n = 4). A similar 89% (n = 3; data not shown) inhibition of the CDCA-invoked current was observed when 10 μM CFTRinh-172 was added to the apical solution before the basolateral membrane permeabilization. In contrast, permeabilization of the apical membrane followed by addition of CDCA did not affect Isc across the basolateral membrane (n = 5; Fig. 4B), In this case, ouabain (100 μM) was added basolaterally to inhibit the activity of the Na+-K+-ATPase pump (13) before the addition of CDCA. In contrast to CDCA, addition of carbachol to the basolateral solution in apically permeabilized cells stimulates Isc, implying that carbachol, but not CDCA, affects a basolateral current, most likely K+ current. Collectively, these inhibitor and permeabilization studies suggest that CDCA acts primarily on the apical Cl− channel CFTR but does not directly stimulate the basolateral K+ channels.
Fig. 4.

Effect of CDCA on Isc in nystatin-permeablized T84 cells. T84 cells grown in Transwells were mounted in Ussing chambers. Representative tracings of Isc show CDCA the effect of CDCA on basolaterally (A; n = 4) or apically (B; n = 5) permeablized T84 cells. A: basolateral membranes of T84 cells were permeabilized with nystatin (200 μg/ml) in the presence of an apical to basolateral Cl− gradient. CDCA (500 μM) was added to the basolateral solution, and after the Isc had stabilized, forskolin (10 μM) was added to the basolateral solution followed by CFTRinh-172 (10 μM) to the apical solution. B: apical membranes of T84 cells were permeabilized with nystatin in the presence of an apical to basolateral K+ gradient. Sequentially ouabain (100 μM), CDCA (500 μM), and carbachol (CCH; 100 μM) were added to the basolateral solution.
Involvement of cAMP signaling in the effect of CDCA.
Our results in Fig. 3C strongly suggest that CDCA stimulates Cl− secretion through CFTR, and since it is well-established that CFTR is activated by the cAMP-PKA pathway, we probed whether this cascade is involved in CDCA action. To examine the involvement of adenylate cyclase, T84 cells were incubated with 50 μM MDL12330A, a known inhibitor of the enzyme (31), for 25 min before the addition of CDCA (500 μM). MDL12330A significantly decreased CDCA-stimulated Isc (ΔIsc CDCA: 21 ± 1.5, n = 3; CDCA + MDL12330A: 12 ± 0.8 μA/cm2, n = 4; Fig. 5A).
Fig. 5.

The role of the cAMP-dependent pathway in the actions of CDCA. A: T84 cells mounted in Ussing chambers were incubated with 50 μM adenylate cyclase inhibitor MDL12330A for 25 min before the basolateral addition of CDCA (500 μM). Bar graph shows the Isc change elicited by CDCA ± MDL12330A. *Statistically significant difference in ΔIsc (P < 0.05) compared with CDCA alone; n = 4. B: T84 cells grown in 96 well-plates were incubated with 0.1% DMSO or 5 μM CDCA or 500 μM CDCA for 20 min, and [cAMP]i was measured using enzyme immunoassay. Bar graph shows the intracellular cAMP concentration ([cAMP]i), expressed in fmole/mg protein. *Statistically significant difference in [cAMP]i (P < 0.05) compared with 0.1% DMSO; n ≥ 4.
In parallel and as shown in Fig. 5B, CDCA treatment (for 20 min) significantly increased intracellular cAMP concentration ([cAMP]i; pmol/mg protein: DMSO: 1.42 ± 0.25, n = 12; 5 μM CDCA: 2.93 ± 0.64, n = 6; 500 μM CDCA: 1.54 ± 0.25, n = 4). Higher concentrations (500 μM) of CDCA gave equivocal results as shown in Fig. 5B. A similar dose dependency was observed when cells were grown on Transwells (data not shown; also see discussion). Forskolin was used as the positive control, and the [cAMP]i after forskolin treatment reached 429 ± 105 pmol/mg protein (n = 6).
Finally, to test the involvement of the cAMP protein kinase (PKA) pathway in the effect of CDCA on Isc, H89 (30 μM) was added to both sides of the monolayer for 30 min before the addition of CDCA. As shown in Fig. 6, pretreatment of T84 cell with H89 caused a 94% reduction in the CDCA elicited response (n = 3). These results suggest that bile acids activate the PKA cascade in T84 cells.
Fig. 6.

Effect of PKA inhibition on CDCA-stimulated Isc in T84 cells. T84 cells mounted in the Ussing chamber were incubated with either 0.1% DMSO or 30 μM H89 for 30 min before the basolateral addition of CDCA (500 μM). Representative tracings of Isc show the effect of CDCA in control (black line) and H89 (gray line)-treated T84 cells; n = 3. Forskolin (FSK; 10 μM) and carbachol (100 μM) were added at the end to test the viability of the cells.
Effect of CDCA on CFTR phosphorylation.
We next examined if exposure to CDCA causes phosphorylation of CFTR by sequential immunoprecipitation and immunoblotting with specific antibodies. As described in materials and methods, T84 cells were treated with DMSO, CDCA (500 μM), or forskolin (10 μM) for 20 min. The cells were lysed and immunoprecipitated with a specific mouse monoclonal antibody against the COOH terminus of human CFTR, and the immunopreciptates resolved on a 7.5% gel by SDS-PAGE. The gels were subjected to immunoblotting with Pan-phospho rabbit antibodies. As shown in Fig. 7A, top, CDCA and forskolin increased the phosphorylation of CFTR. To quantitate this increase by detecting total amount of CFTR, the blots were stripped and reprobed with rabbit polyclonal anti-human CFTR NH2-terminal antibody (Fig. 7A, middle). The bands were quantitated by densitometry and the ratio of phosphorylated CFTR: total CFTR determined in the three samples, with the ratio for DMSO being set at 1. As shown in Fig. 7A, bottom, CDCA caused an approximately twofold increase in CFTR phosphorylation (1.9 ± 0.3), which is similar to the effect of forskolin (2 ± 0.3 vs. DMSO: 1; n = 4; P < 0.05).
Fig. 7.

Effect of CDCA on CFTR phosphorylation in T84 cells. T84 cells grown in 6-well Transwells were treated with 0.1% DMSO, 500 μM CDCA, or 10 μM forskolin basolaterally for 20 min. Immunoprecipitation (IP) and immunoblot were performed as described in materials and methods. A: representative blot shows 1 of 4 experiments. B: Quantification of phosphorylation as shown in A. The CFTR phosphorylation ratio (phospho-CFTR density/total CFTR density) in the DMSO control was set at “1” for all 4 experiments, and used to normalize the data. *Statistically significant difference in phosphorylation (n = 4; P < 0.05) compared with 0.1% DMSO.
Role of microtubule in the effect of CDCA on Isc.
In intestinal epithelial cells, including T84 cells, activation of CFTR by cAMP is accompanied by a recruitment of CFTR to the apical membrane, a process that is dependent on microtubules (6, 32). Thus, if CDCA is acting through a cAMP signaling mechanism, we predict the involvement of microtubules. To test this, we examined the effect of the microtubule disrupting agent, nocodazole on CDCA stimulated Cl− secretion. T84 cells grown in 24-well Transwells were incubated with nocodazole (33 μM) as described in materials and methods. After the incubation, the cells were mounted in Ussing chamber with 33 μM nocodazole in both bathing solutions. The cells were allowed to equilibrate at 37 °C for 30 min, and 500 μM CDCA was added to the basolateral solution. Control Transwells were processed through the same protocol, except that 0.1% DMSO was used instead of nocodazole. As shown in Fig. 8, A and B, nocodazole treatment inhibited CDCA-induced Cl− secretion by 47% (n = 3). The above regimen of nocodazole treatment was sufficient to disrupt polymerized microtubules. As shown in Fig. 8C, there was an increase in the detergent (0.1% Triton X-100) soluble fraction of tubulin in cell lysates of nocodazole-treated cells compared with that of control cells.
Fig. 8.

Role of microtubules in the effect of CDCA on Isc in T84 cells. T84 cells grown in Transwells were pretreated with DMSO (0.1%) or nocodazole (33 μM, bilateral) first on ice for 30 min, and then were mounted in Ussing chambers where they were treated for an additional 30 min at 37°C. CDCA (500 μM) was added to the basolateral solution. Representative tracings of Isc show the effects of CDCA in control (A) and nocodazole (B)-treated T84 cells; n = 3. C: representative blot shows the 0.1% Triton X-100 soluble and insoluble α-tubulin in T84 cells treated with DMSO or nocodazole. Estimated size of the α-tubulin protein is 50 kDa; n = 3.
Role of muscarinic receptor and TGR5 mRNA and protein expression.
The rapidity and side-specific nature of the Isc response to CDCA implies a basolateral membrane-mediated process. It has been show that taurolithocholic acid (TLC) specifically binds to muscarinic M3 receptor and transactivates the epidermal growth factor receptor tyrosine kinase in H508 human colon cancer cells (8). We tested the involvement of muscarinic receptor in the effect of CDCA by exposing the monolayers to bilateral addition of atropine (10 μM) before the addition of CDCA. As shown in Fig. 9A, atropine had no effect on CDCA-stimulated Isc, thereby ruling out the involvement of muscarinic receptors in CDCA action.
Fig. 9.

Function and expression of known bile acid receptors. A: T84 cells mounted in the Ussing chamber were incubated with either 0.1% DMSO or 10 μM atropine for 30 min before the basolateral addition of CDCA (500 μM). Bar graph shows the Isc change elicited by CDCA ± atropine; n = 6. B: representative RT-PCR result shows the presence of TGR5 mRNA in T84 cells. As described in materials and methods and results, 2 different primers were used; TGR5-IE which spans part of the intron between exon 1 and 2 and part of exon 2 and TGR5-E2 which spans only exon 2. TW, Transwell. C: representative blot shows the presence of TGR5 protein in T84 cells. Rat liver, spleen, and distal colon tissues were used as positive controls. Data shown in B and C are representative of three separate experiments. Estimated size of the TGR5 protein is 37 kDa. D: representative RT-PCR result shows the presence of FXR mRNA in T84 cells grown on cell culture dishes or Transwells (n = 3).
The G protein-coupled receptor (GPCR) TGR5 has been implicated in bile acid action in a variety of tissues, including the gallbladder epithelium (21). We probed for the presence of TGR5 mRNA in T84 cells grown in plates or in Transwells. While the gene for TGR5 has two exons, the entire coding sequence resides in exon 2 (16). As described in materials and methods, the primers defining TGR5 IE spans the intron and part of exon 2. As shown in Fig. 9B, left, these primers amplified a product in the DNA samples, but failed to do so in both cDNA samples confirming that the cDNA was not contaminated with DNA. The TGR5 E2 product was visible in both cDNA (from cells either grown on cell culture plates or on Transwell inserts) and DNA samples demonstrating that TGR5 mRNA is present in T84 cells (Fig. 9, right). The PCR product was sequenced, and the nucleotide sequence was 97% identical with human TGR5 (GenBank Accession No. NM_001077194.1). Figure 9C shows that T84 cells express TGR5 protein and it is largely detected in the membrane fraction. Rat tissues were used as positive controls; while rat liver and spleen contain abundant protein, there was modest expression in the rat colon. The estimated size of the TGR5 protein is 37 kDa. The multiple bands seen in the rat liver and T84 samples may be a result of posttranslational modification; the TGR5 protein sequence has at least two putative N-linked glycosylation sites and others (27) have reported multiple bands for TGR5 protein.
Finally, the farnesoid receptor, FXR, is a well-studied bile acid receptor. Employing PCR, we examined the presence of FXR mRNA in T84 cells. As shown in Fig. 9D, FXR transcripts are present in T84 cells grown on cell culture plates and on Transwell inserts.
DISCUSSION
These studies are the first direct demonstration of the involvement of CFTR, the PKA-dependent pathway, and intact microtubules in bile acid-induced Cl− secretion across a human colonic cell line. We also demonstrate that CDCA preferentially acts via the basolateral membrane to activate electrogenic Cl− secretion across the apical membrane.
In addition to their critical role in lipid absorption, bile acids affect intestinal, and colonic epithelial function and integrity including electrolyte transport (1), cell growth, and apoptosis. It is clear that both the response of the colon to bile acids and the signaling pathway(s) employed are influenced by the chemical structure of the bile acid (hydrophobicity and conjugation), the animal species, the developmental age, the tissue segment of the colon, and/or the specific cell line being examined. For example, in mouse (14) and rabbit (36) colonic epithelia, we and others have shown that the endogenous CDCA, DCA, and their taurine conjugates, TCDC and TDC, but neither the trihydroxy cholic acid (CA) nor its taurine conjugate nor the monohydroxy lithocholic acid (LCA) stimulate Cl− secretion. In T84 cells, Keely et al. (20) reported that CDCA and DCA but neither TCA nor LCA stimulate Cl− secretion; we have independently confirmed these findings in these cells and expanded them to test various (5, 50, or 500 μM) concentrations of LCA (n > 3; data not shown). However, there is some variability in the magnitude of response observed in the same cell type, as reported by different laboratories. Thus, while Keely et al. (20) observe that among the bile acid homologues and epimers they tested, DCA to be the most potent, we find CDCA elicits the largest secretory response in T84 cells. Both the Keely group and we find that TDC, TCDC (20), and CDCA (Fig. 2) are more effective in stimulating Cl− secretion from the basolateral side than the apical side (Fig. 2A). However, incubation of T84 cells with cytokines for 24 h elicited a much bigger Isc in response to apical CDCA compared with control monolayers (Fig. 2B).
A tissue that responds prominently to apical exposure to bile acids is the ileum. Thus in distinct contrast to the colon, the trihydroxy bile acid TCA applied apically stimulates Cl− secretion in rat (4) and rabbit ileum (37). This is due to the presence of the sodium-dependent bile acid transporter on the apical membrane (ASBT; Ref. 1). Although the adult colon lacks ASBT (37), three other putative bile acid transporters have been reported to be present in intestinal tissues. Thus the multidrug resistance protein MRP3 has been localized to the basolateral membranes of rat colonocytes, and the organic solute and steroid transporter OSTα-OSTβ have been localized to basolateral membranes of mouse ileum and rat intestine, including the colon. Functional studies in other tissues (e.g., liver) and heterologous expression systems suggest that OSTα-OSTβ chiefly and to a lesser extent MRP3 are involved in the basolateral efflux of bile acids (10). Since there is no compelling evidence for the presence of apical bile acid transporters in the adult colon, and the cytokine regimen caused a 47% decrease in TER, an index of epithelial permeability, it is reasonable to suggest that the inflammatory mediators provide greater access of CDCA to the basolateral membrane by increasing the paracellular permeability of the T84 monolayer. Thus a similar 54% drop in TER (100% TER: 1,000–3,000 Ω/cm2) in T84 cells in response to okadaic acid was accompanied by a more than threefold increase in cumulative [3H]mannitol permeation across monolayers, indicating an increase in paracellular conductance (33).
Of the many studies probing bile acid action, there has been no systematic examination of the various steps of the signaling cascade underlying the stimulation of Cl− secretion in human colonic epithelia. Our studies including the use of adenylate cyclase and protein kinase inhibitors and of [cAMP]i measurement suggest the involvement of the cAMP-PKA pathway. The receptor mechanism(s) by which bile acids achieve this are examined at the end of the discussion. The effects of CDCA on [cAMP]i is interesting and warrants some explanation. Thus, while we were able consistently to measure a significant increase in [cAMP]i in response to 5 μM CDCA, our results with higher doses of CDCA were equivocal (Fig. 5). This is regardless of whether we grow the cells on cell culture dishes or on Transwell inserts. Our current explanation for this is based on a number of reports that spatiotemporal regulation is critical in cell signaling and two examples are provided. Thus adenosine, while acting via A2b receptors, caused a maximal stimulation of CFTR-mediated Cl− secretion in lung epithelial cells, there was no “measurable change” in total cellular [cAMP], leading the authors to suggest a highly localized regulation of CFTR by signaling molecules in apical compartments in these cells (17). In another study, Li et al. (22) elegantly demonstrated, by FRET analyses using CFP-EPAC-YFP-transfected T84 cells, that changes in the levels of cAMP in the vicinity of target proteins, rather than in total intracellular levels, were critical in inducing CFTR-mediated Cl− secretion. Extrapolating these observations to our results, we predict that 500 μM CDCA cause sufficient local changes in [cAMP]i to activate CFTR. Further we predict that our inability to observe changes in total [cAMP]i, in response to 500 μM CDCA, is because CDCA may be activating a global increase in phosphodiesterase activity and thereby masking local spatiotemporal effects. In addition, the consistent increase in total [cAMP]i by 5 μM CDCA was perhaps not sufficiently high in the region of the apical membrane to induce Cl− secretion. We also predict that 5 μM CDCA was not sufficient to activate phosphodiesterase activity. The spatiotemporal coupling may also be relevant in explaining the relative levels of [cAMP]i invoked by forskolin vs. the Isc generated by forskolin or CDCA. We reported similar findings with respect to lubiprostone and forskolin (2). Thus, although there are considerable differences in the levels of cAMP generated in response to forskolin or lubiprostone ([cAMP]i in pmol/mg protein: 250 nM lubiprostone: 1.1 ± 0.2; 10 μM forskolin: 26 ± 1.7), both agents caused similar increases in CFTR-mediated Cl− secretion. Exploring these molecular mechanisms of cAMP generation in detail is the basis of a separate series of ongoing investigations.
The bumetanide, CFTRinh-172, BaCl2 and nystatin results reveal that CDCA causes transepithelial electrogenic secretion, involving an apical membrane ion channel. The inhibitors attenuate secretion whether added to the monolayers before or after CDCA. It is noteworthy that in the present study carbachol but not CDCA appears to activate a basolateral current, most likely a K+ conductance. These results are in contrast to those of Mauricio et al. (25) in the rabbit distal colon, where bile acid activation of Cl− secretion appears to involve the stimulation of basolateral K+ conductances. Using whole cell configurations and patch-clamp methodology, Devor et al. (11) demonstrated activation of both a K+ conductance and a Cl− conductance in response to 0.75 mM TDC. The 50% higher concentration, the use of TDC rather than CDCA, and the use of whole cell current methodology may in part account for the differences in the Devor study and the present study. The fact that the Isc caused by CDCA is almost completely inhibited by the specific CFTRinh-172 strongly suggests that CFTR is the channel mediating CDCA-stimulated Cl− secretion.
The activation of the CFTR channel is initiated by the phosphorylation of the R domain by cAMP-dependent PKA. The phosphorylation of CFTR has been well described for forskolin action, and therefore, we compared the effects of forskolin and CDCA on CFTR phosphorylation. As shown in Fig. 7, forskolin and CDCA had comparable effects on CFTR phosphorylation, further confirming the involvement of CFTR in CDCA action. There are 10 potential sites of phosphorylation by PKA and protein kinase C in the R domain of CFTR. While the phosphorylation of a few sites inhibit CFTR activity, the phosphorylation of most of the PKA consensus sequences leads to activation of CFTR (9). Future studies will probe which sites are selectively phosphorylated by bile acids. In some, but not in all epithelia, PKA activation also increases channel number by recruiting CFTR-bearing endosomes to the apical membrane (28), a process that is dependent on microtubules (6, 32). As shown in Fig. 8, disruption of microtubules was seen in the presence of nocodazole and CDCA, like other cAMP-dependent secretagogues utilizes a microtubule-dependent mechanism in part, to activate Cl− secretion.
To examine the mechanisms by which bile acids interact with cells, considerable focus has been given to the family of steroid nuclear receptors, in particular the farnesoid receptor FXR (1). Depending on the preparation, the EC50 for CDCA is 10–50 μM as measured in cell-free assays. Bile acids can either diffuse into the cell to interact with the receptor or could enter via transporters. Since the adult colon does not have ASBT, as discussed above, other conduits for bile acid transport are the basolaterally located Ostα, Ostβ, and MRP3 (10). Our data show the presence of FXR transcripts in T84 cells (Fig. 9D), and the distribution and roles of MRP3, Ostα, and Ostβ in these cells are not known. Considering the rapid effects (<5 min) of CDCA action on Isc in T84 cells, we postulate that membrane, more than nuclear, receptors are involved in the secretagogue action of bile acids in the colon. Two lines of evidence suggest the involvement of membrane receptors. First, in H508 human colon cancer cells, LCA specifically binds to the muscarinic M3 GPCR and transactivates the EFGR tyrosine kinase (8). However, as shown in Fig. 9A, the colonic Cl− secretory response is not inhibited by atropine and as discussed above, we and others (20) do not observe any effect of LCA on Cl− secretion suggesting that the receptor is not muscarinic M3 GPCR. Second, another GPCR, TGR5, is shown to be bile acid specific, acting via the cAMP pathway to alter metabolic processes in tissues such as brown adipose tissue (19, 24). Our data demonstrate for the first time the presence of TGR5 mRNA and in T84 cells. Since CDCA activates TGR5 in other cell types, it is tempting to propose that CDCA stimulates Cl− secretion via TGR5. However, this needs to be definitively demonstrated, since neither LCA, a potent natural agonist of TGR5, nor CA, stimulates Cl− secretion. Unfortunately, there are no specific available inhibitors of the TGR5 receptor, and future studies will focus on probing for its functional role via small interfering RNA methodology. This will allow us to address a variety of mechanistic questions related to the ontogeny, physiology, and pathophysiology of bile acid action in TGR5−/− mouse models.
In summary, our studies clearly demonstrate for the first time that the dihydroxy bile acid CDCA activates the canonical cAMP-signaling pathway to phosphorylate CFTR, increase its recruitment to the apical membrane involving microtubules and thereby stimulate Cl− secretion in the human colon. The influence of inflammatory cytokines in the sidedness of the response may provide us useful and novel insights into the mechanisms underlying bile acid induced diarrheas in diseased states.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants 1 P01-DK-067887 Project 3 (to M. C. Rao) and DK-71596 (to W. A. Alrefai).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.A., J.S., W.A.A., and M.C.R. conception and design of research; M.A., J.S., and J.D. performed experiments; M.A., J.S., J.D., W.A.A., and M.C.R. analyzed data; M.A., J.S., J.D., W.A.A., and M.C.R. interpreted results of experiments; M.A., J.S., J.D., and M.C.R. prepared figures; M.A., J.S., W.A.A., and M.C.R. drafted manuscript; M.A., J.S., J.D., W.A.A., and M.C.R. edited and revised manuscript; M.A., J.S., J.D., W.A.A., and M.C.R. approved final version of manuscript.
REFERENCES
- 1. Alrefai WA, Gill RK. Bile acid transporters: structure, function, regulation and pathophysiological implications. Pharm Res 24: 1803– 1823, 2007 [DOI] [PubMed] [Google Scholar]
- 2. Ao M, Venkatasubramanian J, Boonkaewwan C, Ganesan N, Syed A, Benya RV, Rao MC. Lubiprostone activates Cl- secretion via cAMP signaling and increases membrane CFTR in the human colon carcinoma cell line, T84. Dig Dis Sci 56: 339– 351, 2011 [DOI] [PubMed] [Google Scholar]
- 3. Barrett KE. Positive and negative regulation of chloride secretion in T84 cells. Am J Physiol Cell Physiol 265: C859– C868, 1993 [DOI] [PubMed] [Google Scholar]
- 4. Bijvelds MJ, Jorna H, Verkade HJ, Bot AG, Hofmann F, Agellon LB, Sinaasappel M, de Jonge HR. Activation of CFTR by ASBT-mediated bile salt absorption. Am J Physiol Gastrointest Liver Physiol 289: G870– G879, 2005 [DOI] [PubMed] [Google Scholar]
- 5. Boonkaewwan C, Ao M, Toskulkao C, Rao MC. Specific immunomodulatory and secretory activities of stevioside and steviol in intestinal cells. J Agric Food Chem 56: 3777– 3784, 2008 [DOI] [PubMed] [Google Scholar]
- 6. Chang SY, Di A, Naren AP, Palfrey HC, Kirk KL, Nelson DJ. Mechanisms of CFTR regulation by syntaxin 1A and PKA. J Cell Sci 115: 783– 791, 2002 [DOI] [PubMed] [Google Scholar]
- 7. Chappe V, Hinkson DA, Zhu T, Chang XB, Riordan JR, Hanrahan JW. Phosphorylation of protein kinase C sites in NBD1 and the R domain control CFTR channel activation by PKA. J Physiol 548: 39– 52, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheng K, Raufman JP. Bile acid-induced proliferation of a human colon cancer cell line is mediated by transactivation of epidermal growth factor receptors. Biochem Pharmacol 70: 1035– 1047, 2005 [DOI] [PubMed] [Google Scholar]
- 9. Dahan D, Evagelidis A, Hanrahan JW, Hinkson DA, Jia Y, Luo J, Zhu T. Regulation of the CFTR channel by phosphorylation. Pflügers Arch 1: S92– 96, 2001 [DOI] [PubMed] [Google Scholar]
- 10. Dawson PA, Hubbert ML, Rao A. Getting the mOST from OST: Role of organic solute transporter, OSTalpha-OSTbeta, in bile acid and steroid metabolism. Biochim Biophys Acta 1801: 994– 1004, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Devor DC, Sekar MC, Frizzell RA, Duffey ME. Taurodeoxycholate activates potassium and chloride conductances via an IP3-mediated release of calcium from intracellular stores in a colonic cell line (T84). J Clin Invest 92: 2173– 2181, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dharmsathaphorn K, Huott PA, Vongkovit P, Beuerlein G, Pandol SJ, Ammon HV. Cl− secretion induced by bile salts. A study of the mechanism of action based on a cultured colonic epithelial cell line. J Clin Invest 84: 945– 953, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Donnellan F, Keating N, Geoghegan P, Murray FE, Harvey BJ, Keely SJ. JNK mitogen-activated protein kinase limits calcium-dependent chloride secretion across colonic epithelial cells. Am J Physiol Gastrointest Liver Physiol 298: G37– G44, 2010 [DOI] [PubMed] [Google Scholar]
- 14. Gelbmann CM, Schteingart CD, Thompson SM, Hofmann AF, Barrett KE. Mast cells and histamine contribute to bile acid-stimulated secretion in the mouse colon. J Clin Invest 95: 2831– 2839, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hofmann AF. The continuing importance of bile acids in liver and intestinal disease. Arch Intern Med 159: 2647– 2658, 1999 [DOI] [PubMed] [Google Scholar]
- 16. Hov JR, Keitel V, Laerdahl JK, Spomer L, Ellinghaus E, ElSharawy A, Melum E, Boberg KM, Manke T, Balschun T, Schramm C, Bergquist A, Weismuller T, Gotthardt D, Rust C, Henckaerts L, Onnie CM, Weersma RK, Sterneck M, Teufel A, Runz H, Stiehl A, Ponsioen CY, Wijmenga C, Vatn MH, Stokkers PC, Vermeire S, Mathew CG, Lie BA, Beuers U, Manns MP, Schreiber S, Schrumpf E, Haussinger D, Franke A, Karlsen TH. Mutational characterization of the bile acid receptor TGR5 in primary sclerosing cholangitis. PLos One 5: e12403, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang P, Lazarowski ER, Tarran R, Milgram SL, Boucher RC, Stutts MJ. Compartmentalized autocrine signaling to cystic fibrosis transmembrane conductance regulator at the apical membrane of airway epithelial cells. Proc Natl Acad Sci USA 98: 14120– 14125, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kanchanapoo J, Ao M, Prasad R, Moore C, Kay C, Piyachaturawat P, Rao MC. Role of protein kinase C-delta in the age-dependent secretagogue action of bile acids in mammalian colon. Am J Physiol Cell Physiol 293: C1851– C1861, 2007 [DOI] [PubMed] [Google Scholar]
- 19. Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, Fukusumi S, Habata Y, Itoh T, Shintani Y, Hinuma S, Fujisawa Y, Fujino M. A G protein-coupled receptor responsive to bile acids. J Biol Chem 278: 9435– 9440, 2003 [DOI] [PubMed] [Google Scholar]
- 20. Keely SJ, Scharl MM, Bertelsen LS, Hagey LR, Barrett KE, Hofmann AF. Bile acid-induced secretion in polarized monolayers of T84 colonic epithelial cells: Structure-activity relationships. Am J Physiol Gastrointest Liver Physiol 292: G290– G297, 2007 [DOI] [PubMed] [Google Scholar]
- 21. Keitel V, Cupisti K, Ullmer C, Knoefel WT, Kubitz R, Haussinger D. The membrane-bound bile acid receptor TGR5 is localized in the epithelium of human gallbladders. Hepatology 50: 861– 870, 2009 [DOI] [PubMed] [Google Scholar]
- 22. Li C, Krishnamurthy PC, Penmatsa H, Marrs KL, Wang XQ, Zaccolo M, Jalink K, Li M, Nelson DJ, Schuetz JD, Naren AP. Spatiotemporal coupling of cAMP transporter to CFTR chloride channel function in the gut epithelia. Cell 131: 940– 951, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Loffing J, Moyer BD, Reynolds D, Stanton BA. PBA increases CFTR expression but at high doses inhibits Cl− secretion in Calu-3 airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 277: L700– L708, 1999 [DOI] [PubMed] [Google Scholar]
- 24. Maruyama T, Miyamoto Y, Nakamura T, Tamai Y, Okada H, Sugiyama E, Nakamura T, Itadani H, Tanaka K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem Biophys Res Commun 298: 714– 719, 2002 [DOI] [PubMed] [Google Scholar]
- 25. Mauricio AC, Slawik M, Heitzmann D, von Hahn T, Warth R, Bleich M, Greger R. Deoxycholic acid (DOC) affects the transport properties of distal colon. Pflügers Arch 439: 532– 540, 2000 [DOI] [PubMed] [Google Scholar]
- 26. McKay DM, Baird AW. Cytokine regulation of epithelial permeability and ion transport. Gut 44: 283– 289, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Poole DP, Godfrey C, Cattaruzza F, Cottrell GS, Kirkland JG, Pelayo JC, Bunnett NW, Corvera CU. Expression and function of the bile acid receptor GpBAR1 (TGR5) in the murine enteric nervous system. Neurogastroenterol Motil 22: 814–825, e227–818, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rao M, Sarathy (nee Verkatasubramanian) J, Ao M. Intestinal Water and Electrolyte Transport in Health and Disease Colloquium Series on Integrated Systems Physiology: From Molecule to Function to Disease, Lecture #31, edited by Granger D, Granger J. New York: Morgan and Claypool Life Sciences, 2012 [Google Scholar]
- 29. Robb BW, Matthews JB. Bile salt diarrhea. Curr Gastroenterol Rep 7: 379– 383, 2005 [DOI] [PubMed] [Google Scholar]
- 30. Saksena S, Gill RK, Tyagi S, Alrefai WA, Sarwar Z, Ramaswamy K, Dudeja PK. Involvement of c-Src and protein kinase C delta in the inhibition of Cl(−)/OH- exchange activity in Caco-2 cells by serotonin. J Biol Chem 280: 11859– 11868, 2005 [DOI] [PubMed] [Google Scholar]
- 31. Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, Pellicciari R, Auwerx J, Schoonjans K. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab 10: 167– 177, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tousson A, Fuller CM, Benos DJ. Apical recruitment of CFTR in T-84 cells is dependent on cAMP and microtubules but not Ca2+ or microfilaments. J Cell Sci 109: 1325– 1334, 1996 [DOI] [PubMed] [Google Scholar]
- 33. Tripuraneni J, Koutsouris A, Pestic L, De Lanerolle P, Hecht G. The toxin of diarrheic shellfish poisoning, okadaic acid, increases intestinal epithelial paracellular permeability. Gastroenterology 112: 100– 108, 1997 [DOI] [PubMed] [Google Scholar]
- 34. Venglarik CJ, Bridges RJ, Frizzell RA. A simple assay for agonist-regulated Cl and K conductances in salt-secreting epithelial cells. Am J Physiol Cell Physiol 259: C358– C364, 1990 [DOI] [PubMed] [Google Scholar]
- 35. Venkatasubramanian J, Rao MC, Sellin JH. Intestinal electrolyte absorption and secretion. In: Sleisenger and Fordtran's Gastrointestinal and Liver Disease, edited by Feldman M, Friedman LS, Brandt LJ. Philadelphia, PA: Saunders Elsevier, 2010, p. 1675–1694 [Google Scholar]
- 36. Venkatasubramanian J, Selvaraj N, Carlos M, Skaluba S, Rasenick MM, Rao MC. Differences in Ca2+ signaling underlie age-specific effects of secretagogues on colonic Cl− transport. Am J Physiol Cell Physiol 280: C646– C658, 2001 [DOI] [PubMed] [Google Scholar]
- 37. Weihrauch D, Kanchanapoo J, Ao M, Prasad R, Piyachaturawat P, Rao MC. Weanling, but not adult, rabbit colon absorbs bile acids: flux is linked to expression of putative bile acid transporters. Am J Physiol Gastrointest Liver Physiol 290: G439– G450, 2006 [DOI] [PubMed] [Google Scholar]
- 38. Yu JZ, Dave RH, Allen JA, Sarma T, Rasenick MM. Cytosolic Gαs acts as an intracellular messenger to increase microtubule dynamics and promote neurite outgrowth. J Biol Chem 284: 10462– 10472, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]