

Abstract
The voltage-gated Na channel isoform 1.5 (NaV1.5) is the pore forming α-subunit of the voltage-gated cardiac Na channel, which is responsible for the initiation and propagation of cardiac action potentials. Mutations in the SCN5A gene encoding NaV1.5 have been linked to changes in the Na current leading to a variety of arrhythmogenic phenotypes, and alterations in the NaV1.5 expression level, Na current density, and/or gating have been observed in acquired cardiac disorders, including heart failure. The precise mechanisms underlying these abnormalities have not been fully elucidated. However, several recent studies have made it clear that NaV1.5 forms a macromolecular complex with a number of proteins that modulate its expression levels, localization, and gating and is the target of extensive post-translational modifications, which may also influence all these properties. We review here the molecular aspects of cardiac Na channel regulation and their functional consequences. In particular, we focus on the molecular and functional aspects of Na channel phosphorylation by the Ca/calmodulin-dependent protein kinase II, which is hyperactive in heart failure and has been causally linked to cardiac arrhythmia. Understanding the mechanisms of altered NaV1.5 expression and function is crucial for gaining insight into arrhythmogenesis and developing novel therapeutic strategies.
Keywords: Na channel, CaMKII, phosphorylation, heart failure, arrhythmia
the cardiac Na channel, encoded by the gene SCN5A, is activated at negative membrane voltages and is responsible for the generation of the rapid upstroke of the cardiac action potential (AP). The α-subunit of the voltage-gated cardiac Na channel isoform (NaV1.5) forms a macromolecular complex through interactions with many accessory proteins (75), regulatory proteins (132), and other ion channels (77, 105) that alter its expression, trafficking, localization, and gating [reviewed in Abriel (1)] (Fig. 1). Recent evidence also suggests that multiple pools of NaV1.5 may exist in cardiomyocytes, and these channel populations may be differentially regulated (94, 109). Whereas most of the Na channels inactivate rapidly (within a few milliseconds), prolonged membrane depolarization during the AP plateau causes slow/intermediate (hundreds of milliseconds) inactivation of some channels, which recover to a closed conformation as the cell membrane repolarizes during diastole. In physiological situations, Na channel activation and inactivation properties are tightly regulated, thus maintaining cardiac excitability and ensuring propagation of the electrical impulse. However, alterations in channel function (e.g., due to gene mutations or post-translational modifications) may profoundly affect cardiac electrophysiology and arrhythmogenesis. Herein, we review post-translational regulation of NaV1.5. While we describe the molecular and functional aspects of post-translational regulation by PKA, PKC, oxidative stress, Ca, and CaM, we focus on Na channel phosphorylation by Ca/CaM-dependent protein kinase II (CaMKII). We further use CaMKII modulation as a model to understand how Na channel dysfunction may be implicated in acquired arrhythmias.
Fig. 1.
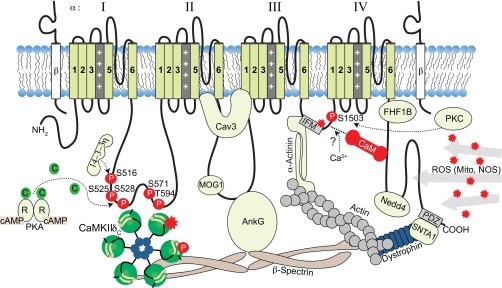
Na channel as a macromolecular signaling complex. AnKG, ankyrin-G; FHF1B, fibrobast growth factor homologous factor 1; MOG1, multicopy suppressor of Gsp1; Nedd4, neural precursor cell expressed developmentally downregulated protein 4; C, catalytic; R, regulatory; P, phosphorylation; Cav3, caveolin-3; IFM, Ile-Phe-Met; ROS, reactive oxygen species; Mito, mitochondria; NOS, nitric oxide synthase; PDZ, postsynaptic density protein/Drosophila disk large tumor suppressor/zonula occludens-1; SNTA1, α1-syntrophin.
Na Channel Alterations in Inherited SCN5A Channelopathies and Acquired Diseases
A number of mutations in the gene SCN5A have been described, which cause enhanced or reduced channel function and are linked to cardiac disorders including long QT syndrome (LQTS), Brugada syndrome (BrS), conduction defects, sinus dysfunction, and familial atrial fibrillation (AF) (101). These have tremendously aided our understanding of the regulation of expression, localization, and function of NaV1.5 and suggest that its role is not limited to AP initiation and conduction, but it may also have more subtle functions and be involved in repolarization abnormalities. In addition, mutations disrupting Na channel protein interactions (within the macromolecular signaling complex modulating the localization and biophysical properties of NaV1.5) have also been associated with LQTS and BrS [reviewed by Wilde and Brugada (132)].
These channelopathies provided useful models for understanding the molecular mechanisms whereby pharmacological interventions or disease states provoke cardiac arrhythmias. For example, LQT3 syndrome is caused by gain-of-function defects in Na channel inactivation, which lead to delays in ventricular repolarization that result in the prolongation of the QT interval. Numerous mutations induce a sustained (late Na current, INa,L) component of inward current (∼1% of the peak current) throughout the depolarization period. The role of this small current in prolonging repolarization and generating cardiac arrhythmias has been validated in quantitative models of the AP (27) and is further illustrated in the next subsection.
On the other hand, BrS is associated with loss-of-function mutations and is characterized by normal QT intervals and ST segment elevation in the right precordial leads, which mimics ischemic ECG manifestations and predisposition to malignant ventricular tachyarrhythmias. The pathophysiological mechanism is thought to involve heterogeneous loss of the AP dome between the epicardium and endocardium, which creates a transmural voltage gradient during ST segment (36, 137) or conduction delay in the right ventricular outflow tract (76). SCN5A mutations have also been reported leading to combinations of these phenotypes, known as overlap syndromes (100). Intriguingly, a single human mutation at 1795InsD in SCN5A is linked to simultaneous LQT3 and BrS features (124). Mutation-induced slowing of INa recovery from inactivation, increase in intermediate inactivation, and hyperpolarizing shift in channel availability all reduce INa (loss of function) and underlie the BrS-like symptoms (slow conduction) of patients at higher heart rates. In addition, this 1795InsD mutation causes an increase in INa,L. At slow heart rates where INa recovery from inactivation may be more complete and APs are intrinsically longer, INa,L-dependent AP prolongation is responsible for LQTS (28).
Emerging evidence also suggests that Na channel gating alterations are involved in widespread acquired diseases, e.g., drug-induced LQTS, cardiac ischemia, heart failure (HF), and AF. For example, CaMKII, which is upregulated and more active in HF (5), has been shown to regulate Na channel gating and almost exactly phenocopies the spectrum of gating changes seen for the combined LQTS/BrS phenotype due to 1795InsD (see Fig. 2). Also, the generation of reactive oxygen species (ROS) in conditions of increased oxidative stress (such as during ischemia, HF, and AF) correlates with CaMKII-mediated reduced INa availability, leading to impairment of cardiac conduction following myocardial infarction (25, 46) and enhancement of late INa and consequent arrhythmias (128). In this context, post-translational modifications of NaV1.5 may constitute an acquired arrhythmogenic Na channel defect that could affect millions of people (as compared with the relatively small number of individuals affected by the mechanistically informative genetic Na channel mutations).
Fig. 2.

Effects of CaMKIIδC hyperactivity (or mutation 1795insD) on Na current (INa) gating. SSI and activation voltage dependence (A), entry of Na channels into intermediate inactivation (B), recovery from inactivation (C), and late INa (INa,L; D) are assessed with the voltage clamp protocols shown in insets in control (solid lines) and CaMKIIδC overexpression/1795insD mutation (dashed lines).
Late Na Current
The late Na current is carried by a fraction of Na channels that remain active throughout the AP plateau, rather than quickly inactivating as the majority of Na channels do. These channels undergo special modes of gating (burst and late-scattered openings) that confer INa,L slow inactivation and voltage dependence similar to the fast component (71). INa,L is increased in a number of LQT3 mutations and acquired diseases, including HF, where its inactivation kinetics are even slower than normal hearts (69, 71). INa,L alterations may be acute and result from post-translational modification of NaV channels, or chronic, and involve changes in expression of NaV and its modulatory proteins.
The molecular details of INa,L and its modulation are not fully understood. A significant (∼50%) contribution of noncardiac Na channel isoforms to INa,L has been reported in healthy canine myocytes (15), and it has been proposed that its enhancement in pathological conditions could be due to an increase in neuronal Na channel isoforms with higher fractional INa,L (135). In fact, specific NaV1.8 inhibition in mice and rabbits is antiarrhythmic (138). Modulation of Na channels could also underlie augmentation of INa,L in disease. For example, Maltsev et al. (66) showed that the β1-subunit (but not β2) modulates INa,L produced by heterologously expressed NaV1.5 by slowing its decay and increasing its amplitude relative to the peak current. Recently, they showed that reduced expression of either β1- or β2-subunits caused a significant loss of function or gain of function of INa,L, respectively, in both normal and failing canine myocytes (78). However, there are contrasting results on the modulation of INa,L by β-subunits in heterologous and native systems and whether they modulate similarly cardiac and noncardiac isoforms. Furthermore, interaction with the cytoskeleton, regulatory kinases (as discussed here) and phosphatases, trafficking proteins, and extracellular matrix proteins may all modulate INa,L in different pathological conditions (71).
Although small in magnitude compared with the peak Na current (∼1%), INa,L may disrupt the delicate balance between depolarizing and repolarizing currents during the AP plateau, leading to AP prolongation and increasing the propensity to ventricular arrhythmia (79). Indeed, a pathological increase in INa,L has been associated with arrhythmogenic phenotypes in inherited and acquired cardiac diseases including LQT3, HF, myocardial infarction, and AF. Physiological INa,L has been shown to contribute to reverse-rate dependence of AP duration (APD) and beat-to-beat variability caused by K current inhibitors in rabbit hearts (134). It has also been suggested that its inhibition may diminish the rate-dependent prolongation of the APD/QT interval caused by either drugs or pathological conditions that decrease rapidly activating K current and may decrease the occurrence of slow rate- or pause-triggered cardiac arrhythmias. Conversely, INa,L enhancement in HF has been predicted to exacerbate reverse frequency dependence of APD (115), which may increase the proarrhythmic risk. In addition, the increased Na influx via INa,L may partly be responsible for altered global Na and Ca homeostasis in cardiac hypertrophy (29) and failure (111), although the precise mechanism (longer APD vs. higher intracellular Na concentration) and the quantitative aspects of INa,L contribution are debatable (38, 128). In fact, a tetrodotoxin-sensitive increase in intracellular Na concentration has been reported in HF even in resting cardiac myocytes (where the Na channels are supposedly closed) (31).
Post-translational Modulation of NaV1.5
Regulation by PKA and caveolin-3 complex.
Initial reports on β-adrenergic regulation of cardiac INa were controversial. Several groups reported a decrease of cardiac INa in response to PKA activation (22). However, subsequent studies convincingly demonstrated that PKA potentiates INa, possibly involving both a fast saturable and a slow unsaturable component. The fast component involves direct channel phosphorylation events regulating the kinetics and voltage dependence of channel gating. The most consistent fast effects were a negative shift in both steady-state inactivation (SSI) and activation and slowed recovery from inactivation (less available but more active channels) (141). As proposed by Zhou et al. (141), the negative shift in inactivation and use of more depolarized holding potentials (where Na channels may not have been fully available) could explain the discrepancies in early results. Subsequent studies that used more negative holding potentials (to ensure full availability of Na channels) observed potentiation of INa. These observations may have important physiological consequences for the increase in cardiac conduction velocity observed with sympathetic stimulation (where INa is thought to be increased) and the genesis of re-entrant arrhythmias in ischemic myocardium, which is often associated with depolarized diastolic membrane potentials (86).
The slow component of PKA-dependent INa potentiation is due to enhanced trafficking and insertion of additional functional channels into the membrane, as revealed in a series of studies with recombinant channels expressed in oocytes by the Murray group (41, 140, 141). Although it was demonstrated that the I–II cytoplasmic linker loop is critical for this effect (141), mutation of five putative PKA sites in this loop (including S483/S571/S593) failed to abolish PKA-dependent potentiation of rat INa (107, 140). Murphy and colleagues identified two sites on rat NaV1.5 at positions S526 (525 in human) and S529 (528 in human) that are phosphorylated by PKA both in vitro and in vivo (83) (Fig. 1). PKA potentiation of INa was subsequently determined to be dependent on PKA phosphorylation of S525 and S528 and the presence of three endoplasmic reticulum (ER) retention signals on the I–II cytoplasmic linker loop (140). A S525A/S528A double phosphomutant or mutation of all three putative ER retention signals (with Arg-X-Arg at 533–535 playing the most prominent role) abolished PKA effects on INa (140). This favors a model whereby PKA phosphorylation of S525 and S528 (and possibly other proteins) recruits binding of protein partners that mask the ER retention signals of intracellular channel reserves to facilitate forward trafficking of channels to the membrane.
This concept of intracellular storage pools of Na channels was originally proposed by Catterall and colleagues (106) for neurons (where PKA has opposite effects). It is supported by studies in mammalian cells, including dog cardiomyocytes, which demonstrated that intracellular pools of NaV1.5 exist in the ER and subsarcolemmal space that can be recruited to the membrane in response to PKA activation (41, 142). The intracellular pool hypothesis is further substantiated by the observation that NaV1.5 and β-adrenergic receptors colocalize to caveolin domains that participate in membrane trafficking (139). Indeed, it has been shown that NaV1.5 associates with caveolin 3 (Cav3, Fig. 1) (139), and this interaction facilitates direct (PKA independent) Gαs protein stimulation of cardiac INa (62, 89). Furthermore, mutations in Cav3 that disrupt its interaction with NaV1.5 are reported to result in increased INa,L, which is the basis for arrhythmogenic LQT9 (123).
Regulation by PKC and glycerol-3-phosphate dehydrogenase 1-like protein complex.
Analogous to neuronal NaV1.2 channels, PKC decreases cardiac INa in native and heterologous cell systems (98). This INa decrease is dependent on voltage and phosphorylation of S1505 (rodent; S1503 human) in the NaV1.5 III–IV inactivation loop (Fig. 1) (99). In cell attached current recordings of Chinese hamster cells expressing rat NaV1.5, the PKC activator 1-oleoyl-2-acetylglycerol (10 μM) resulted in a voltage-dependent decrease in INa and a 15mV negative shift in SSI that were reversed by a PKC inhibitor peptide or mutation of S1505 to a nonphosphorylatable alanine (99). The combined effects of a decrease in maximal INa and a negative shift of the SSI curve likely explain the voltage dependence of PKC-induced INa reduction, with a greater decrease observed at depolarized potentials.
The importance of PKC in regulating INa was further demonstrated through the discovery that inherited mutations in glycerol-3-phosphate (G3P) dehydrogenase 1-like protein (GPD1L) are linked to the reduction in INa amplitude seen in some forms of BrS [A280V (61)] and Sudden Infant Death Syndrome [mutation E83K (121)] [see (132) for review]. Although the function of GPD1L is unknown, the related enzyme GPD1 catalyzes the NAD+-dependent reversible conversion of G3P to dihydroxyacetone phosphate and is an important metabolic link between glycolysis and triglyceride synthesis. The A280V and E83K mutations cause a loss of GPD1L enzymatic activity and decrease INa when coexpressed with wild-type (WT) NaV1.5 in HEK293 cells (120). Importantly, both WT and mutant GPD1L-glutathione-S-transferase (GST) fusions pull down NaV1.5, which suggests that the effect of GPD1L mutations on INa is through enzymatic function and not binding. The proposed pathway involves PKC, because when WT GPD1L and NaV1.5 are coexpressed in HEK293 cells, 1-oleoyl-2-acetylglycerol or G3P (a GPD1 substrate) cause INa reduction, which is reversed with the serine/threonine kinase inhibitor staurosporine or mutation of S1503 to nonphosphorylatable alanine. Thus loss of function of GPD1L activity results in accumulation of G3P and phospholipid that activate PKC to phosphorylate S1503 and decrease INa (120). Interestingly, NADH mass action block of GPD1L also decreased INa, while there was no significant change with NAD+.
Studies by Liu et al. (60) further suggested that INa might be regulated directly by pyridine nucleotides, such as NADH. They found a twofold increase in intracellular NADH concentration when the A280V GPD1L mutant was adenovirally transfected into HEK293 cells stably expressing NaV1.5. Moreover, there was a dose-dependent decrease in INa with intracellular application of NADH delivered by patch pipette. The effect of NADH was rapid (suggesting a post-translational effect), did not appreciably alter INa gating or mRNA levels and was reproducible in neonatal myocytes. The NADH-induced INa decrease required both PKC and ROS, and ROS was downstream of PKC indicating the channel may be directly oxidized. The reported PKC effects (and the GPD1L-dependent pathway) connect the metabolic state of the cell to INa and cardiac excitability and may be crucial in cardiac ischemia and failure, which are characterized by increased metabolic stress (11, 112) and PKC upregulation (130). Of clinical relevance, mouse hearts perfused with external lactate/pyruvate to increase intracellular NADH concentration had enhanced inducible ventricular tachycardia with programmed electrical stimulation. Conversely, increased extracellular NAD+ concentration was antiarrhythmic in SCN5A+/− heterozygous mice that have decreased INa and increased propensity to inducible ventricular tachycardia (60).
GPD1L mutations are associated with a PKC-dependent decrease in the number of functional channels at the membrane. Mutation A280V was shown to decrease cell surface expression of NaV1.5 by immunocytochemistry and confocal microscopy (61). Additionally, flow cytometry measurements of HEK293 cells showed decreased NaV1.5 cell surface expression when coexpressed with A280V or E83K mutant GPD1L (120). This was reversed by mutation of S1503 on NaV1.5 to alanine or addition of staurosporine to inhibit PKC.
Analogous PKC-dependent effects were obtained independent of GPD1L mutations. Hallaq et al. (40) monitored the movement of green fluorescent protein (GFP)-tagged NaV1.5 (COOH-terminus) in HEK293 cells compared with hemagglutinin-tagged NaV1.5 immobilized at the membrane using confocal microscopy (40). In response to PKC activators, they observed a decrease in NaV1.5-GFP at the membrane, which was blocked by PKC inhibition, mutation of S1503 to alanine, or ROS inhibition. Fluorescence recovery after photobleach studies further identified that channels have decreased mobility within the membrane with PKC stimulation and, conversely, an increase with PKA stimulation. Peak INa was decreased at all test potentials after a 30-min exposure to the PKC activator phorbol 12-myristate 13-acetate (40). In contrast to the above experiments with relative long (30 min) drug exposures, Liu et al. (60) did not see a change in NaV1.5 surface expression, assayed by biotinylation and Western blot of surface NaV1.5 protein or confocal experiments of NaV1.5-GFP at the membrane, upon acute (2–10 min) treatment with pyruvate/lactate (to increase NADH) or phorbol 12-myristate 13-acetate (to activate PKC) (60). Decreased NaV1.5 cell surface expression and hence functional INa may be particularly relevant for chronic channel remodeling in HF, where PKC is increased (130).
Regulation by oxidation/nitrosylation: mitochondria and syntrophin complex.
Metabolic state is intimately coupled to INa function and relevant to our understanding of arrhythmogenesis in ischemia and HF (11, 86, 112), where both NADH and ROS are increased. In an elegant study, the Dudley group determined that mitochondria are the main source of the NADH-dependent ROS that decreases INa in HEK293 cells and rat neonatal ventricular myocytes (59). With the use of specific inhibitors of enzymes in the mitochondrial electron transport chain, the mechanism was further refined by identifying complexes I and III as the main sources of NADH-dependent ROS and the mitochondrial inner membrane anion channel as the main site of ROS release (59).
The studies of Liu and colleagues suggest that NADH directly activates PKC to increase mitochondrial ROS. ROS may have direct effects on the channel or may act through other downstream effectors [e.g., constitutive activation of CaMKII (34), lipoxidation products (84)]. Direct oxidation of methionine or cysteine residues on NaV1.5 is associated with INa,L resulting from slowed inactivation and a hyperpolarizing shift in availability that decreases peak INa (129). In a study of several NaV isoforms (NaV1.2, 1.5, 1.7, 1.4), mutation of multiple methionine residues to leucines, including M1305L on the Ile-Phe-Met (IFM) inactivation motif, significantly attenuated but did not completely abolish the ROS-induced slowing of inactivation (53). The decrease in peak INa observed with ROS may also be influenced by the recent observation that ROS can negatively regulate SCN5A gene expression by promoting nuclear localization of the Foxo1 transcription factor and its binding to the SCN5A promoter to suppress transcription (72). This may have implications for the NaV1.5 mRNA decrease observed in some disease models (see Tables).
Endogenous sources of ROS other than mitochondria also exist in cardiac myocytes, including nitric oxide (NO) produced by NO synthases (NOSs). Interestingly, NO has been associated with an increase in INa,L that is the proposed arrhythmogenic mechanism behind a missense mutation in syntrophin, A390V, causing LQT12 (116). α1-Syntrophin is a dystrophin-associated scaffold protein that contains multiple protein interaction motifs. It associates with the neuronal NOS (nNOS), the plasmalemmal Ca-ATPase (PMCA), and the COOH-terminus of NaV1.5 through postsynaptic density protein/Drosophila disk large tumor supressor/zonula occludens-1 protein (PDZ) domain interactions. Coimmunoprecipitations from mouse cardiac homogenates or pull downs with GST tagged NaV1.5 COOH-terminus as bait suggest nNOS, PMCA, α1-syntrophin, and NaV1.5 interact to form a signaling complex (see Fig. 1). The A390V mutation on syntrophin is in the binding region for PMCA. In HEK293 cells, expression of this mutant syntrophin disrupted pull down of PMCA and a biotin switch assay revealed increased direct S-nitrosylation of NaV1.5. Thus it was determined that A390V syntrophin fails to associate with PMCA4b, thus releasing PMCA4b-dependent inhibition of nNOS and promoting S-nitrosylation of NaV1.5. NO increases peak and late INa, and this was reversed by inhibiting NOS and recapitulated with the NO donors (+)-(E)-4-ethyl-2-[(E)-hydroxyimino]-5-nitro-3-hexenamide (NOR3) and S-nitrosoglutathione (GSNO). The positive shift in inactivation and increased window current (gain-of-function effects) induced by the A390V syntrophin mutant may explain the INa alterations. Furthermore, a recent study suggests that the molecular mechanism for a mutation in Cav3 (F97C) resulting in increased late INa and LQT9 (see Regulation by PKA and Cav3 complex) may also involve increased direct S-nitrosylation of NaV1.5 (24) analogous to the above A390V syntrophin mutant that results in LQT12.
In contradiction to the above, Ahmmed et al. (4) showed that extracellularly applied NO in native guinea pig and mouse cardiomyocytes decreased INa through combined cGMP (PKG) and cAMP (PKA) pathways (4). However, release of caged intracellular NO in ventricular myocytes increased INa,L independent of guanlyate cyclase in another study (2). Thus direct oxidation or S-nitrosylation of NaV1.5 may be partly responsible for the increased INa,L observed by some groups in diseased human or animal cardiac tissue (see Table 1).
Table 1.
INa changes in heart disease
| Model | Species | Cell Type | INa, A/F | SSI | SSA | INaL | INa Kinetics | SCN5A mRNA | Nav1.5 Protein | Comments (References) |
|---|---|---|---|---|---|---|---|---|---|---|
| HCM | Human | V | ↑ | ↔ | ↔ | Mild to moderate HF, NYHA class II–III (29) | ||||
| CM/CHD ± HF | Human | V | ↔ | ↔ | Development and recovery from inactivation ↔ | Window current detected, no Δ in HF (102) | ||||
| Explanted HF | Human | LV | ↔ | Myocytes from midmyocardial wall (50) | ||||||
| Explanted HF | Human | LV, RV | ↑ | (91) | ||||||
| Explanted end-stage HF | Human | LV, RV | ↓ | No Δ with LVADs; CaMKIIγ and δ ↔ in HF LVs; CaMKIIδ ↑ twofold in RV; CaMKIIδ ↓ to 40% of controls with LVAD (17) | ||||||
| Explanted HF | Human | V | ↑Truncation ↓native | ↓ protein expression and current in heterologous system with truncation variants (108) | ||||||
| Arrhythmogenic cardiomyopathy | Human | LV, RV | ↓ ICC | (87) | ||||||
| Explanted/pacing | Human/dog | V | ↓ | ↔ | ↔ | ↑ | ↔ | No Δ in Nav 1.1, 1.3, β1- and β2-subunits; ↑ cell capacitance in HF (119) | ||
| Explanted/pacing | Human/dog | V | ↑ | Slower INaL decay | (69, 70) | |||||
| Multiple coronary microembolizations | Dog | LV | ↓ | ↔ | ↔ | ↑ | ↔ | ↓ | ↓ Peak reversed by carvedilol; no Δ in β1 mRNA, β1 or β2 protein; ranolazine decreased APD, EADs, APD beat-to-beat variability and dispersion (68, 118) | |
| Pacing-induced HF | Dog | V | ↔ | ↔ | Rate and voltage dependence of INa decay ↔ | Ito downregulation explains APD prolongation (51) | ||||
| Ventricular tachypacing | Dog | P | ↓ | ↓ | Slowed conduction and AP rate of rise (64) | |||||
| 5-day infarction/ischemia | Dog | EBZ | ↓↓ | ← | ↔← | Slower recovery from inactivation and enhanced development of inactivation | Abnormal Nav1.5 cell surface staining | Reentrant excitation; ventricular tachycardia; slowed AP rate of rise (9, 10, 97) | ||
| Pressure/volume overload | Rabbit | V | ↔ | ↔ | ↑Heart weight, longer QRS duration, slower conduction velocity (131) | |||||
| Aortic banding | Guinea pig | LV | ↑ | ↔ | ↔ | Time course of INa inactivation and recovery ↔ | Increase in INa density with cardiac hypertrophy (at 4 wk) is attenuated with progression to cardiac failure (at 8 wk) (3) | |||
| Post-MI | Rat | LV | Slower decay | ↔ | 18% TTX-sensitive component of APD prolongation; ↑Nav1.1 mRNA and protein expression (45) | |||||
| Siderotic heart disease | Gerbil/neonatal rat | V | ↓ | ← | ↔ | Time course of INa activation and inactivation ↔; slower recovery from inactivation | ↔ | ↔Single channel currents, ↓AP overshoot and duration (57) | ||
| RV pressure overload | Rat | LV, RV | ↔ | ↑RV | ↔ | CTEPH model; reduced CX43; trend to INa density ↓ in both LV and RV (42) | ||||
| Volume overload | Rat | SAN | ↓ | ↓ | Nav1.1 and Nav1.6 primary isoforms, Nav1.5 and Nav1.7 weakly expressed, Nav1.2 and Nav1.3 not expressed; ↓ HR and ↑ sinus node recovery time (32) |
APD, action potential (AP) duration; CM, cardiomyopathy; CHD, congenital heart disease; CTEPH, chronic thromboembolic pulmonary hypertension; CX43, connexin 43; EADs, early afterdepolarizations; EBZ, epicardial border zone; HCM, hypertrophic cardiomyopathy; HF, heart failure; HR, heart rate; ICC, immunocytochemistry; INa, Na current; INa,L, late INa; Ito, transient outward K current; LV, left ventricle; LVAD, LV assist device; MI, myocardial infarction; NaV1.5, voltage-gated Na channel isoform 1.5; NYHA, New York Heart Association; P, Purkinje; RV, right ventricle; SAN, sinoatrial node; SSA, steady-state activation; SSI, steady-state inactivation; TTX, tetrodotoxin; V, ventricle.
Regulation by CaM.
The regulation of NaV by Ca, either directly or indirectly through CaM or CaMKII, has been a fervent area of research with groups reporting conflicting results. CaM was the first of these studied. Yeast two-hybrid screens demonstrated that CaM directly interacts with an IQ motif in the COOH-terminus of neuronal NaV1.2 (81) and cardiac NaV1.5 (113). Additionally, GST-fusion proteins of the WT COOH-terminal domain of both NaV1.5 and the skeletal muscle NaV1.4 pull-down CaM (30), demonstrating CaM binding to these regions, and mutation of the IQ domain prevents CaM pull down (30, 54). As with CaV1.2 (95), the NaV COOH-terminus binds both Ca/CaM and apoCaM with different affinities (20, 54). In whole cell patch-clamp experiments of TSA201 cells transfected with NaV1.5, Tan et al. (113) showed that a peptide antagonist of Ca-dependent CaM binding (peptide 290–309) delivered by patch pipette caused a +6 mV depolarizing shift in inactivation (CaM hyperpolarizes SSI) and decreased intermediate inactivation. Deschenes et al. (30), however, showed that CaM shifted SSI to negative potentials in both a mouse myoblast cell line and HEK293 cells expressing NaV1.4, but observed no effect of CaM on NaV1.5. Thus the effects of CaM may be NaV isoform dependent.
NaV1.5 may also be directly regulated by Ca. A pair of EF hand-like domains are present in the NaV1.5 COOH-terminus just upstream from the IQ domain (Glu1773-Asp1852) (133). Enhancement of intrinsic tyrosine or tryptophan residue fluorescence in the IQ or EF hand-like domains, respectively, was used as an assay for Ca binding the COOH-terminus (54). It was found that Ca binds to CaM in complex with the NaV1.5 (or NaV1.2) COOH-terminus but not to the COOH-terminus itself. This is in direct contradiction to studies by the Balser and Chazin groups that report the first EF hand-like domain in the COOH-terminus directly binds Ca, which destabilizes INa inactivation and shifts SSI to depolarized potentials (133). Nuclear magnetic resonance studies by the same group revealed the local structure of the COOH-terminal EF hand-like domain is capable of binding Ca (21).
Differences in experimental design, cell types, Ca buffering conditions, and the addition of fluoride to internal pipette solutions likely all contribute to these discrepant early studies. In particular, fluoride is a known Ca chelator and phosphatase inhibitor that could unmask basal phosphorylation effects. Additionally, peptide 290–309, which was used in early studies to disrupt CaM binding (113, 133), was originally developed as a CaMKII inhibitor that disrupts Ca/CaM-dependent activation of the kinase (93). This raises the interesting possibility that the effects of CaM reported in these studies may have intersected with CaMKII effects by inhibiting basal CaMKII phosphorylation. In fact, exogenous Ca/CaM enhanced slow inactivation (a reported effect of CaMKII) in one study using fluoride and peptide 290–309 (113), but not in another study that omitted these (30). Despite the discrepancies above, arrhythmogenic mutations in or around the COOH-terminal IQ and EF hand-like domains support a critical role for these domains in regulation of INa inactivation (26).
So what is CaM doing on the NaV COOH-terminus? In contrast to earlier studies, more recent studies have implicated the COOH-terminus in stabilization of the fast inactivation gate through interaction with the III–IV loop (82) and IQ-bound Ca/CaM binding this same loop (96). Inherited mutations in both the III–IV linker and COOH-terminus that are associated with INa,L and arrhythmia (26) support the novel idea that CaM could potentially influence fast inactivation (103). A GST construct comprising the III–IV loop pulls down a NaV1.5 COOH-terminus/CaM complex in Ca but not in EGTA solution (54). This interaction required CaM and was absent in an IQ/AA mutant, although CaM was not pulled down, suggesting it may be displaced. Recently, the Ahern group (103) captured a crystal structure of the NaV1.5 COOH-terminus in complex with the III–IV linker via a Ca/CaM bridge. They further identified two aromatic tyrosine residues (Tyr1494/95) on the III–IV loop, which are critical for its interaction with the C-lobe of CaM (104). It is interesting to speculate that the Ahern crystal structure may have captured a snapshot of Ca/CaM bound transiently to the III–IV loop before it is displaced to allow fast inactivation to proceed normally through interactions with the COOH-terminus and occlusion of the α-pore. Displaced Ca/CaM may function to activate CaMKII or remove latent regulation of INa inactivation by apoCaM bound to the IQ motif (16). These questions remain unresolved.
What are the potential functional consequences of CaM binding the COOH-terminus and how does this relate to the III–IV loop? Electrophysiological studies of INa in NaV1.5 COOH-terminal (including the IQ motif) LQTS mutants reveal INa,L potentially related to perturbed interactions of the COOH-terminus with the III–IV loop, i.e., fast inactivation gate (37, 54). This might explain the arrhythmogenic phenotype in inherited mutations involving the IQ motif (and possibly also the III–IV loop). As for the potential role of CaM to modulate fast inactivation in physiological conditions, one hypothesis is that high-frequency stimulation that elevates the local [Ca] around the channel results in resident apo-CaM binding Ca, CaM lobe switching, and destabilization of inactivation (122) (see Fig. 1). Ca remains bound to CaM to disrupt the interaction of the COOH-terminus with the III–IV loop and impair fast inactivation. Thus the availability curve is shifted to depolarized potentials and more Na channels will be available to drive the next AP upstroke. Although purely speculative, such a mechanism for channel facilitation may be particularly important for repetitive firing during rapid heart rates, e.g., in the response to exercise. It is intriguing to consider the possibility that CaM serves a physiological role in transducing Ca signals to enhance Na channel availability through impairment of fast inactivation, whereas CaMKII plays a pathophysiological role in enhancing slow inactivation and stabilizing the inactivated state (as illustrated below).
Regulation by CaMKII.
A number of studies have aimed at elucidating the functional effects of CaMKII-mediated NaV1.5 phosphorylation (14). In HEK293 cells expressing NaV1.5, inhibition of CaM kinase with 10 μM KN93 (but not its inactive analog KN92) slowed INa decay, shifted SSI to depolarized potentials, and slowed entry into inactivated states (30). On the other hand, 100 nM autocamtide-2-related inhibitory peptide (AIP) in the pipette had no effect on gating, but intracellular [AIP] might have been limiting (IC50 for CaMKII inhibition ∼40 nM). As discussed below, these CaMK effects are consistent with more recent studies analyzing CaMKII effects on INa gating.
In a seminal study by Wagner et al. (126), CaMKII was shown to associate with and phosphorylate NaV1.5 in cardiac myocytes. CaMKIIδC overexpression, either chronically in transgenic (TG) mice or by acute adenoviral overexpression in rabbit, caused simultaneous gain- and loss-of-function effects on native INa. Specifically, in myocytes CaMKII shifted SSI to negative potentials (Fig. 2A), enhanced accumulation of intermediate/slow inactivation (Fig. 2B), slowed recovery from inactivation (Fig. 2C), slowed fast INa inactivation, and increased INa,L (Fig. 2D). Moreover, all of these effects could be acutely reversed with CaMKII inhibition, thus strongly arguing for specific CaMKII-dependent modulation of cardiac Na channels. CaMKIIδC overexpressing mice also displayed enhanced propensity to tachyarrhythmias. Importantly, these effects phenocopy the inherited 1795insD mutation on NaV1.5 that in patients results in combined LQTS and BrS effects (124). Moreover, Maltsev et al. (67) tested the inhibition of CaMKII and found that CaMKII slows the decay of INa,L, more so in failing versus nonfailing canine myocytes.
Aiba et al. (6) subsequently used GST fusion constructs of the intracellular regions of NaV1.5 to narrow down CaMKII-dependent phosphorylation to the I–II loop (6). However, the reported INa functional effects of CaMKII phosphorylation (positive shift in SSI, faster recovery from inactivation, decreased entry into intermediate or slow inactivation, and increased INa,L) were largely inconsistent with those described by Wagner et al. (126) and other groups (8, 47). These discrepancies may be due to Aiba and colleagues' use of the CaMKIIα isoform (vs. CaMKIIδC), delivery method (dialysis via patch pipette vs. adenovirus or transgenesis), different Ca buffering, and use of fluoride in pipette solutions.
Through alanine scanning of phosphorylation sites fitting the traditional CaMKII phosphorylation motif, RXXS/T, Hund et al. (47) identified S571 as a CaMKII target site in vitro (47). In a HEK293 heterologous cell system expressing NaV1.5, CaMKII shifts the SSI of WT NaV1.5 to negative potentials. This effect on channel inactivation [observed in all studies, except Aiba et al. (6)] was abolished when S571 was mutated to a nonphosphorylatable alanine and mimicked when S571 was mutated to a phosphomimetic glutamate residue. Furthermore, the authors demonstrated a role for βIV-spectrin in targeting CaMKII to NaV1.5 at the intercalated discs. In a more recent study by the same group (56), it was further demonstrated that S571 is phosphorylated in the border zone of infarcted canine hearts, is slightly increased in human HF, and is phosphorylated in mouse hearts following acute stimulation by isoproterenol (with phosphatase inhibitors), but not in mice overexpressing the CaMKII inhibitor AC3I. Two rare, negatively charged arrhythmogenic point mutations at A572D and Q573E functionally mimic the effect of CaMKII phosphorylation at the adjacent S571 residue, suggesting negative charge within this region may confer similar effects on channel biophysics. Although a direct link between S571 and increased INa,L has not yet been established, prolonged APs and triggered activity were observed with arrhythmia variants A572D and Q573E, expressed in neonatal myocytes and simulation studies. However, a study by Tester et al. (114) demonstrated that the inherited A572D mutation, although originally identified in LQTS genetic screens, is not functionally different from WT channels and is not proarrhythmic by itself.
Our group was the first to show that autophosphorylated CaMKII binds the I–II loop, and we confirmed this loop as the primary CaMKII phosphorylation target in vitro (8). Using peptide spot arrays of the entire I–II loop, we systematically identified CaMKII phosphorylation sites by P32 incorporation with in vitro kinase assays. We found that S516 and T594 are specifically phosphorylated by CaMKII but found no evidence for phosphorylation at the S571 site identified in the Hund study. In HEK293 cells expressing NaV1.5α, we demonstrated that CaMKIIδC shifts INa SSI to negative potentials and enhances accumulation of intermediate inactivation (consistent with the previously reported CaMKIIδC effects). These effects are reversed by AIP or mutation of any of the reported CaMKII targets (T594, S516, or S571) to nonphosphorylatable alanine. T594E and S516E, but not S571E, charged glutamate phosphomimetics recapitulate CaMKII effects even in the absence of exogenous CaMKII and presence of AIP. Interestingly, a recent phosphoproteomic study suggested that S571 is phosphorylated at baseline in unstimulated mouse NaV1.5α, whereas phosphorylation at S516 and T594 was not seen (74). CaMKII phosphorylation at T594 and S516 awaits further confirmation in native myocytes, and more studies are needed to understand the relative contributions of T594, S516, and S571 to INa gating changes in pathological conditions.
Functional Consequences for Acquired Arrhythmias: CaMKII as a Model
Because CaMKII can cause both LQTS-like INa,L and loss-of-function BrS-like effects, we sought to investigate how its effects contribute to arrhythmogenesis (38). Indeed, acquired forms of altered NaV1.5 function attributable to post-translational modifications (e.g., phosphorylation or oxidation) may increase the risk of arrhythmia and sudden cardiac death during ischemia-reperfusion or HF. Furthermore, CaMKII phosphorylates numerous Ca handling proteins as well as sarcolemmal K channels (Fig. 4) (13). This in turn can influence myocyte Ca regulation and synergize with INa gating effects in arrythmogenesis.
Fig. 4.

Arrhythmogenic mechanisms of CaMKIIδC-mediated INa regulation. Gain (+) and loss (−) of function effects of CaMKIIδC phosphorylation are indicated with upward and downward triangles, respectively. NCX, Na/Ca exchanger; NKA, Na-K-ATPase; NaV1.5, voltage-gated Na channel isoform 1.5; RyR, ryanodine receptor; PLB, phospholamban; SERCA, sarco(endo)plasmic reticulum Ca ATPase; APD, AP duration.
CaMKII-mediated gain-of-function effects, i.e., the late or persistent inward current, may delay repolarization, prolong APD (Fig. 3B), and alter intracellular Na and Ca homeostasis, potentially predisposing to arrhythmogenesis [via early and delayed afterdepolarizations (EADs and DADs, respectively); Figs. 3, A and B, and 4]. Our modeling study (38) predicted that CaMKII-mediated enhancement in INa,L could cause AP prolongation, especially at slow pacing rates (consistent with a LQT3 phenotype). A mechanistic link between H2O2 (CaMKII mediated)-dependent increase of late INa, AP prolongation, and EADs has been established in experimental (136) and simulation (43) studies. Edwards et al. (33) reported that myocytes from TG mice overexpressing CaMKIIδC (before pronounced HF remodeling) are susceptible to EADs, which are dependent on sarcoplasmic reticulum (SR) Ca release caused by pronounced isoproterenol-induced increases in Ca transient amplitude and APD. Computational modeling also suggested that the mechanism of AP prolongation and EAD initiation involves recruitment of INa,L secondary to SR Ca release-dependent augmentation of inward Na/Ca exchange current. Prolonged APs and cellular arrhythmias (both EADs and DADs) have been reported in human cardiomyocytes from patients with hypertrophic cardiomyopathy and attributed to CaMKII-mediated INa,L increase (29). Disrupting spectrin-mediated molecular anchoring of CaMKII at NaV1.5 has been shown to remove the effects of CaMKII on INa and reduce EAD propensity (47). By enhancing SR Ca loading and promoting spontaneous intracellular Ca waves, H2O2-induced EADs were also shown to cause DADs (136). Recently, the generation of ROS under conditions of increased oxidative stress, such as during HF, was shown to correlate with CaMKII-mediated augmentation of late INa and consequent arrhythmias (128).
Fig. 3.

Consequences of CaMKIIδC hyperactivity on cardiac function. A: delayed afterdepolarizations (DADs). B: early afterdepolarizations (EADs). C: transmural dispersion of repolarization. D: slowing of action potential (AP) rate of rise and decreased AP amplitude. Endo, endocardium; Epi, epicardium; bpm, beats/min.
The loss-of-function effects (decreased availability, enhanced intermediate inactivation, and slowed recovery there from) may favor a BrS-like phenotype (Fig. 4). Our model (38) predicted that these alterations induced by CaMKII could reduce the velocity of AP upstroke (Fig. 3D) and slow conduction and are likely to be exacerbated at high heart rates (Fig. 3D) whereby the diastolic interval is reduced and Na channels have less time to recover. Increased post-repolarization refractoriness in the infarct border zone [where CaMKII activity is enhanced (25, 46)], attributed to INa remodeling, has been proposed as a mechanism for slow conduction and arrhythmogenesis (18). Indeed, simulations showed CaMKII hyperactivity in the infarct border zone, due primarily to increased oxidation, causes INa-mediated reduction of AP upstroke (46) and conduction velocity (25), increase in effective refractory period, and increased susceptibility to formation of conduction block at the border zone margin, which predisposes to reentrant arrhythmias (25).
CaMKII also mediates Ca current facilitation, which enhances peak L-type Ca current and slows inactivation (gain of function) and tends to prolong APD (along with INa,L) (38). CaMKII also alters both fast and slow transient outward K current (Ito; enhanced recovery from inactivation) and inward rectifier K current (127) in ways that shorten APD (Fig. 4). Simulations predicted that with transmural heterogeneity of Ito and Ito downregulation in HF, the net effect of CaMKII would be to shorten epicardial APD and prolong endocardial APD, thus accentuating the normal transmural dispersion of repolarization (Fig. 3C) (38), an effect known to be proarrhythmic (7).
As discussed, recent computational myocytes models have incorporated elements of the CaMKII signaling cascade and provided new insights regarding the role of CaMKII in regulating cardiomyocyte contractility and excitability in health and disease. Whereas much progress has been made to determine and model the kinetics of phosphorylation of various CaMKII targets (110), the kinetics of CaMKII-mediated phosphorylation and dephosphorylation of Na channels is unknown. Thus these models are somewhat limited in assuming steady-state fractional phosphorylation of Na channels rather than dynamic transitions between unphosphorylated and phosphorylated Na channels (43). However, the dynamics of CaMKII-dependent Na channel phosphorylation may be critical for its arrhythmogenic consequences and associated potential treatment. Recently, Moreno et al. (80) developed a computational approach to study the interaction of kinetics of the blockers flecainide and lidocaine with cardiac Na channels. They used the model to predict the drug effects on human ventricular cellular and tissue electrical activity and in the setting of one common arrhythmia trigger, spontaneous ventricular ectopy. The model predicted that clinically relevant concentrations of the antiarrhythmic drugs flecainide and lidocaine would exacerbate, rather than ameliorate, arrhythmia. On the other hand, therapies that target late INa, such as block by ranolazine, may have promise in treating patients that are at risk for arrhythmia (65, 125).
Pathological Changes in Na Current
Studies of the INa changes occurring in cardiac pathologies/disease (summarized in Tables 1 and 2) are less abundant than those characterizing inherited NaV1.5 channelopathies. This is largely due to the difficulty in obtaining human donor transplant tissue suitable for electrophysiology (or biochemistry), as well as the cost and complexity of animal models. Moreover, the results of such studies are often variable and difficult to interpret. For example, human explant studies have wide variability in age, sex, ethnicity, severity of disease, and low sample number and are further complicated by patient history (e.g., pharmacological treatments that can alter INa). Animal models (Table 1) may have important species differences, and TG mice with altered genetics (Table 2) may have compensatory ion channel remodeling effects that complicate the interpretation of INa functional effects.
Table 2.
INa changes in TG mice with HF
| Model | Cell Type | INa, A/F | SSI | SSA | INaL | INa Kinetics | SCN5A mRNA | Nav1.5 Protein | Comments (References) |
|---|---|---|---|---|---|---|---|---|---|
| CaMKIIδc-OE | V | ↔ | ← | ↔ | ↑ | Slower recovery from inactivation and enhanced development of inactivation | ↑ | Trend to ↓ INa density; TG mice exhibit longer QRS intervals and monomorphic and polymorphic VT upon programmed electrical stimulation (126) | |
| Muscle LIM protein MLP−/− | V | ↓ | ← | → | Slowed inactivation | ↓ | ↑ APD and EAD propensity, ↓ AP amplitude and rate of depolarization, lower Nav1.5 protein molecular weight (deglycosylation) (117) | ||
| Unilateral nephrectomy/DOCA implantation, salt water substitution | V | ↓ | ↔ | ↔ | Reducing mitochondrial ROS by application of NAD+, mitoTEMPO, PKC inhibitors, or PKA activators, restored INa (58) | ||||
| Inducible CX43-KO | LV, RV | ↓ | ↓ | Slower conduction and longer QRS in pacing induced VT+ mice (49) | |||||
| PKP2 ± ARVC | V | ↓ | ← | ↔ | Slower recovery from inactivation | ↔ ICC | Abnormal ultrastructure; exaggerated INa decrease and slowed conduction w/flecainide, arrhythmia and SCD (19) | ||
| ACE 8/8-OE | ↔ | ↔ ← | ← | Cardia-specific (α-MHC) overexpression; results in fourfold increase in ANG II (48, 52) | |||||
| Calcineurin-OE | V | ↓↓ | ↔ | ↔ | ↔ | ↔ ICC | Hypertrophy, ↓ rate of AP rise, progressive heart block, SCD (39) | ||
| Snail ± OE/DCM | V | ↓ | ↓ | DCM, ECG abnormalities, conduction defects; Homozygous mice lethal (44) | |||||
| CSQ-OE | V | ↓↓ | → | → | DCM, hypertrophy, ↑ cell capacitance, ↑ PR and QT interval, conduction block (55) | ||||
| Mdx (5cv) | V | ↓ | ↔ | ↓ | Dystrophin deficient; impaired conduction (35) | ||||
| AnkB−/− KO | V | ↓ | ← | ← | ↑ | Slowed recovery from inactivation | Prolonged APD; impaired QT-rate adaptation; slowed HR; longer single Na channel mean open time; late single channel openings (23) |
ACE, angiotensin-converting enzyme; ANG II, angiotensin II; ARVC, arrhythmogenic RV cardiomyopathy; CSQ, calsequestrin; DCM, dilated cardiomyopathy; KO, knockout; MHC, myosin heavy chain; MLP, muscle LIM protein; OE, overexpression; PKP2, plakophilin-2; TG, transgenic; VT, ventricular tachycardia.
Na channel expression and functional alterations in HF are complex and may affect cardiac electrophysiology in various ways. Despite the above limitations, a few common observations in the reported results emerge. First, many studies report a decrease in functional INa density (9, 10, 57, 68, 97, 118, 119). Second, INa,L has been reported in both canine (68–70, 118) and human HF (119), as well as in failing mouse cardiomyocytes (126). Both observations could be the result of various complex post-translational modifications of NaV1.5, including phosphorylation by CaMKII. Moreover, both would be expected to contribute to arrhythmias as illustrated in Figs. 3 and 4 for CaMKII-dependent NaV1.5 gain- and loss-of-function effects: the reduced INa could lead to conduction slowing and ventricular arrhythmias based on reentrant mechanisms, whereas increased INa,L may delay repolarization, prolong APD, alter intracellular Na and Ca homeostasis, and potentially predispose to arrhythmogenesis (EADs, DADs). As reported for K channels (73), a decrease in functional current density could be the result of either a decrease in peak conductance, an active decrease in channel number at the membrane, or myocyte hypertrophy without compensatory increases in channel expression (85). Most studies do not distinguish between these. Thus the decrease in INa current densities reported in Tables 1 and 2 must be interpreted cautiously. However, a few studies have reported changes in NaV1.5 protein and/or mRNA levels (Tables 1 and 2) that coexist and are consistent with a decrease in INa density. More controlled experiments in animal disease models are needed to further refine the molecular mechanisms of INa changes in cardiac disease and their relative contributions to arrhythmias.
Summary and Concluding Remarks
We reviewed here the molecular and functional aspects of cardiac Na channel modulation and its causal link to cardiac arrhythmias. Interestingly, PKA, PKC, and CaMKII similarly shift INa availability to negative potentials. Although not yet identified, it is probable that additional putative PKC sites exist on the NaV1.5 I–II loop (as for PKA and CaMKII) that also decrease availability. Thus kinase phosphorylation may act in a concerted manner through the I–II loop to enhance inactivation under various stimuli. A negative shift in INa availability would be particularly detrimental in ischemic conditions, which are associated with depolarized resting membrane potentials (86).
Chronic phosphorylation by either PKA or PKC alters channel surface levels, but in antagonistic ways. In the case of PKA, the increase in myocardial conduction velocity associated with sympathetic stimulation implicates an increase in channel density at the membrane. In contrast, PKC decreases the number of functional channels at the membrane, and this may be particularly relevant for chronic channel remodeling as in HF. In fact, PKC isoforms are upregulated with increased activity in HF (88, 130). More studies are needed to confirm this regulation in cardiomyocytes and determine if CaMKII similarly alters channel surface expression.
Additionally, PKC regulation appears to be intimately intertwined with metabolism, since mutations in GPD1L that increase both NADH and ROS decrease INa in a PKC-dependent manner. It is still unclear which mode of PKC activation (increased G3P and phospholipids or direct activation by NADH) is the most relevant, but there is good evidence that direct phosphorylation of NaV1.5 by PKC at S1503 (and likely other sites) decreases INa. Likewise, the exact mode of regulation that leads to the decrease in INa observed with PKC activation is unclear (ROS vs. direct PKC phosphorylation). Is the effect of ROS direct or indirect via activation of other downstream proteins, such as CaMKII? Indeed, it is conceivable that increased PKC activity fuels an increase in mitochondrial ROS, which constitutively activate CaMKII to regulate INa. Both CaMKII and PKC appeared to be involved in the Ca-dependent increase in INa,L observed in rabbit cardiomyocytes (63).
Interestingly, the PKA phosphorylation sites at S525/S528 and the ER retention motif at 533–535 all neighbor a CaMKII phosphorylation site identified at S516 (8) and a methylated Arg at position 513 (12) (of the same CaMKII motif). The close proximity of these PKA and CaMKII phosphorylation motifs and their similar functional consequences for channel inactivation suggest that this region of the I–II loop is important for channel gating and conformationally/structurally accessible to regulation in the fully folded channel complex. Although the Catterall group (92) recently solved the crystal structure for a bacterial voltage-gated Na channel, this channel lacks the regulatory cytoplasmic loops present on mammalian channels. Regarding CaMKII sites identified on NaV1.5 thus far, a recent phosphoproteomic study of unstimulated, native mouse NaV1.5α showed basal phosphorylation of S571 and no phosphorylation at T594 or S516 (74). Although the S516 site is conserved in mouse, the critical Arg at P-3 (R513) of the CaMKII motif is not (14). Peptides containing T594 were not recovered by mass spectrometry. Furthermore, PKA and CaMKII regulation of NaV1.5 both appear to require multiple phosphorylation events on the channel, analogous to the graded regulation that has been observed for some K channels (90).
Collectively, these studies highlight the important role post-translational modifications play in both acute INa gating changes and chronic regulation of NaV1.5 surface expression. Much work remains to fully unravel the complexities of post-translational regulation of NaV1.5 and its role in cardiac physiology and disease.
GRANTS
This study was supported by the National Institutes of Health grants P01-HL-080101, R01-HL-105242, and training grant T32-GM-099608 and the Fondation Leducq Transatlantic CaMKII Alliance.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
A.W.H. and E.G. conception and design of research; A.W.H. and E.G. interpreted results of experiments; A.W.H. and E.G. prepared figures; A.W.H. and E.G. drafted manuscript; A.W.H., D.M.B., and E.G. approved final version of manuscript; D.M.B. edited and revised manuscript.
REFERENCES
- 1.Abriel H. Cardiac sodium channel Na(v)1.5 and interacting proteins: Physiology and pathophysiology. J Mol Cell Cardiol 48: 2–11, 2010 [DOI] [PubMed] [Google Scholar]
- 2.Ahern GP, Hsu SF, Klyachko VA, Jackson MB. Induction of persistent sodium current by exogenous and endogenous nitric oxide. J Biol Chem 275: 28810–28815, 2000 [DOI] [PubMed] [Google Scholar]
- 3.Ahmmed GU, Dong PH, Song G, Ball NA, Xu Y, Walsh RA, Chiamvimonvat N. Changes in Ca2+ cycling proteins underlie cardiac action potential prolongation in a pressure-overloaded guinea pig model with cardiac hypertrophy and failure. Circ Res 86: 558–570, 2000 [DOI] [PubMed] [Google Scholar]
- 4.Ahmmed GU, Xu Y, Hong Dong P, Zhang Z, Eiserich J, Chiamvimonvat N. Nitric oxide modulates cardiac Na+ channel via protein kinase A and protein kinase G. Circ Res 89: 1005–1013, 2001 [DOI] [PubMed] [Google Scholar]
- 5.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res 97: 1314–1322, 2005 [DOI] [PubMed] [Google Scholar]
- 6.Aiba T, Hesketh GG, Liu T, Carlisle R, Villa-Abrille MC, O'Rourke B, Akar FG, Tomaselli GF. Na+ channel regulation by Ca2+/calmodulin and Ca2+/calmodulin-dependent protein kinase II in guinea-pig ventricular myocytes. Cardiovasc Res 85: 454–463, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antzelevitch C. Role of spatial dispersion of repolarization in inherited and acquired sudden cardiac death syndromes. Am J Physiol Heart Circ Physiol 293: H2024–H2038, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ashpole NM, Herren AW, Ginsburg KS, Brogan JD, Johnson DE, Cummins TR, Bers DM, Hudmon A. Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. J Biol Chem 287: 19856–19869, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baba S, Dun W, Boyden PA. Can PKA activators rescue Na+ channel function in epicardial border zone cells that survive in the infarcted canine heart? Cardiovasc Res 64: 260–267, 2004 [DOI] [PubMed] [Google Scholar]
- 10.Baba S, Dun W, Cabo C, Boyden PA. Remodeling in cells from different regions of the reentrant circuit during ventricular tachycardia. Circulation 112: 2386–2396, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barth AS, Tomaselli GF. Cardiac metabolism and arrhythmias. Circ Arrhythm Electrophysiol 2: 327–335, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beltran-Alvarez P, Pagans S, Brugada R. The cardiac sodium channel is post-translationally modified by arginine methylation. J Proteome Res 10: 3712–3719, 2011 [DOI] [PubMed] [Google Scholar]
- 13.Bers DM, Grandi E. Calcium/calmodulin-dependent kinase II regulation of cardiac ion channels. J Cardiovasc Pharmacol 54: 180–187, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bers DM, Herren AW. Na+ channel I–II loop mediates parallel genetic and phosphorylation-dependent gating changes. Circulation 126: 2042–2046, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biet M, Barajas-Martinez H, Ton AT, Delabre JF, Morin N, Dumaine R. About half of the late sodium current in cardiac myocytes from dog ventricle is due to non-cardiac-type Na+ channels. J Mol Cell Cardiol 53: 593–598, 2012 [DOI] [PubMed] [Google Scholar]
- 16.Biswas S, DiSilvestre D, Tian Y, Halperin VL, Tomaselli GF. Calcium-mediated dual-mode regulation of cardiac sodium channel gating. Circ Res 104: 870–878, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borlak J, Thum T. Hallmarks of ion channel gene expression in end-stage heart failure. FASEB J 17: 1592–1608, 2003 [DOI] [PubMed] [Google Scholar]
- 18.Cabo C, Boyden PA. Electrical remodeling of the epicardial border zone in the canine infarcted heart: a computational analysis. Am J Physiol Heart Circ Physiol 284: H372–H384, 2003 [DOI] [PubMed] [Google Scholar]
- 19.Cerrone M, Noorman M, Lin X, Chkourko H, Liang FX, van der Nagel R, Hund T, Birchmeier W, Mohler P, van Veen TA, van Rijen HV, Delmar M. Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc Res 95: 460–468, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chagot B, Chazin WJ. Solution NMR structure of Apo-calmodulin in complex with the IQ motif of human cardiac sodium channel NaV1.5. J Mol Biol 406: 106–119, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chagot B, Potet F, Balser JR, Chazin WJ. Solution NMR structure of the C-terminal EF-hand domain of human cardiac sodium channel NaV1.5. J Biol Chem 284: 6436–6445, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chandra R, Chauhan VS, Starmer CF, Grant AO. β-Adrenergic action on wild-type and KPQ mutant human cardiac Na+ channels: shift in gating but no change in Ca2+:Na+ selectivity. Cardiovasc Res 42: 490–502, 1999 [DOI] [PubMed] [Google Scholar]
- 23.Chauhan VS, Tuvia S, Buhusi M, Bennett V, Grant AO. Abnormal cardiac Na+ channel properties and QT heart rate adaptation in neonatal ankyrin(B) knockout mice. Circ Res 86: 441–447, 2000 [DOI] [PubMed] [Google Scholar]
- 24.Cheng J, Valdivia CR, Vaidyanathan R, Balijepalli RC, Ackerman MJ, Makielski JC. Caveolin-3 suppresses late sodium current by inhibiting nNOS-dependent S-nitrosylation of SCN5A. J Mol Cell Cardiol pii: S0022–S2828, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Christensen MD, Dun W, Boyden PA, Anderson ME, Mohler PJ, Hund TJ. Oxidized calmodulin kinase II regulates conduction following myocardial infarction: a computational analysis. PLoS Comput Biol 5: e1000583, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clancy CE, Kass RS. Inherited and acquired vulnerability to ventricular arrhythmias: cardiac Na+ and K+ channels. Physiol Rev 85: 33–47, 2005 [DOI] [PubMed] [Google Scholar]
- 27.Clancy CE, Rudy Y. Linking a genetic defect to its cellular phenotype in a cardiac arrhythmia. Nature 400: 566–569, 1999 [DOI] [PubMed] [Google Scholar]
- 28.Clancy CE, Rudy Y. Na+ channel mutation that causes both Brugada and long-QT syndrome phenotypes: a simulation study of mechanism. Circulation 105: 1208–1213, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coppini R, Ferrantini C, Yao L, Fan P, Del Lungo M, Stillitano F, Sartiani L, Tosi B, Suffredini S, Tesi C, Yacoub M, Olivotto I, Belardinelli L, Poggesi C, Cerbai E, Mugelli A. Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation 127: 575–584, 2013 [DOI] [PubMed] [Google Scholar]
- 30.Deschenes I, Neyroud N, DiSilvestre D, Marban E, Yue DT, Tomaselli GF. Isoform-specific modulation of voltage-gated Na+ channels by calmodulin. Circ Res 90: E49–E57, 2002 [DOI] [PubMed] [Google Scholar]
- 31.Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM. Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation 105: 2543–2548, 2002 [DOI] [PubMed] [Google Scholar]
- 32.Du Y, Huang X, Wang T, Han K, Zhang J, Xi Y, Wu G, Ma A. Downregulation of neuronal sodium channel subunits Nav1.1 and Nav16 in the sinoatrial node from volume-overloaded heart failure rat. Pflügers Arch 454: 451–459, 2007 [DOI] [PubMed] [Google Scholar]
- 33.Edwards AG, Hake J, Heller Brown J, McCulloch A. Calcium calmodulin dependent protein kinase ii overexpression predisposes myocytes to isoproterenol-induced early afterdepolarizations before the onset of heart failure. Circulation 124: A14642, 2011 [Google Scholar]
- 34.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O'Donnell SE, Aykin-Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133: 462–474, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gavillet B, Rougier JS, Domenighetti AA, Behar R, Boixel C, Ruchat P, Lehr HA, Pedrazzini T, Abriel H. Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ Res 99: 407–414, 2006 [DOI] [PubMed] [Google Scholar]
- 36.Gima K, Rudy Y. Ionic current basis of electrocardiographic waveforms: a model study. Circ Res 90: 889–896, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Glaaser IW, Osteen JD, Puckerin A, Sampson KJ, Jin X, Kass RS. Perturbation of sodium channel structure by an inherited long QT syndrome mutation. Nat Commun 3: 706, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grandi E, Puglisi JL, Wagner S, Maier LS, Severi S, Bers DM. Simulation of Ca-calmodulin-dependent protein kinase II on rabbit ventricular myocyte ion currents and action potentials. Biophys J 93: 3835–3847, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo J, Zhan S, Somers J, Westenbroek RE, Catterall WA, Roach DE, Sheldon RS, Lees-Miller JP, Li P, Shimoni Y, Duff HJ. Decrease in density of INa is in the common final pathway to heart block in murine hearts overexpressing calcineurin. Am J Physiol Heart Circ Physiol 291: H2669–H2679, 2006 [DOI] [PubMed] [Google Scholar]
- 40.Hallaq H, Wang DW, Kunic JD, George AL, Jr, Wells KS, Murray KT. Activation of protein kinase C alters the intracellular distribution and mobility of cardiac Na+ channels. Am J Physiol Heart Circ Physiol 302: H782–H789, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hallaq H, Yang Z, Viswanathan PC, Fukuda K, Shen W, Wang DW, Wells KS, Zhou J, Yi J, Murray KT. Quantitation of protein kinase A-mediated trafficking of cardiac sodium channels in living cells. Cardiovasc Res 72: 250–261, 2006 [DOI] [PubMed] [Google Scholar]
- 42.Hardziyenka M, Campian ME, Verkerk AO, Surie S, van Ginneken AC, Hakim S, Linnenbank AC, de Bruin-Bon HA, Beekman L, van der Plas MN, Remme CA, van Veen TA, Bresser P, de Bakker JM, Tan HL. Electrophysiologic remodeling of the left ventricle in pressure overload-induced right ventricular failure. J Am Coll Cardiol 59: 2193–2202, 2012 [DOI] [PubMed] [Google Scholar]
- 43.Hashambhoy YL, Winslow RL, Greenstein JL. CaMKII-dependent activation of late INa contributes to cellular arrhythmia in a model of the cardiac myocyte. Conf Proc IEEE Eng Med Biol Soc 2011: 4665–4668, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hesse M, Kondo CS, Clark RB, Su L, Allen FL, Geary-Joo CT, Kunnathu S, Severson DL, Nygren A, Giles WR, Cross JC. Dilated cardiomyopathy is associated with reduced expression of the cardiac sodium channel Scn5a. Cardiovasc Res 75: 498–509, 2007 [DOI] [PubMed] [Google Scholar]
- 45.Huang B, El-Sherif T, Gidh-Jain M, Qin D, El-Sherif N. Alterations of sodium channel kinetics and gene expression in the postinfarction remodeled myocardium. J Cardiovasc Electrophysiol 12: 218–225, 2001 [DOI] [PubMed] [Google Scholar]
- 46.Hund TJ, Decker KF, Kanter E, Mohler PJ, Boyden PA, Schuessler RB, Yamada KA, Rudy Y. Role of activated CaMKII in abnormal calcium homeostasis and INa remodeling after myocardial infarction: insights from mathematical modeling. J Mol Cell Cardiol 45: 420–428, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hund TJ, Koval OM, Li J, Wright PJ, Qian L, Snyder JS, Gudmundsson H, Kline CF, Davidson NP, Cardona N, Rasband MN, Anderson ME, Mohler PJ. A beta(IV)-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J Clin Invest 120: 3508–3519, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iravanian S, Sovari AA, Lardin HA, Liu H, Xiao HD, Dolmatova E, Jiao Z, Harris BS, Witham EA, Gourdie RG, Duffy HS, Bernstein KE, Dudley SC., Jr Inhibition of renin-angiotensin system (RAS) reduces ventricular tachycardia risk by altering connexin43. J Mol Med (Berl) 89: 677–687, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jansen JA, Noorman M, Musa H, Stein M, de Jong S, van der Nagel R, Hund TJ, Mohler PJ, Vos MA, van Veen TA, de Bakker JM, Delmar M, van Rijen HV. Reduced heterogeneous expression of Cx43 results in decreased Nav1.5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional Cx43 knockout mice. Heart Rhythm 9: 600–607, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaab S, Dixon J, Duc J, Ashen D, Nabauer M, Beuckelmann DJ, Steinbeck G, McKinnon D, Tomaselli GF. Molecular basis of transient outward potassium current downregulation in human heart failure: a decrease in Kv4.3 mRNA correlates with a reduction in current density. Circulation 98: 1383–1393, 1998 [DOI] [PubMed] [Google Scholar]
- 51.Kaab S, Nuss HB, Chiamvimonvat N, O'Rourke B, Pak PH, Kass DA, Marban E, Tomaselli GF. Ionic mechanism of action potential prolongation in ventricular myocytes from dogs with pacing-induced heart failure. Circ Res 78: 262–273, 1996 [DOI] [PubMed] [Google Scholar]
- 52.Kasi VS, Xiao HD, Shang LL, Iravanian S, Langberg J, Witham EA, Jiao Z, Gallego CJ, Bernstein KE, Dudley SC., Jr Cardiac-restricted angiotensin-converting enzyme overexpression causes conduction defects and connexin dysregulation. Am J Physiol Heart Circ Physiol 293: H182–H192, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kassmann M, Hansel A, Leipold E, Birkenbeil J, Lu SQ, Hoshi T, Heinemann SH. Oxidation of multiple methionine residues impairs rapid sodium channel inactivation. Pflügers Arch 456: 1085–1095, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim J, Ghosh S, Liu H, Tateyama M, Kass RS, Pitt GS. Calmodulin mediates Ca2+ sensitivity of sodium channels. J Biol Chem 279: 45004–45012, 2004 [DOI] [PubMed] [Google Scholar]
- 55.Knollmann BC, Knollmann-Ritschel BE, Weissman NJ, Jones LR, Morad M. Remodelling of ionic currents in hypertrophied and failing hearts of transgenic mice overexpressing calsequestrin. J Physiol 525, 483–498, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koval OM, Snyder JS, Wolf RM, Pavlovicz RE, Glynn P, Curran J, Leymaster ND, Dun W, Wright PJ, Cardona N, Qian L, Mitchell CC, Boyden PA, Binkley PF, Li C, Anderson ME, Mohler PJ, Hund TJ. Ca2+/calmodulin-dependent protein kinase II-based regulation of voltage-gated Na+ channel in cardiac disease. Circulation 126: 2084–2094, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kuryshev YA, Brittenham GM, Fujioka H, Kannan P, Shieh CC, Cohen SA, Brown AM. Decreased sodium and increased transient outward potassium currents in iron-loaded cardiac myocytes. Implications for the arrhythmogenesis of human siderotic heart disease. Circulation 100: 675–683, 1999 [DOI] [PubMed] [Google Scholar]
- 58.Liu M, Gu L, Sulkin MS, Liu H, Jeong EM, Greener I, Xie A, Efimov IR, Dudley SC., Jr Mitochondrial dysfunction causing cardiac sodium channel downregulation in cardiomyopathy. J Mol Cell Cardiol 54: 25–34, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu M, Liu H, Dudley SC., Jr Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ Res 107: 967–974, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu M, Sanyal S, Gao G, Gurung IS, Zhu X, Gaconnet G, Kerchner LJ, Shang LL, Huang CL, Grace A, London B, Dudley SC., Jr Cardiac Na+ current regulation by pyridine nucleotides. Circ Res 105: 737–745, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, Viswanathan PC, Pfahnl AE, Shang LL, Madhusudanan M, Baty CJ, Lagana S, Aleong R, Gutmann R, Ackerman MJ, McNamara DM, Weiss R, Dudley SC., Jr Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation 116: 2260–2268, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lu T, Lee HC, Kabat JA, Shibata EF. Modulation of rat cardiac sodium channel by the stimulatory G protein alpha subunit. J Physiol 518: 371–384, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ma J, Luo A, Wu L, Wan W, Zhang P, Ren Z, Zhang S, Qian C, Shryock JC, Belardinelli L. Calmodulin kinase II and protein kinase C mediate the effect of increased intracellular calcium to augment late sodium current in rabbit ventricular myocytes. Am J Physiol Cell Physiol 302: C1141–C1151, 2012 [DOI] [PubMed] [Google Scholar]
- 64.Maguy A, Le Bouter S, Comtois P, Chartier D, Villeneuve L, Wakili R, Nishida K, Nattel S. Ion channel subunit expression changes in cardiac Purkinje fibers: a potential role in conduction abnormalities associated with congestive heart failure. Circ Res 104: 1113–1122, 2009 [DOI] [PubMed] [Google Scholar]
- 65.Maier LS. A novel mechanism for the treatment of angina, arrhythmias, and diastolic dysfunction: inhibition of late INa using ranolazine. J Cardiovasc Pharmacol 54: 279–286, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maltsev VA, Kyle JW, Undrovinas A. Late Na+ current produced by human cardiac Na+ channel isoform Nav1.5 is modulated by its beta1 subunit. J Physiol Sci 59: 217–225, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maltsev VA, Reznikov V, Undrovinas NA, Sabbah HN, Undrovinas A. Modulation of late sodium current by Ca2+, calmodulin, and CaMKII in normal and failing dog cardiomyocytes: similarities and differences. Am J Physiol Heart Circ Physiol 294: H1597–H1608, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maltsev VA, Sabbab HN, Undrovinas AI. Down-regulation of sodium current in chronic heart failure: effect of long-term therapy with carvedilol. Cell Mol Life Sci 59: 1561–1568, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maltsev VA, Sabbah HN, Higgins RS, Silverman N, Lesch M, Undrovinas AI. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation 98: 2545–2552, 1998 [DOI] [PubMed] [Google Scholar]
- 70.Maltsev VA, Silverman N, Sabbah HN, Undrovinas AI. Chronic heart failure slows late sodium current in human and canine ventricular myocytes: implications for repolarization variability. Eur J Heart Fail 9: 219–227, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maltsev VA, Undrovinas A. Late sodium current in failing heart: friend or foe? Prog Biophys Mol Biol 96: 421–451, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mao W, You T, Ye B, Li X, Dong HH, Hill JA, Li F, Xu H. Reactive oxygen species suppress cardiac NaV1.5 expression through Foxo1. PLoS One 7: e32738, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marionneau C, Brunet S, Flagg TP, Pilgram TK, Demolombe S, Nerbonne JM. Distinct cellular and molecular mechanisms underlie functional remodeling of repolarizing K+ currents with left ventricular hypertrophy. Circ Res 102: 1406–1415, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marionneau C, Lichti CF, Lindenbaum P, Charpentier F, Nerbonne JM, Townsend RR, Merot J. Mass spectrometry-based identification of native cardiac Nav1.5 channel alpha subunit phosphorylation sites. J Proteome Res 11: 5994–6007, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Meadows LS, Isom LL. Sodium channels as macromolecular complexes: implications for inherited arrhythmia syndromes. Cardiovasc Res 67: 448–458, 2005 [DOI] [PubMed] [Google Scholar]
- 76.Meregalli PG, Wilde AA, Tan HL. Pathophysiological mechanisms of Brugada syndrome: depolarization disorder, repolarization disorder, or more? Cardiovasc Res 67: 367–378, 2005 [DOI] [PubMed] [Google Scholar]
- 77.Milstein ML, Musa H, Balbuena DP, Anumonwo JM, Auerbach DS, Furspan PB, Hou L, Hu B, Schumacher SM, Vaidyanathan R, Martens JR, Jalife J. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc Natl Acad Sci USA 109: E2134–E2143, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mishra S, Undrovinas NA, Maltsev VA, Reznikov V, Sabbah HN, Undrovinas A. Post-transcriptional silencing of SCN1B and SCN2B genes modulates late sodium current in cardiac myocytes from normal dogs and dogs with chronic heart failure. Am J Physiol Heart Circ Physiol 301: H1596–H1605, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Moreno JD, Clancy CE. Pathophysiology of the cardiac late Na current and its potential as a drug target. J Mol Cell Cardiol 52: 608–619, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moreno JD, Zhu ZI, Yang PC, Bankston JR, Jeng MT, Kang C, Wang L, Bayer JD, Christini DJ, Trayanova NA, Ripplinger CM, Kass RS, Clancy CE. A computational model to predict the effects of class I anti-arrhythmic drugs on ventricular rhythms. Sci Transl Med 3: 98ra83, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mori M, Konno T, Ozawa T, Murata M, Imoto K, Nagayama K. Novel interaction of the voltage-dependent sodium channel (VDSC) with calmodulin: does VDSC acquire calmodulin-mediated Ca2+-sensitivity? Biochemistry 39: 1316–1323, 2000 [DOI] [PubMed] [Google Scholar]
- 82.Motoike HK, Liu H, Glaaser IW, Yang AS, Tateyama M, Kass RS. The Na+ channel inactivation gate is a molecular complex: a novel role of the COOH-terminal domain. J Gen Physiol 123: 155–165, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Murphy BJ, Rogers J, Perdichizzi AP, Colvin AA, Catterall WA. cAMP-dependent phosphorylation of two sites in the alpha subunit of the cardiac sodium channel. J Biol Chem 271: 28837–28843, 1996 [DOI] [PubMed] [Google Scholar]
- 84.Nakajima T, Davies SS, Matafonova E, Potet F, Amarnath V, Tallman KA, Serwa RA, Porter NA, Balser JR, Kupershmidt S, Roberts LJ., 3rd Selective gamma-ketoaldehyde scavengers protect Nav1.5 from oxidant-induced inactivation. J Mol Cell Cardiol 48: 352–359, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nattel S. Effects of heart disease on cardiac ion current density versus current amplitude: important conceptual subtleties in the language of arrhythmogenic ion channel remodeling. Circ Res 102: 1298–1300, 2008 [DOI] [PubMed] [Google Scholar]
- 86.Nattel S, Maguy A, Le Bouter S, Yeh YH. Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev 87: 425–456, 2007 [DOI] [PubMed] [Google Scholar]
- 87.Noorman M, Hakim S, Kessler E, Groeneweg JA, Cox MG, Asimaki A, van Rijen HV, van Stuijvenberg L, Chkourko H, van der Heyden MA, Vos MA, de Jonge N, van der Smagt JJ, Dooijes D, Vink A, de Weger RA, Varro A, de Bakker JM, Saffitz JE, Hund TJ, Mohler PJ, Delmar M, Hauer RN, van Veen TA. Remodeling of the cardiac sodium channel, connexin43, and plakoglobin at the intercalated disk in patients with arrhythmogenic cardiomyopathy. Heart Rhythm 10: 412–419, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Palaniyandi SS, Sun L, Ferreira JC, Mochly-Rosen D. Protein kinase C in heart failure: a therapeutic target? Cardiovasc Res 82: 229–239, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Palygin OA, Pettus JM, Shibata EF. Regulation of caveolar cardiac sodium current by a single Gsalpha histidine residue. Am J Physiol Heart Circ Physiol 294: H1693–H1699, 2008 [DOI] [PubMed] [Google Scholar]
- 90.Park KS, Mohapatra DP, Misonou H, Trimmer JS. Graded regulation of the Kv2.1 potassium channel by variable phosphorylation. Science 313: 976–979, 2006 [DOI] [PubMed] [Google Scholar]
- 91.Partemi S, Batlle M, Berne P, Berruezo A, Campos B, Mont L, Riuro H, Roig E, Perez-Villa F, Ortiz J, Pascali VL, Oliva A, Brugada R, Brugada J. Analysis of the arrhythmogenic substrate in human heart failure. Cardiovasc Pathol 22: 133–140, 2013 [DOI] [PubMed] [Google Scholar]
- 92.Payandeh J, Gamal El-Din T.M, Scheuer T, Zheng N, Catterall WA. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 486: 135–139, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Payne ME, Fong YL, Ono T, Colbran RJ, Kemp BE, Soderling TR, Means AR. Calcium/calmodulin-dependent protein kinase II. Characterization of distinct calmodulin binding and inhibitory domains. J Biol Chem 263: 7190–7195, 1988 [PubMed] [Google Scholar]
- 94.Petitprez S, Zmoos AF, Ogrodnik J, Balse E, Raad N, El-Haou S, Albesa M, Bittihn P, Luther S, Lehnart SE, Hatem SN, Coulombe A, Abriel H. SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1.5 in cardiomyocytes. Circ Res 108: 294–304, 2011 [DOI] [PubMed] [Google Scholar]
- 95.Pitt GS. Calmodulin and CaMKII as molecular switches for cardiac ion channels. Cardiovasc Res 73: 641–647, 2007 [DOI] [PubMed] [Google Scholar]
- 96.Potet F, Chagot B, Anghelescu M, Viswanathan PC, Stepanovic SZ, Kupershmidt S, Chazin WJ, Balser JR. Functional interactions between distinct sodium channel cytoplasmic domains through the action of calmodulin. J Biol Chem 284: 8846–8854, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pu J, Boyden PA. Alterations of Na+ currents in myocytes from epicardial border zone of the infarcted heart. A possible ionic mechanism for reduced excitability and postrepolarization refractoriness. Circ Res 81: 110–119, 1997 [DOI] [PubMed] [Google Scholar]
- 98.Qu Y, Rogers J, Tanada T, Scheuer T, Catterall WA. Modulation of cardiac Na+ channels expressed in a mammalian cell line and in ventricular myocytes by protein kinase C. Proc Natl Acad Sci USA 91: 3289–3293, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Qu Y, Rogers JC, Tanada TN, Catterall WA, Scheuer T. Phosphorylation of S1505 in the cardiac Na+ channel inactivation gate is required for modulation by protein kinase C. J Gen Physiol 108: 375–379, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Remme CA, Wilde AA, Bezzina CR. Cardiac sodium channel overlap syndromes: different faces of SCN5A mutations. Trends Cardiovasc Med 18: 78–87, 2008 [DOI] [PubMed] [Google Scholar]
- 101.Ruan Y, Liu N, Priori SG. Sodium channel mutations and arrhythmias. Nat Rev Cardiol 6: 337–348, 2009 [DOI] [PubMed] [Google Scholar]
- 102.Sakakibara Y, Furukawa T, Singer DH, Jia H, Backer CL, Arentzen CE, Wasserstrom JA. Sodium current in isolated human ventricular myocytes. Am J Physiol Heart Circ Physiol 265: H1301–H1309, 1993 [DOI] [PubMed] [Google Scholar]
- 103.Sarhan MF, Tung CC, Van Petegem F, Ahern CA. Crystallographic basis for calcium regulation of sodium channels. Proc Natl Acad Sci USA 109: 3558–3563, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sarhan MF, Van Petegem F, Ahern CA. A double tyrosine motif in the cardiac sodium channel domain III–IV linker couples calcium-dependent calmodulin binding to inactivation gating. J Biol Chem 284: 33265–33274, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sato PY, Coombs W, Lin X, Nekrasova O, Green KJ, Isom LL, Taffet SM, Delmar M. Interactions between ankyrin-G, plakophilin-2, and connexin43 at the cardiac intercalated disc. Circ Res 109: 193–201, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schmidt JW, Catterall WA. Biosynthesis and processing of the alpha subunit of the voltage-sensitive sodium channel in rat brain neurons. Cell 46: 437–444, 1986 [DOI] [PubMed] [Google Scholar]
- 107.Schreibmayer W, Frohnwieser B, Dascal N, Platzer D, Spreitzer B, Zechner R, Kallen RG, Lester HA. Beta-adrenergic modulation of currents produced by rat cardiac Na+ channels expressed in Xenopus laevis oocytes. Receptors Channels 2: 339–350, 1994 [PubMed] [Google Scholar]
- 108.Shang LL, Pfahnl AE, Sanyal S, Jiao Z, Allen J, Banach K, Fahrenbach J, Weiss D, Taylor WR, Zafari AM, Dudley SC., Jr Human heart failure is associated with abnormal C-terminal splicing variants in the cardiac sodium channel. Circ Res 101: 1146–1154, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shy D, Gillet L, Abriel H. Cardiac sodium channel NaV1.5 distribution in myocytes via interacting proteins: the multiple pool model. Biochim Biophys Acta 1833: 886–894, 2013 [DOI] [PubMed] [Google Scholar]
- 110.Soltis AR, Saucerman JJ. Synergy between CaMKII substrates and beta-adrenergic signaling in regulation of cardiac myocyte Ca2+ handling. Biophys J 99: 2038–2047, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sossalla S, Maurer U, Schotola H, Hartmann N, Didie M, Zimmermann WH, Jacobshagen C, Wagner S, Maier LS. Diastolic dysfunction and arrhythmias caused by overexpression of CaMKIIdelta(C) can be reversed by inhibition of late Na+ current. Basic Res Cardiol 106: 263–272, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85: 1093–1129, 2005 [DOI] [PubMed] [Google Scholar]
- 113.Tan HL, Kupershmidt S, Zhang R, Stepanovic S, Roden DM, Wilde AA, Anderson ME, Balser JR. A calcium sensor in the sodium channel modulates cardiac excitability. Nature 415: 442–447, 2002 [DOI] [PubMed] [Google Scholar]
- 114.Tester DJ, Valdivia C, Harris-Kerr C, Alders M, Salisbury BA, Wilde AA, Makielski JC, Ackerman MJ. Epidemiologic, molecular, and functional evidence suggest A572D-SCN5A should not be considered an independent LQT3-susceptibility mutation. Heart Rhythm 7: 912–919, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Trenor B, Cardona K, Gomez JF, Rajamani S, Ferrero JM, Jr, Belardinelli L, Saiz J. Simulation and mechanistic investigation of the arrhythmogenic role of the late sodium current in human heart failure. PLoS One 7: e32659, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ueda K, Valdivia C, Medeiros-Domingo A, Tester DJ, Vatta M, Farrugia G, Ackerman MJ, Makielski JC. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci USA 105: 9355–9360, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ufret-Vincenty CA, Baro DJ, Lederer WJ, Rockman HA, Quinones LE, Santana LF. Role of sodium channel deglycosylation in the genesis of cardiac arrhythmias in heart failure. J Biol Chem 276: 28197–28203, 2001 [DOI] [PubMed] [Google Scholar]
- 118.Undrovinas AI, Maltsev VA, Sabbah HN. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: role of sustained inward current. Cell Mol Life Sci 55: 494–505, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, Kamp TJ, Makielski JC. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol 38: 475–483, 2005 [DOI] [PubMed] [Google Scholar]
- 120.Valdivia CR, Ueda K, Ackerman MJ, Makielski JC. GPD1L links redox state to cardiac excitability by PKC-dependent phosphorylation of the sodium channel SCN5A. Am J Physiol Heart Circ Physiol 297: H1446–H1452, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, Ackerman MJ. Molecular and functional characterization of novel glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) mutations in sudden infant death syndrome. Circulation 116: 2253–2259, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Van Petegem F, Lobo PA, Ahern CA. Seeing the forest through the trees: towards a unified view on physiological calcium regulation of voltage-gated sodium channels. Biophys J 103: 2243–2251, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, Tester DJ, Balijepalli RC, Foell JD, Li Z, Kamp TJ, Towbin JA. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation 114: 2104–2112, 2006 [DOI] [PubMed] [Google Scholar]
- 124.Veldkamp MW, Viswanathan PC, Bezzina C, Baartscheer A, Wilde AA, Balser JR. Two distinct congenital arrhythmias evoked by a multidysfunctional Na+ channel. Circ Res 86: E91–E97, 2000 [DOI] [PubMed] [Google Scholar]
- 125.Verrier RL, Kumar K, Nieminen T, Belardinelli L. Mechanisms of ranolazine's dual protection against atrial and ventricular fibrillation. Europace 15: 317–324, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest 116: 3127–3138, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wagner S, Hacker E, Grandi E, Weber SL, Dybkova N, Sossalla S, Sowa T, Fabritz L, Kirchhof P, Bers DM, Maier LS. Ca/calmodulin kinase II differentially modulates potassium currents. Circ Arrhythm Electrophysiol 2: 285–294, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T, Anderson ME, Grandi E, Bers DM, Backs J, Belardinelli L, Maier LS. Reactive oxygen species-activated Ca/calmodulin kinase IIdelta is required for late INa augmentation leading to cellular Na and Ca overload. Circ Res 108: 555–565, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang GK, Wang SY. Modifications of human cardiac sodium channel gating by UVA light. J Membr Biol 189: 153–165, 2002 [DOI] [PubMed] [Google Scholar]
- 130.Wang J, Liu X, Sentex E, Takeda N, Dhalla NS. Increased expression of protein kinase C isoforms in heart failure due to myocardial infarction. Am J Physiol Heart Circ Physiol 284: H2277–H2287, 2003 [DOI] [PubMed] [Google Scholar]
- 131.Wiegerinck RF, Verkerk AO, Belterman CN, van Veen TA, Baartscheer A, Opthof T, Wilders R, de Bakker JM, Coronel R. Larger cell size in rabbits with heart failure increases myocardial conduction velocity and QRS duration. Circulation 113: 806–813, 2006 [DOI] [PubMed] [Google Scholar]
- 132.Wilde AA, Brugada R. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel. Circ Res 108: 884–897, 2011 [DOI] [PubMed] [Google Scholar]
- 133.Wingo TL, Shah VN, Anderson ME, Lybrand TP, Chazin WJ, Balser JR. An EF-hand in the sodium channel couples intracellular calcium to cardiac excitability. Nat Struct Mol Biol 11: 219–225, 2004 [DOI] [PubMed] [Google Scholar]
- 134.Wu L, Ma J, Li H, Wang C, Grandi E, Zhang P, Luo A, Bers DM, Shryock JC, Belardinelli L. Late sodium current contributes to the reverse rate-dependent effect of IKr inhibition on ventricular repolarization. Circulation 123: 1713–1720, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Xi Y, Wu G, Yang L, Han K, Du Y, Wang T, Lei X, Bai X, Ma A. Increased late sodium currents are related to transcription of neuronal isoforms in a pressure-overload model. Eur J Heart Fail 11: 749–757, 2009 [DOI] [PubMed] [Google Scholar]
- 136.Xie LH, Chen F, Karagueuzian HS, Weiss JN. Oxidative-stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ Res 104: 79–86, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation 100: 1660–1666, 1999 [DOI] [PubMed] [Google Scholar]
- 138.Yang T, Atack TC, Stroud DM, Zhang W, Hall L, Roden DM. Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ Res 111: 322–332, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yarbrough TL, Lu T, Lee HC, Shibata EF. Localization of cardiac sodium channels in caveolin-rich membrane domains: regulation of sodium current amplitude. Circ Res 90: 443–449, 2002 [DOI] [PubMed] [Google Scholar]
- 140.Zhou J, Shin HG, Yi J, Shen W, Williams CP, Murray KT. Phosphorylation and putative ER retention signals are required for protein kinase A-mediated potentiation of cardiac sodium current. Circ Res 91: 540–546, 2002 [DOI] [PubMed] [Google Scholar]
- 141.Zhou J, Yi J, Hu N, George AL, Jr, Murray KT. Activation of protein kinase A modulates trafficking of the human cardiac sodium channel in Xenopus oocytes. Circ Res 87: 33–38, 2000 [DOI] [PubMed] [Google Scholar]
- 142.Zimmer T, Biskup C, Dugarmaa S, Vogel F, Steinbis M, Bohle T, Wu YS, Dumaine R, Benndorf K. Functional expression of GFP-linked human heart sodium channel (hH1) and subcellular localization of the a subunit in HEK293 cells and dog cardiac myocytes. J Membr Biol 186: 1–12, 2002 [DOI] [PubMed] [Google Scholar]