

Abstract
Dicoumarols and coumarin derivatives have shown a variety of pharmaceutical activities and have been found to be potent inhibitor for the NAD(P)H-dependent flavoproteins. In this report, dicoumarol and its derivatives containing the substituted benzene ring at the methylenebis position were synthesized and evaluated for their antibacterial activity against gram-positive bacteria: Staphylococcus aureus and Bacillus subtilis, and gram-negative bacteria: Escherichia coli and Klebsiella sp. The results showed that the synthesized dicoumarols affect cell growth but are selective against gram-positive over gram-negative bacterial cells. However, for most derivatives, the substitution of steric bulky benzene group on the methylenebis position appears to decrease in the efficacy of antibacterial effect. This finding is roughly described by the predicted poorer docked structure of the derivatives to a homology model of S. aureus flavoprotein. 3D-QSAR study highlighted structural features around the substituted benzene ring of dicoumarols as the antibacterial activity. CoMFA and CoMSIA contour maps support the idea that steric repulsion at the para position could diminish the antibacterial activity. The results of this study provide a better understanding of the molecular basis for the antibacterial activity of dicoumarols.
1. Introduction
Dicoumarol (3,3′-methylene-bis-4-hydroxycoumarin) is a naturally coumarin-based compound which has long been used as an oral anticoagulant drug. It is metabolically produced from coumarin which was first isolated from both of the Tonka bean (Dipteryx odorata) and the sweet clover (Melilotus alba and Melilotus officinalis) [1]. It is now known to be present in many other plants. Dicoumarol derivative, warfarin (3-(α-acetonyl benzyl)-4-hydroxycoumarin), is commonly used as a natural anticoagulant for the prevention and treatment of excessive blood-clotting disorder [2]. Many dicoumarols and coumarin derivatives have also shown a variety of pharmaceutical activities such as antiinflammatory, antibacterial, antiviral, anticancer, anti-HIV, and antiproliferative properties [3–9]. Therefore, dicoumarols have received much attention for medical and pharmaceutical applications.
Dicoumarols and coumarin-based inhibitors exhibit a broad spectrum of activity against gram-positive bacteria [10, 11]. They have been shown to impede the growth of several bacteria strains, for instance, Staphylococcus aureus, Bacillus anthracis, and Streptococcus pyogenes. Another interesting property of dicoumarols lies in the effective anticancer activity [12]. Particularly, several evidences have shown that dicoumarol appears to be the most potent inhibitor which competes with NAD(P)H coenzyme for binding to the two-electron reduction quinone oxidoreductase [13], nitroreductase [14], azo-dyes azoreductase [15], and a ubiquitous flavoprotein found widely in various organisms. This flavoprotein is an antioxidant enzyme which plays a role in cellular protection by preventing the formation of free radical oxygen/nitrogen such as superoxide (O2 −), hydroxyl radical (HO·), and nitric oxide (NO·). These reactive oxygen and nitrogen species (ROS and RNS) are toxic to cells. Inhibition of the redox flavoprotein by dicoumarol generates a high level of ROS and RNS which increase cellular toxicity and inhibit cell division. Thus, we hypothesize that the antiproliferative mechanism of action of dicoumarol might be via a major suppression of NAD(P)H-dependent flavoprotein activity. This hypothesis also relies on the available 3D crystal structures of dicoumarol-oxidoreductase complexes such as human NQO1 (pdb 2F1O), nitroreductase (pdb 1OOQ), and azoreductase (pdb 2Z9C) from Escherichia coli. From the crystallographic data, these proteins share great similarities in the secondary structure organization and the tertiary fold. They are homodimeric proteins with similar quaternary structure arrangement of the two monomers, providing two catalytic sites at the dimer interface. In each binding pocket, dicoumarol and flavin molecules interact with the surrounding residues of both subunits. Based on these structural studies, the results provided useful information about the potential and common target proteins for dicoumarol.
Due to the evolution of antibiotic resistance to existing drugs, the development of novel antibacterial drugs is essential. In search for more active compounds, it has been shown that the substitution of phenyl ring at the methylenebis position of dicoumarol exhibits interesting biological activity [16]. In this study, we focused on the synthesis and the antibacterial activity of a series of dicoumarol derivatives containing 3,3′-phenylmethylene-bis-4-hydroxycoumarins. Dicoumarols have been synthesized from the condensation reaction of 4-hydroxycoumarin with benzaldehydes (Figure 1). The synthesized compounds were tested for antibiotic susceptibility against gram-positive S. aureus and gram-negative E. coli bacteria through disk diffusion assay. The antibacterial activity tested against E. coli cells was used as a control. Since their antibacterial mode of action is still poorly understood, we made the use of existing protein sequences and structural information available in databases to identify the potential antibiotic target. A molecular modeling approach was employed to reveal the inhibition mechanism of the synthesized compounds on the NAD(P)H-dependent flavoprotein, a hypothetical protein target for dicoumarol. In the study, molecular docking was used to predict the binding mode of dicoumarol to the protein target and a 3D-QSAR approach was carried out for the analysis of structural features of dicoumarols as antibacterial activity. Understanding the molecular mechanism of drug action can provide the basis for the rational design of effective drugs.
Figure 1.
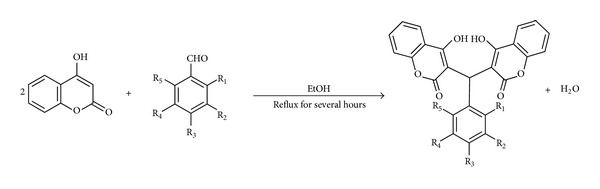
Synthesis of dicoumarol derivatives via condensation of 4-hydroxycoumarin with ortho-, meta-, and para-substituted benzaldehydes.
2. Materials and Methods
2.1. Synthesis of Dicoumarols
All the chemicals and reagents were of analytical grade and were purchased from Sigma-Aldrich. All solvents used in the study were purified using standard methodology except for those which were reagent grade. The synthesis of dicoumarol and its derivatives was based on the previously described method [5]. Dicoumarol was synthesized by dissolving 4-hydroxycoumarin 1.00 g in 300 mL of boiling water; the solution was allowed to cool to 70°C and 10 mL of 40% aqueous formaldehyde was added with stirring. The mixture was then chilled. The crude product was then filtered off and washed well with water. The compound was obtained by recrystallization with ethanol.
A series of nineteen dicoumarols with various phenyl-based derivatives were synthesized by mixing 4-hydroxycoumarin with aromatic aldehydes containing different groups in ortho, meta, or para with the 2 : 1 ratio of molar equivalent (see Figures S1 and S2 in Supplementary Material available online at http://dx.doi.org/10.1155/2013/178649). The mixture was dissolved in ethanol. Each solution was refluxed until the solid began to precipitate. After cooling, the product was filtered and recrystallized with appropriate solvent. The purified compounds were identified by NMR. 1H NMR spectra were recorded on a Varian 400 MHz NMR spectrometer. The NMR samples were dissolved in deuterated chloroform (CDCl3) with tetramethylsilane (TMS) as an internal reference. The chemical shifts were assigned by a comparison with residue solvent protons.
2.2. Antibacterial Susceptibility Testing
The biological activity was undertaken by disk diffusion method [17]. The purified dicoumarol compounds were tested with gram-positive Staphylococcus aureus ATCC-6538 and Bacillus subtilis ATCC-6633, while gram-negative Escherichia coli ATCC-25922 and Klebsiella sp. TISTR-1843. A stock solution was prepared by dissolving 10 mg of the tested sample in 1000 μL of DMSO solvents. A 20 μL of each preparation was dropped into a disk of 6 mm in diameter. After incubation at 37°C for 24 hours, diameter of clear zone was measured. For positive control, penicillin G against S. aureus and E. coli and chloramphenicol against B. subtilis and Klebsiella sp. were used, while DMSO was used as negative control. The antibiotic susceptibility tests were undertaken twice.
2.3. Molecular Modeling
2.3.1. Comparative Modeling of a Protein Target from S. aureus
First, we used protein sequences from the three crystal structures human NQO1 (pdb 2F1O), nitroreductase (pdb 1OOQ), and azoreductase (pdb 2Z9C) from E. coli as primary information to identify homologous proteins from S. aureus. NCBI BLAST search for NAD(P)H-dependent flavodoxin-like proteins, a hypothetical target for dicoumarols, was performed through identified protein-encoding genes in the S. aureus genome. The resulting S. aureus flavoprotein that is showed to be the most similar to the reference protein sequences and complete sequence information was chosen as a putative dicoumarol-binding target protein for building a structure model. Based on the BLAST results, a comparative model of the S. aureus homodimeric protein was generated using the crystal structure of E. coli azoreductase as template. Sequence alignment and comparative modeling were carried out using Discovery Studio 2.5 (Accelrys Inc., San Diego, CA, USA). Subsequently, the best S. aureus protein structure with the lowest DOPE (the density optimisation potential energy) score was selected and subjected to structure validation using PROCHECK [18]. Then, energy minimization of the comparative protein structure was performed in the presence of dicoumarol and flavin mononucleotide (FMN) using AMBER 10 software package [19]. The structure of the protein-dicoumarol-FMN complex was prepared by superimposing the model onto the template structure. The force field parameters of FMN were taken from Lugsanangarm et al. [20], while those of dicoumarol were developed using the same protocol as described in the literature [20].
2.3.2. Molecular Docking
To evaluate the binding mode of dicoumarol with the surrounding residues of the S. aureus flavoprotein, the flexible docking was performed using AutoDock Vina [21]. 3D structures of all synthesized dicoumarol derivatives were built and optimized at the semiempirical AM1 level using the Gaussian 03 program [22]. For the target protein, we removed the dicoumarol molecule and all the hydrogen atoms of the energy-minimized comparative model, while the FMN molecule remained in the binding pocket of the receptor during the docking simulation. Gasteiger partial charges, atom types, and polar hydrogen were assigned to the protein and all ligands using AutoDock Tools (ADT) [23]. The torsional bonds of ligands were set free. The center of the 40 × 40 × 40 Å grid box was estimated from the dicoumarol position present in the crystal structure. The exhaustiveness parameter was set to 64. Top ten docked conformations from each calculation were ranked according to the binding energy and clustered on the basis of root-mean-square deviation (RMSD) of the ligand atoms. For each compound, the docked poses that adopt an orientation similar to that of the experimental X-ray structure (RMSD <3Å) were chosen as a representative bound conformation. Structure analysis and visualization were performed using VMD software [24].
2.3.3. D-QSAR Models
All the synthesized dicoumarol derivatives and their inhibition zone were used to construct CoMFA and CoMSIA models using the program SYBYL (Tripos International, Missouri, USA). The training set of 14 dicoumarol derivatives was selected on the basis of structural diversity and wide range of activities and the test set contains 5 compounds (Table 1). Model building of the compounds was already described in the previous section (molecular docking). Atomic charges of the compounds were assigned according to Gasteiger-Huckel method [25]. Structural alignment of the compounds was performed using the SYBYL automatic alignment feature. For CoMFA, steric and electrostatic fields of the compounds were automatically computed using the standard CoMFA method with default parameters. Analogously, the calculated CoMSIA fields are associated with five descriptors including steric, electrostatic, hydrophobic, hydrogen bond donor, and hydrogen bond acceptor. The partial least squares (PLS) analysis was carried out using the “leave-one-out” cross-validation protocol to choose the optimum number of components (ONC) that does not exceed one-third of the number of the training compounds but yields the highest cross-validated correlation coefficient (q 2). Subsequently, non-cross-validation analysis was performed with the same ONC to calculate the conventional correlation coefficient (r 2), standard error of estimation (SEE), and F values.
Table 1.
Reaction yield and antibacterial properties of dicoumarols.
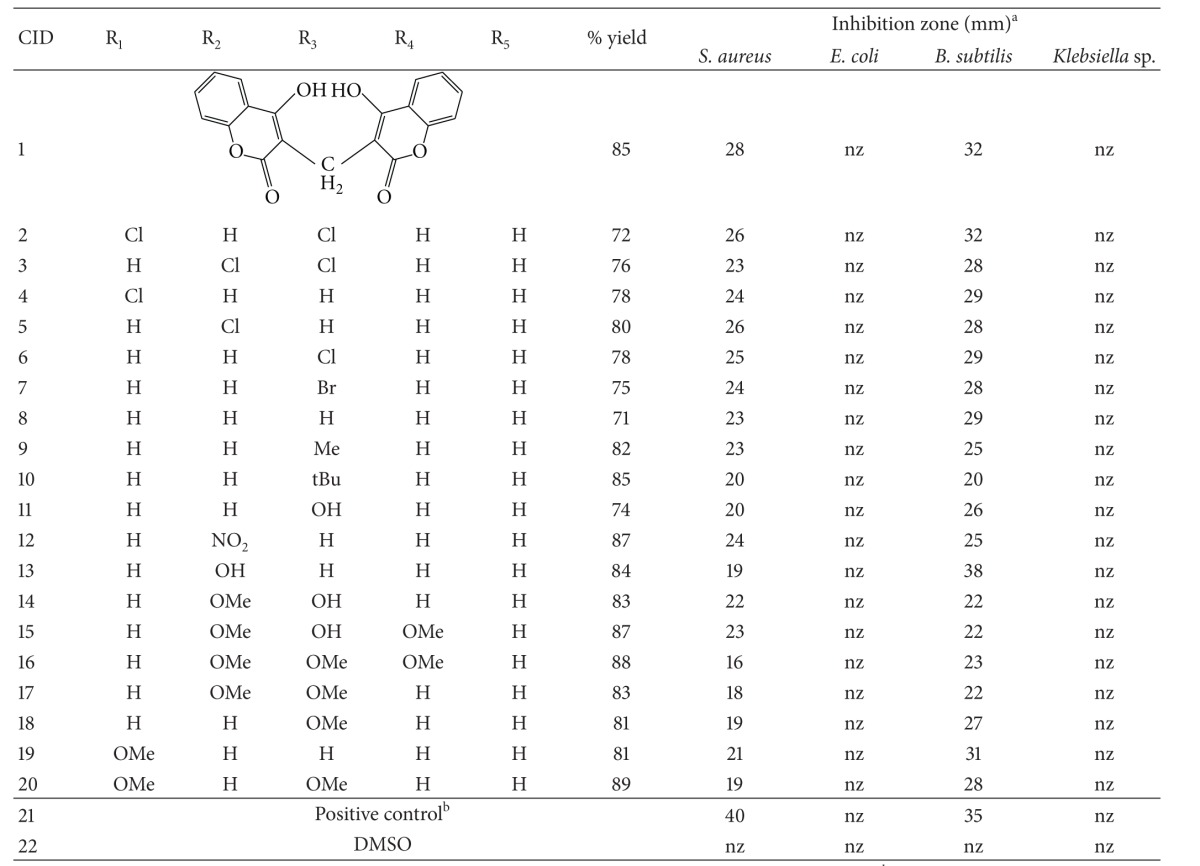
|
Note: athe antibiotic tests were undertaken twice. Average values are reported. nz: no inhibition zone; nf: not found. bPositive control is penicillin G for S. aureus and E. coli, while chloramphenicol was used for B. subtilis and Klebsiella sp.
3. Results and Discussion
3.1. Synthesis of Dicoumarols
Dicoumarol and nineteen dicoumarol derivatives were synthesized from the condensation reaction between two-mole equivalent of 4-hydroxycoumarin and one-mole equivalent of corresponding aldehydes. In preparation of dicoumarol, formaldehyde rapidly reacted with 4-hydroxycoumarin in hot water. The compound derivatives were from the condensation of 4-hydroxycoumarin with different substituted aromatic aldehydes in ethanol for 24 hours. This method is useful for aromatic aldehydes because the insoluble products were easily separated by filtration.
The synthesized compounds have an excellent yield. The yield of all dicoumarol derivatives was found to be in the range of 70–90% (Table 1). Most of dicoumarols were soluble in CDCl3; DMSO was used as a solvent for dicoumarols which were insoluble in CDCl3. For dicoumarol moiety, 2H integration of H-5 was shown as doublet around 7.80–8.10 ppm and 2H integration of H-6 was seen as triplet at 7.50–7.60 ppm. The overlapped 4H integration of H-7 and H-8 at 7.20–7.45 ppm was detected. The typical 1H integration of CH methylene bridge was observed in a range between 3.85 and 6.70 ppm depending on the R substitution at the bridge carbon. Characteristic spectroscopic data of the synthesized compounds are provided in the supplementary data.
3.2. Antibacterial Susceptibility Testing
Two gram-positive bacteria, S. aureus, and B. subtilis, and two gram-negative bacteria, E. coli and Klebsiella sp., were selected for the assay of cell-growth inhibition. Data of antibacterial effects of tested compounds are presented in Table 1. The results showed that all the synthesized compounds had no inhibitory activity against E. coli and Klebsiella sp. strains whereas their antibacterial effects were found for S. aureus and B. subtilis. The antibacterial activities of the dicoumarol derivatives against S. aureus are in a range of the clear zone from 16 to 26 mm. Although the antibacterial susceptibility was not great when compared to penicillin G (inhibition zone = 40 mm), the halide-substituted benzene ring of the derivatives (CID 2, 3, 4, 5, and 6) had comparable activity to the parent dicoumarol (CID 1). The derivative CID 16 had the lowest inhibitory activity. For B. subtilis, the results show that CID 13 exhibits the greatest antibacterial activity with an average clear zone of 38 mm. Again the derivatives containing halide group showed the activity comparable to the parent dicoumarol, especially CID 2 exhibiting the equivalent zone of the inhibition (32 mm). The tertiary butyl group substituent at para position (CID 10) showed the lowest activity that is 20 mm. It appears that the antibacterial effect of the dicoumarol derivatives against B. subtilis and S. aureus has a similar trend. The steric or bulky hydrophobic properties at the para position tend to decrease the inhibition zone of dicoumarol, whilst the halide-substituted benzene ring of the derivatives (CID 2, 3, 4, 5, and 6) tends to increase the activity.
The results are consistent with the previously observed inhibition of dicoumarols that act selectively against gram-positive microorganisms [10, 26]. One of the possible explanations is due to a permeability barrier to dicoumarol molecule because structure and properties of cell membranes between the two bacterial cells are different. The presence of outer membrane of peptidoglycan in E. coli may prevent the transport of dicoumarols into the cell. There is yet no evidence for the relation between molecular properties of the tested compounds and their transport across the membrane. Our results seem to be likely the case, but the study is currently limited to test such hypothesis.
Among the active compounds, the results show that the parent dicoumarol exhibits the high antibacterial activity with an average clear zone of 28 mm for S. aureus and of 32 mm for B. subtilis. In case of S. aureus, addition of the benzene ring at the methylene bridge position does not increase the activity. Analysis of substitution effect on the antibacterial activity against S. aureus revealed that the compounds containing halide and nitro groups exhibited the inhibition zone in a range of 24–26 mm, whereas those containing the methyl, methoxy, and hydroxyl groups had a narrower clear zone of 18–23 mm. This indicated that the presence of electron withdrawing group in benzene ring gave a more potent inhibitor compared to the substituted electron donating groups. Particularly, the antibacterial activity of the compound with three methoxy substitutions (CID 16) was the lowest among the active compounds. A fairly similar substitution effect on the activity against B. subtilis was observed, except for CID 13 (38 mm) and 19 (31 mm). Nevertheless, from the results, adding steric or bulky hydrophobic properties at the para position tends to diminish the antibacterial inhibition zone of both S. aureus and B. subtilis cells.
3.3. Exploring Putative Target and Binding Site for Dicoumarols
Next, we attempted to explain the observed decrease in the efficacy of derivatives with steric bulk at the para position of the phenyl ring in terms of the structure property and the binding of the dicoumarols. Initially, we looked for the potential dicoumarol target in DrugBank (http://www.drugbank.ca), a database containing some information on drug targets. In search for potential targets of dicoumarols, we found vitamin K epoxide reductase complex subunit 1, NAD(P)H dehydrogenase [quinone], quinone oxidoreductase, and cytochrome P450 2C9. Among these proteins, the 3D crystal structure is available only for a class of quinone or NAD(P)H-dependent oxidoreductase in complex with dicoumarol.
A further analysis of the NAD(P)H-dependent oxidoreductase proteins from S. aureus genome through NCBI database identified potential antibacterial targets including quinone reductase, nitroreductase, flavin reductase, and FMN oxidoreductase. The hit proteins from the search were subsequently screened to identify the protein target, of which the 3D structure is available as a template. It appears that an FMN-dependent NADH-azoreductase had similar sizes and homology suited for further investigation through a molecular modeling approach.
A comparative model of NAD(P)H-dependent flavoprotein from S. aureus (A9635) was constructed based on the 3D structure of dicoumarol-binding azoreductase (2Z9C) with 34% of sequence identity (Figure 2). The model is a homodimer of ternary complex containing the enzyme, FMN, and dicoumarol (Figure 3). Each monomer contains 206 out of 232 residues of the full-length protein. Dicoumarol is located at the dimer interface and interacts with active site residues of both chains.
Figure 2.
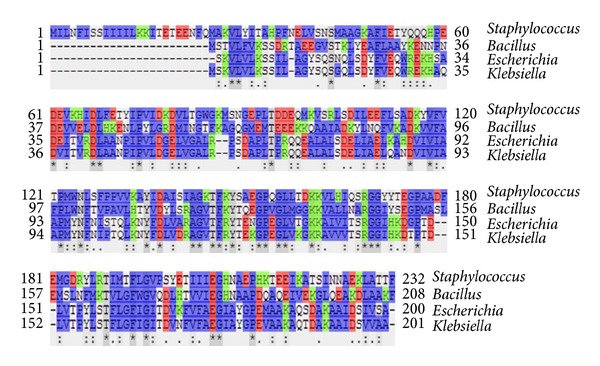
Multiple sequence alignment of azoreductase from Staphylococcus aureus, Bacillus subtilis, Escherichia coli, and Klebsiella sp. Amino acid residues are colored to indicate their similarity: blue = hydrophobic, red = acidic, and green = basic residues. For comparative model, the sequence identity with the template is of 34%.
Figure 3.
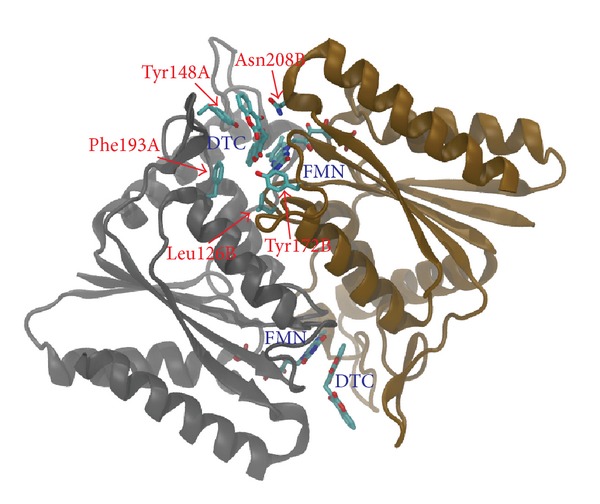
Comparative model of S. aureus azoreductase in complex with FMN and dicoumarol. The protein model is a homodimer composed of two ligand-binding sites located at the dimer interface. FMN, dicoumarol (DTC), and the conserved binding residues are shown in stick representation.
To further validate the quality of the homology model, dicoumarol was docked into the enzyme binding pocket. The results show that one of the ten energetically most favorable docking poses adopted ligand orientation similar to that of the comparative model of the ternary complex but differed slightly in the depth of the ligand position within the binding pocket (Figure 4(a)). The docking model revealed sandwich-stacking interactions of dicoumarol with the FMN cofactor and the residues Phe146A, Tyr148A, Phe193A, Gly170B, and Asn208B (“A” and “B” indicate the protein chain) which are mainly hydrophobic (Figure 4(c)). Particularly, one of coumarin rings stacks between the isoalloxazine ring of FMN and the sidechain of Tyr148A. The pi-pi stacking motif is similar to the binding mode of conserved residues found in the X-ray structures of human NQO1 quinone reductase and E. coli azoreductase. Thus, the conserved binding mode suggested that the S. aureus azoreductase might be a potential protein target for antiproliferative activity of dicoumarols.
Figure 4.
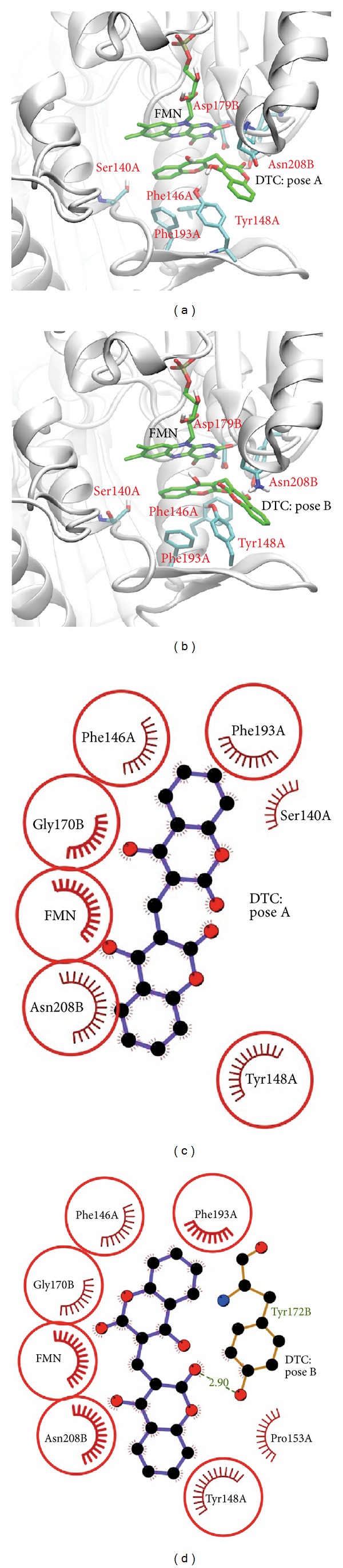
Docked orientations and interactions of dicoumarol in the enzyme binding sites. (a) and (b) compare the ligand poses A and poses B, respectively. The surrounding conserved residues, FMN, and dicoumarol molecules are in stick representation. (c) and (d) are schematic 2D diagrams of protein-ligand binding poses A and B, respectively. Spoked arcs represent residues making hydrophobic contacts with the ligand, whereas dashed lines represent hydrogen bonds between the atoms involved. The 2D representation was created using the program LigPlot [27].
Further inspection of docking results illustrated an interesting binding mode of dicoumarols. Given the structure of the binding site, the two coumarin rings possibly adopted two alternative orientations, denoted as pose A and pose B. As described in the previous paragraph, the ligand orientation of pose A is similar to that of the crystal structures. Pose B shows a very similar binding pose in a way that one of the two coumarin rings flips approximately 180 degrees with respect to pose A (Figure 4). The binding mode of pose B revealed a similar pi-pi stacking motif, with additional hydrogen bonding between the hydroxyl hydrogen of the flipped coumarin ring and Tyr172B sidechain. Considering molecular shape of the ligands, the two binding poses are similar. Ring flipping of the ligands has no obvious effect on the ring stacking interactions. This can be seen as the binding energies between the two poses are not significantly different (Table 2). At this point, we cannot distinguish between these possibilities; however, the previous study by Nolan et al. also observed similar docked poses of dicoumarols in the active site of human NQO1 [16].
Table 2.
Binding energy of dicoumarols.
| CID | Binding energy (kcal/mole) |
|
|---|---|---|
| Pose A | Pose B | |
| 1 | −9.3 | −9.3 |
| 2 | nf | −8.9 |
| 3 | −9.0 | −8.8 |
| 4 | −9.4 | −9.6 |
| 5 | −9.0 | −8.7 |
| 6 | −8.9 | nf |
| 7 | −8.5 | −8.5 |
| 8 | −8.8 | −8.8 |
| 9 | −8.1 | −9.5 |
| 10 | −8.4 | −8.5 |
| 11 | −9.6 | −9.3 |
| 12 | −9.0 | −8.8 |
| 13 | nf | −8.6 |
| 14 | −8.2 | −8.2 |
| 15 | −8.6 | −8.5 |
| 16 | nf | −8.4 |
| 17 | nf | −8.5 |
| 18 | −8.3 | nf |
| 19 | −8.2 | nf |
| 20 | −9.4 | −9.6 |
Note: nf: not found.
The docking studies of 19 dicoumarol derivatives suggested that the S. aureus azoreductase model possesses a large binding cavity and can accommodate larger ligands. For all docked conformations, these compounds possess a similar orientation of coumarin rings with RMSD <2Å (Figure 5). Most compounds bind in the binding pocket with a more shallow position than that previously observed for the docked poses of the parent dicoumarol. A shift in position of the derivatives may be due to steric hindrance of the bulky benzene ring. By comparing the resulting ligand poses, the majority of the binding modes of all derivatives have fallen into two binding poses A and B similar to docking results of the parent compound. Both binding poses share common interactions as the coumarin ring forms pi-pi stacking with the FMN cofactor and Tyr148A. Inspection of these two ligand-receptor complexes identified several contact residues including Lys86A, Ser140A, Ile141A, Phe146A, Tyr148A, Phe36B, His206B, and Asn208B. It should be noted that the interactions of these residues are predominantly hydrophobic.
Figure 5.
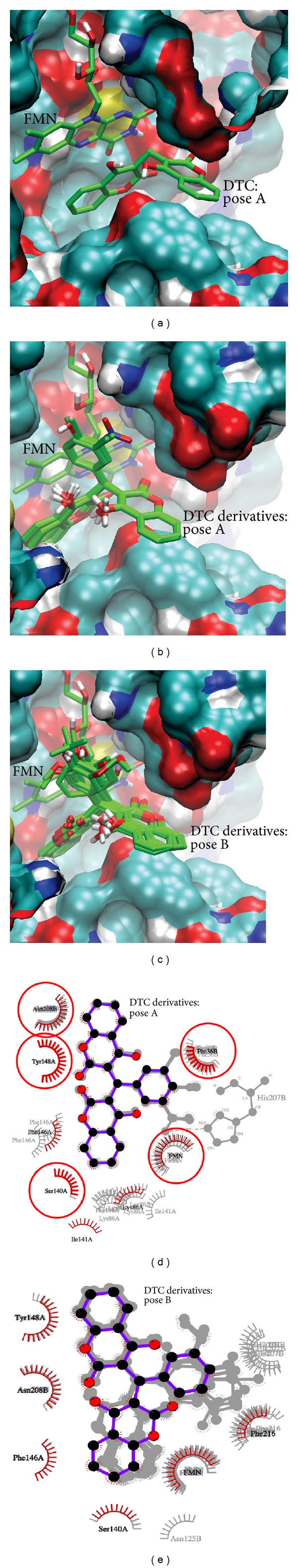
Docked orientations and interactions of dicoumarol derivatives in the enzyme binding site. (b) and (c) are the docking poses A and B of compound derivatives in comparison with pose A of dicoumarol (a) in the pocket (surface representation). (c) and (d) are schematic 2D diagrams of protein-ligand binding poses A and B, respectively.
Analysis of docking results suggests that the parent compound produced, on average, good binding energy when compared to its derivatives, except for CID 4, 11, and 20 (Table 2). The good agreement between the experimental activity and the docking supports the idea that adding a benzene ring at the methylenebis position could reduce antibacterial activity. However, no clear correlation could be established between the calculated binding energy and experimentally determined inhibition zone for the tested compounds.
It should be noted that the relation between the experimental work and molecular docking is speculative. There is no direct evidence that the derivatives indeed inhibit or are binding to the target enzyme. The results could be greatly strengthened if one can experimentally prove that this is truly the case. At the time being, we have limited resources for the validation.
3.4. Analysis of 3D-QSAR Results
In an attempt to establish a relationship between the structure and activities, we performed CoMFA and CoMSIA analysis using the inhibition zone of S. aureus. Statistical results of CoMFA and CoMSIA studies are summarized in Table 3. For the CoMFA model, PLS analysis showed a q 2 value of 0.694 with 2 components, r 2 value of 0.894 with SEE of 0.929, and F value of 46.3. The steric and electrostatic contributions were 59.2% and 40.8%, respectively. This indicated that the steric properties of the substituents have more impact on the antibacterial activity than the electrostatic properties.
Table 3.
Statistical results of CoMFA and CoMSIA models.
| Models | Descriptors | Statistical parameters | |||||
|---|---|---|---|---|---|---|---|
| q 2 | r 2 | N | SEE | F | Field contribution (%) | ||
| CoMFA | S/E | 0.694 | 0.894 | 2 | 0.929 | 46.3 | 59.2/40.8 |
| CoMSIA | S/E/H/D/A | 0.683 | 0.977 | 4 | 0.479 | 94.8 | 10.3/25.6/19.9/31.7/9.5 |
S: steric field; E: electrostatic field; H: hydrophobic field; D: donor; A: acceptor; q 2: cross-validated correlation coefficient; r 2: non-cross-validated correlation coefficient; N: optimal number of components; SEE: standard error of estimate; F: F test.
The PLS results of CoMSIA analysis showed a q 2 of 0.683 with an optimized component number of 4. The non-cross-validation analysis of CoMSIA models gave a high r 2 value of 0.977 with a SEE of 0.479 and F value of 94.8. The predominant field contributions from CoMSIA were electrostatic (25.6%) and hydrogen bond donor (31.7%), implying the importance of the electrostatic and hydrogen bond donor of the substituent for the activity. The other contributions, steric (10.3%), hydrophobic (19.9%), and the hydrogen bond acceptor (9.5%), gave minor effects.
The predicted inhibition zone versus their experimental data of training set and test set compounds are illustrated in Figure 6. Although both models gave a fairly good quality of the activity prediction with q 2 > 0.6 for the training set, the predictive quality of the test set is not great. An average deviation between the predicted and experimental inhibition zone was about 4 mm (Table 4). Particularly, the activities of the two CID, 2 and 18, were poorly predicted. Compound 2 has the highest value in the set, whereas compound 18 exhibits almost the lowest activity.
Figure 6.
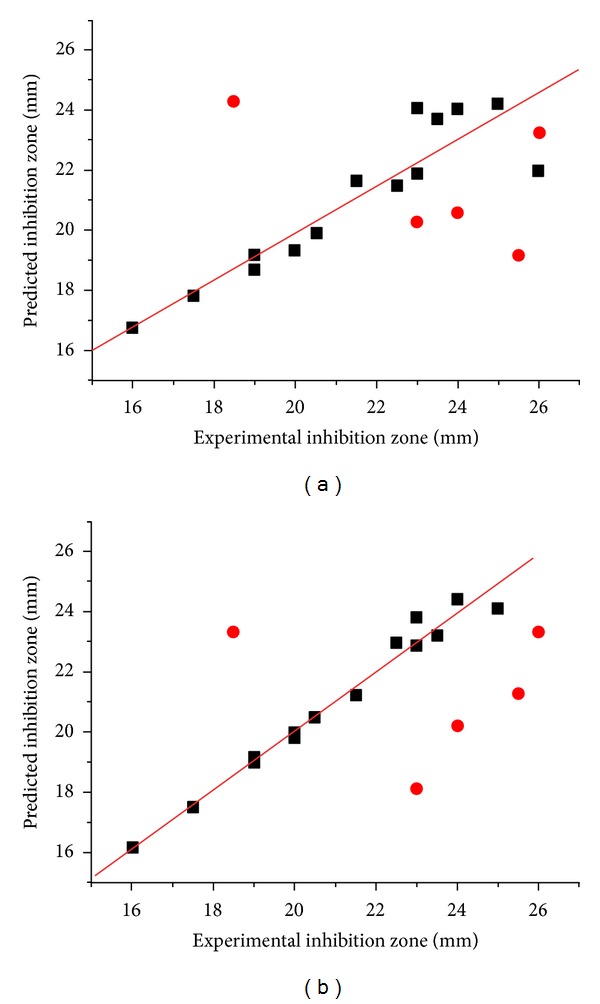
Plots between the experimental and predicted biological activities of training (filled square) and test set (filled circle): (a) CoMFA plot and (b) CoMSIA plot.
Table 4.
Experimental and predicted activities of test set compounds.
| CID | Inhibition zone (mm) | CoMFA | CoMSIA | ||
|---|---|---|---|---|---|
| Predicted (mm) | Residual (mm) | Predicted (mm) | Residual (mm) | ||
| 2 | 26 | 19 | 7 | 21 | 5 |
| 4 | 24 | 21 | 3 | 20 | 4 |
| 5 | 26 | 23 | 3 | 23 | 3 |
| 15 | 23 | 20 | 3 | 18 | 5 |
| 18 | 19 | 24 | 5 | 23 | 4 |
|
| |||||
| Avg | 4 | Avg | 4 | ||
The effects of field contributions are illustrated by CoMFA and CoMSIA contour maps (Figures 7(a)-7(b)). Our interpretation is focused on the effects of substituted phenyl ring to the antibacterial activity. For CoMFA, a green contour was found near the plane of substituted phenyl ring of compound, while a yellow contour was located in the vicinity of the R3 substituent. This indicates that the bulky substituents at the para position were unfavorable and might have negative effect on the activity of the compounds. Therefore, the activity of the CID 10, 16, 17, 18, and 20 is relatively low. For electrostatic maps, a red contour was found around the R1-position. This corresponds well with the –OMe substitution at ortho position of CID 19 and 20 with an electron-donating group and hence these two compounds exhibit moderate and low activities, respectively.
Figure 7.
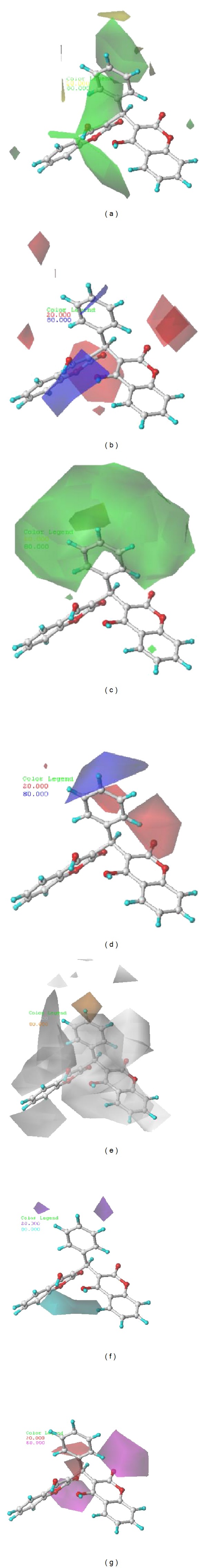
The stdev*coeff. contour maps. (a) and (b) represent CoMFA steric and electrostatic, respectively. (c) to (f) represent CoMSIA steric, electrostatic, hydrophobic, hydrogen bond donor, and hydrogen bond acceptor, respectively. Steric field is represented by green and yellow contour maps. Electrostatic field is represented by blue and red contour maps. Hydrophobic field is represented by orange and white contour maps. Hydrogen bond donor field is represented by cyan and purple contour maps. Hydrogen bond acceptor field is represented by magenta and red contour maps. All the contours represented the default 80% and 20% level contributions for favorable and unfavorable regions, respectively.
For CoMSIA contour maps, steric and electrostatic are similar to those of CoMFA as shown by a yellow contour at the para position (inside a big green contour) and a red contour at the R1-position (Figures 7(c)-7(d)). Hydrophobic contour map from CoMSIA is shown in Figure 7(e). An orange contour found at the R3-position suggests that a hydrophobic substituent at this site is expected to improve the activity. For hydrogen bond donor contour maps (Figure 7(f)), small purple contour located near the R2 (or R4) substituent indicated that the position requires the hydrogen bond acceptor substituent. A magenta contour for hydrogen bond acceptor group suggested that the R1 substituent is in favor of hydrogen bond acceptor (Figure 7(g)).
4. Conclusion
We describe the synthesis of several 3,3′-phenylmethylene-bis-4-hydroxycoumarins containing ortho, meta, and para substitutions on the benzene ring and their antibacterial activity against S. aureus, B. subtilis, E. coli, and Klebsiella sp. The selectivity of these compounds for gram-positive bacteria is clearly demonstrated. The compounds are reasonable, although not great, inhibitors of the growth of the gram-positive bacteria compared to the parent dicoumarol compound. For most compounds, the substitutions of bulky benzene group on the methylenebis position do not significantly increase in inhibitory activity. This finding is broadly explained by the predicted poorer docked structure of the derivatives to a homology model of S. aureus flavoprotein. The QSAR results illustrate a relationship between the antibacterial activity and dicoumarol derivatives. Both CoMFA and CoMSIA models provided chemical and structural information insight into the effects of steric, electrostatic, hydrophobic, hydrogen-bond donor, and acceptor fields around the substituted benzene ring of dicoumarol on biological activity. The findings of the study provide a better understanding of the inhibition mechanism of dicoumarols and may be useful guiding structural design of more effective antibacterial agents.
Supplementary Material
All spectroscopic evidences and methodology for synthesis of twenty compounds were shown in this supplementary material. The 1H NMR spectra of the twenty synthesized compounds were obtained on a Varian spectrometer that was operated at 400 MHz of proton frequency. The NMR samples were dissolved in CDCl3 with tetramethylsilane (TMS) as an internal reference or DMSO-d6. For compound 1 was synthesized by dissolving 4-hydroxycoumarin 1.00 g (2.97 mmol) in 300 mL of boiling water, the solution was allowed to cool to 70°C and 10 mL of 40% aqueous formaldehyde was added with stirring. The mixture was then chilled, the crude product was filtered off and washed well with water, dried and recrystallized with EtOH.
A series of nineteen dicoumarols (compound 2-20) with various phenyl derivatives were synthesized by mixing 4-hydroxycoumarin (15 mmol) and selected aromatic aldehydes (7.5 mmol) with different substituents at ortho, meta or para (2:1 ratio of molar equivalent). The mixture was dissolved in EtOH 45 mL. Each solution was refluxed approximately 24 hours until the solid began to precipitate. After cooling, the product was filtered and recrystallized with EtOH. The purified compounds were identified by NMR spectrometer.
Acknowledgments
The authors thank Dr. Somsak Pianwanit for guiding QSAR method and Dr. T. Palaga for helpful suggestions. This work is supported by the National Research University Project of CHE (HR1155A), the 90th Anniversary of Chulalongkorn University Fund, Ratchadaphiseksomphot Endowment Fund (RES560530217-HR), and the 72nd anniversary of H.M. King Bhumibol Adulyadej. National Electronics and Computer Technology Center (NECTEC), Computational Chemistry Unit Cell (CCUC), and The Thai Government Stimulus Package 2 (TKK2555) are gratefully acknowledged for supporting computational facilities.
Abbreviations
- NAD(P)H:
The reduced form of nicotinamide adenine dinucleotide (phosphate)
- PDB:
Protein data bank
- FMN:
Flavin mononucleotide
- NQO1:
Quinone oxidoreductase 1
- QSAR:
Quantitative structure-activity relationship
- NCBI:
National Center for Biotechnology Information
- BLAST:
Basic local alignment search tool
- DMSO:
Dimethyl sulfoxide
- CoMFA:
Comparative molecular field analysis
- CoMSIA:
Comparative molecular similarity indices analysis.
References
- 1.Stahmann MA, Huebner CF, Link KP. Studies on the hemorrhagic sweet clover disease. V. Identification and synthesis of the hemorrhagic agent. The Journal of Biological Chemistry. 1941;138(2):513–527. [Google Scholar]
- 2.Holbrook AM, Pereira JA, Labiris R, et al. Systematic overview of warfarin and its drug and food interactions. Archives of Internal Medicine. 2005;165(10):1095–1106. doi: 10.1001/archinte.165.10.1095. [DOI] [PubMed] [Google Scholar]
- 3.Adami E, Uberti EM, Turba C. Experimental and statistical data on the analgesic action of 4-hydroxycoumarin. Archivio Italiano di Scienze Farmacologiche. 1959;9(1):61–69. [PubMed] [Google Scholar]
- 4.Chohan ZH, Shaikh AU, Rauf A, Supuran CT. Antibacterial, antifungal and cytotoxic properties of novel N-substituted sulfonamides from 4-hydroxycoumarin. Journal of Enzyme Inhibition and Medicinal Chemistry. 2006;21(6):741–748. doi: 10.1080/14756360600810340. [DOI] [PubMed] [Google Scholar]
- 5.Hamdi N, Puerta MC, Valerga P. Synthesis, structure, antimicrobial and antioxidant investigations of dicoumarol and related compounds. European Journal of Medicinal Chemistry. 2008;43(11):2541–2548. doi: 10.1016/j.ejmech.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 6.Luchini AC, Rodrigues-Orsi P, Cestari SH, et al. Intestinal anti-inflammatory activity of coumarin and 4-hydroxycoumarin in the trinitrobenzenesulphonic acid model of rat colitis. Biological and Pharmaceutical Bulletin. 2008;31(7):1343–1350. doi: 10.1248/bpb.31.1343. [DOI] [PubMed] [Google Scholar]
- 7.Mahajan DH, Pannecouque C, de Clercq E, Chikhalia KH. Synthesis and studies of new 2-(coumarin-4-yloxy)-4,6-(substituted)-s- triazine derivatives as potential anti-HIV agents. Archiv der Pharmazie. 2009;342(5):281–290. doi: 10.1002/ardp.200800149. [DOI] [PubMed] [Google Scholar]
- 8.Stern P, Dezelic M, Kosak R. Analgesic & antipyretic effects of vitamin K & dicumarol with special reference to 4-hydroxycoumarin. Naunyn-Schmiedebergs Archiv für Experimentelle Pathologie und Pharmakologie. 1957;232(1):356–359. [PubMed] [Google Scholar]
- 9.Velasco-Velázquez MA, Agramonte-Hevia J, Barrera D, et al. 4-hydroxycoumarin disorganizes the actin cytoskeleton in B16-F10 melanoma cells but not in B82 fibroblasts, decreasing their adhesion to extracellular matrix proteins and motility. Cancer Letters. 2003;198(2):179–186. doi: 10.1016/s0304-3835(03)00333-1. [DOI] [PubMed] [Google Scholar]
- 10.Goth A. The antibacterial properties of dicumarol. Science. 1945;101(2624, article 382) doi: 10.1126/science.101.2624.383. [DOI] [PubMed] [Google Scholar]
- 11.Dadák V, Hoďák K. Some relations between the structure and the antibacterial activity of natural coumarins. Experientia. 1966;22(1):38–39. doi: 10.1007/BF01897757. [DOI] [PubMed] [Google Scholar]
- 12.Cresteil T, Jaiswal AK. High levels of expression of the NAD(P)H: quinone oxidoreductase (NQO1) gene in tumor cells compared to normal cells of the same origin. Biochemical Pharmacology. 1991;42(5):1021–1027. doi: 10.1016/0006-2952(91)90284-c. [DOI] [PubMed] [Google Scholar]
- 13.Asher G, Dym O, Tsvetkov P, Adler J, Shaul Y. The crystal structure of NAD(P)H quinone oxidoreductase 1 in complex with its potent inhibitor dicoumarol. Biochemistry. 2006;45(20):6372–6378. doi: 10.1021/bi0600087. [DOI] [PubMed] [Google Scholar]
- 14.Johansson E, Parkinson GN, Denny WA, Neidle S. Studies on the nitroreductase prodrug-activating system. Crystal structures of complexes with the inhibitor dicoumarol and dinitrobenzamide prodrugs and of the enzyme active form. Journal of Medicinal Chemistry. 2003;46(19):4009–4020. doi: 10.1021/jm030843b. [DOI] [PubMed] [Google Scholar]
- 15.Ito K, Nakanishi M, Lee W-C, et al. Expansion of substrate specificity and catalytic mechanism of azoreductase by X-ray crystallography and site-directed mutagenesis. Journal of Biological Chemistry. 2008;283(20):13889–13896. doi: 10.1074/jbc.M710070200. [DOI] [PubMed] [Google Scholar]
- 16.Nolan KA, Doncaster JR, Dunstan MS, et al. Synthesis and biological evaluation of coumarin-based inhibitors of NAD(P)H: quinone oxidoreductase-1 (NQO1) Journal of Medicinal Chemistry. 2009;52(22):7142–7156. doi: 10.1021/jm9011609. [DOI] [PubMed] [Google Scholar]
- 17.Cruickshank RD, Duguid JP, Marmion BP, Swain RHA. Medical Microbiology. 12th edition. Vol. 2. Edinburgh, Scotland: Churchill Livingstone; 1975. (The Practice of Medical Microbiology). [Google Scholar]
- 18.Laskowski RA, Rullmann JAC, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. Journal of Biomolecular NMR. 1996;8(4):477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 19.Case DA, TAD, Cheatham TE, III, Simmerling CL, et al. AMBER 10. San Francisco, Calif, USA: University of California, San Francisco; 2008. [Google Scholar]
- 20.Lugsanangarm K, Pianwanit S, Kokpol S, Tanaka F. Homology modelling and molecular dynamics simulations of wild type and mutated flavodoxins from Desulfovibrio vulgaris (Miyazaki F): insight into FMN-apoprotein interactions. Molecular Simulation. 2011;37(14):1164–1178. [Google Scholar]
- 21.Trott O, Olson AJ. Software news and update AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frisch MJ, GWT, Schlegel HB, Scuseria GE, et al. Gaussian 03. Pittsburgh, Pa, USA: Gaussian; 2003. [Google Scholar]
- 23.Morris GM, Ruth H, Lindstrom W, et al. Software news and updates AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. Journal of Computational Chemistry. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. Journal of Molecular Graphics. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 25.Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity-a rapid access to atomic charges. Tetrahedron. 1980;36(22):3219–3228. [Google Scholar]
- 26.Smyth T, Ramachandran VN, Smyth WF. A study of the antimicrobial activity of selected naturally occurring and synthetic coumarins. International Journal of Antimicrobial Agents. 2009;33(5):421–426. doi: 10.1016/j.ijantimicag.2008.10.022. [DOI] [PubMed] [Google Scholar]
- 27.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Engineering. 1995;8(2):127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
All spectroscopic evidences and methodology for synthesis of twenty compounds were shown in this supplementary material. The 1H NMR spectra of the twenty synthesized compounds were obtained on a Varian spectrometer that was operated at 400 MHz of proton frequency. The NMR samples were dissolved in CDCl3 with tetramethylsilane (TMS) as an internal reference or DMSO-d6. For compound 1 was synthesized by dissolving 4-hydroxycoumarin 1.00 g (2.97 mmol) in 300 mL of boiling water, the solution was allowed to cool to 70°C and 10 mL of 40% aqueous formaldehyde was added with stirring. The mixture was then chilled, the crude product was filtered off and washed well with water, dried and recrystallized with EtOH.
A series of nineteen dicoumarols (compound 2-20) with various phenyl derivatives were synthesized by mixing 4-hydroxycoumarin (15 mmol) and selected aromatic aldehydes (7.5 mmol) with different substituents at ortho, meta or para (2:1 ratio of molar equivalent). The mixture was dissolved in EtOH 45 mL. Each solution was refluxed approximately 24 hours until the solid began to precipitate. After cooling, the product was filtered and recrystallized with EtOH. The purified compounds were identified by NMR spectrometer.