

Abstract
During angiotensin II (ANG II)-dependent hypertension, ANG II stimulates, while hypertension inhibits, Na+ transporter activity to balance Na+ output to input. This study tests the hypothesis that ANG II infusion activates Na+ transporters in the distal nephron while inhibiting transporters along the proximal nephron. Male Sprague-Dawley rats were infused with ANG II (400 ng·kg−1·min−1) or vehicle for 2 wk. Kidneys were dissected (cortex vs. medulla) or fixed for immunohistochemistry (IHC). ANG II increased mean arterial pressure by 40 mmHg, urine Na+ by 1.67-fold, and urine volume by 3-fold, evidence for hypertension and pressure natriuresis. Na+ transporters' abundance and activation [assessed by phosphorylation (-P) or proteolytic cleavage] were measured by immunoblot. During ANG II infusion Na+/H+ exchanger 3 (NHE3) abundance decreased in both cortex and medulla; Na-K-2Cl cotransporter 2 (NKCC2) decreased in medullary thick ascending loop of Henle (TALH) and increased, along with NKCC2-P, in cortical TALH; Na-Cl cotransporter (NCC) and NCC-P increased in the distal convoluted tubule; and epithelial Na+ channel subunits and their cleaved forms were increased in both cortex and medulla. Like NKCC2, STE20/SPS1-related proline alanine-rich kinase (SPAK) and SPAK-P were decreased in medulla and increased in cortex. By IHC, during ANG II NHE3 remained localized to proximal tubule microvilli at lower abundance, and the differential regulation of NKCC2 and NKCC2-P in cortex versus medulla was evident. In summary, ANG II infusion increases Na+ transporter abundance and activation from cortical TALH to medullary collecting duct while the hypertension drives a natriuresis response evident as decreased Na+ transporter abundance and activation from proximal tubule through medullary TALH.
Keywords: pressure natriuresis, NHE3, NCC, NKCC2, SPAK, ENaC
hypertension is the leading cause of stroke and cardiovascular diseases and is the leading risk factor for global disease burden (20). Evidence suggests that the renin-angiotensin system (RAS) is activated during hypertension since inhibitors of ANG II production or action are effective at lowering blood pressure in most hypertensives. Chronic ANG II infusion, a well-studied model that mimics the RAS activation of Goldblatt hypertension (11, 13), stimulates both Na+ reabsorption and vascular contractility, which elevates both extracellular fluid volume (ECFV) and blood pressure, triggering a pressure-natriuresis response to normalize ECFV (13). How the ANG II-stimulated antinatriuretic responses and the hypertension-stimulated natriuretic responses are integrated along the nephron during chronic ANG II infusion has not been investigated systematically; however, studies of specific regions provide evidence for increased abundance and/or activity of distal convoluted tubule (DCT) NCC and cortical collecting duct (CD) epithelial sodium channel (ENaC) (3, 6, 11, 13, 41, 49), and decreased abundance of proximal tubule (PT) NHE3 and TALH NKCC2 (13).
Recent studies have demonstrated activation of sodium transporters by phosphorylation and by proteolytic cleavage. SPAK and the oxidative-stress responsive kinase 1 (OSR1) are homologous kinases that can phosphorylate and, thus, activate NKCC2 and NCC (34, 35). ANG II infusion increases total abundance and phosphorylation of SPAK, but not OSR1 (6, 11, 41), and SPAK, in turn, is regulated by With-No-Lysine (WNK) kinases (6). The ANG II-dependent activation of NCC and SPAK is independent of aldosterone stimulation (41) but depends on an intact intrarenal RAS (11). ENaCs (from late DCT through CD) are activated by extracellular proteolytic cleavage of the α and γ subunits by tubular fluid proteases (16, 29) and ANG II infusion increase α and γ proteolysis (11).
In this study we tested the hypothesis that during ANG II-dependent hypertension there is nephron region-specific stimulation of sodium transporters by ANG II balanced by nephron-specific inhibition of sodium transporters by hypertension, evidenced by respective increases and decreases in transporter abundance, phosphorylation, and cleavage. The results indicate that during ANG II hypertension there is inhibition of transporters from PT through medullary TALH (inhibition of cortical and medullary NHE3, medullary NKCC2, SPAK, and Na,K-ATPase) and stimulation of transporters from cortical TALH through medullary CD (stimulation of cortical NKCC2, NCC, and SPAK and both cortical and medullary ENaC). These findings define the regions that provoke the rise in Na+ reabsorption, extracellular fluid volume (ECFV), and blood pressure and also define the regions where these elevations in ECFV and/or blood pressure can override the effects of ANG II to suppress Na+ reabsorption and normalize ECFV.
MATERIALS AND METHODS
Animal protocols.
All animal procedures were approved by the Institutional Animal Care and Use Committee of the Keck School of Medicine of the University of Southern California and were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Male Sprague-Dawley rats (225–250 g body wt) obtained from Harlan Laboratories (San Diego, CA) were anesthetized intramuscularly with 200 μl of ketamine and xylazine (mixed at a 1:1 volume ratio), randomized to two groups (n = 8 each), and implanted with osmotic minipumps (Alzet, model 2002, Cupertino, CA) subcutaneously containing either vehicle (5% acetic acid, control) or ANG II (400 ng·kg−1·min−1; Sigma; ANG II infused). Infusion was continued for 14 days during which the rats had free access to normal vivarium diet and drinking water.
Physiological measurements.
Rats were placed in metabolic cages overnight (16 h) for urine collection both before minipump implantation and before they were euthanized. Urine volumes were measured by graduated cylinders, urinary [Na+] and [K+] were measured by flame photometry (Radiometer FLM3), and osmolality was measured with an osmometer (Precision Systems, μOsmette). Urinary angiotensinogen was assessed by immunoblot in a constant fraction (0.02%) of the overnight urine volume.
After 2 wk of ANG II or vehicle infusion, rats were anesthetized intraperitoneally with Inactin (125 mg/kg; Sigma), body temperature was maintained thermostatically at 37°C, and cannulas were inserted into the jugular vein for fluid infusion (0.9% NaCl + 4% BSA, 50 μl/min) to maintain euvolemia and into the carotid artery for blood pressure recording. Mean arterial pressure (MAP) was calculated as the sum of one-third systolic blood pressure and two-thirds diastolic blood pressure. After stable blood pressure was recorded, renal arteries were clamped, kidneys excised and placed in iced saline; a blood sample was collected from the carotid cannula, and hearts were removed, flushed with PBS, blotted and weighed. Plasma samples were prepared from the blood sample by centrifugation for electrolyte measurements.
Homogenate preparation and quantitative immunoblotting.
Kidney cortex and medulla were immediately dissected manually and separately diced and homogenized as described in detail previously (27), quick frozen in aliquots in liquid N2; protein concentration was determined by BCA assay (Pierce Thermo, Rockford, IL). Cortical and medullary homogenates were denatured in SDS-PAGE sample buffer for 20 min at 60°C (27). To verify uniform protein concentration, 10 μg of protein from each sample was resolved by SDS-PAGE, stained with Coomassie blue, and multiple random bands quantified and determined to be uniform (if not, protein reassessed and gel rerun). For immunoblot, each sample was run at both one and one-half amounts to verify linearity of the detection system on each immunoblot (only one amount is shown in figures). Antibodies used in this study, dilutions, and vendors are catalogued in Table 1. Blots were never stripped and reprobed. Signals were detected with Odyssey Infrared Imaging System (Li-COR) and quantified by accompanying software. Arbitrary density units collected were normalized to mean intensity of control group, defined as 1.0. Since the samples were run twice (at 1 and 0.5), the normalized values were averaged and mean values compiled for statistical analysis.
Table 1.
Immunoblot and immunofluorescence antibody details
| Protein/Lane, μg |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Antibody (Ab) Target | Primary Ab Supplier | Cortex | Medulla | Ab host | Dilution | Incubation Time | Secondary Ab Supplier | Ab Host and Target | Dilution | Incubation Time |
| Immunoblots | ||||||||||
| AMPK | Cell Signaling | 80 | 37 | Rb | 1:1,500 | O/N | Invitrogen | GAR680 | 1:5,000 | 1 h |
| AMPKpT172 | Cell Signaling | 150 | 37 | Rb | 1:1,500 | 2 h | Invitrogen | GAR680 | 1:5,000 | 1 h |
| Angiotensinogen | Sernia | Sheep | 1:5,000 | O/N | Invitrogen | DAS680 | 1:5,000 | 1 h | ||
| NCC | McDonough | 60 | Rb | 1:5,000 | 2 h | Invitrogen | GAR680 | 1:5,000 | 1 h | |
| NCCpS71 | Loffing (Zurich) | 30 | Rb | 1:5,000 | 2 h | Invitrogen | GAR680 | 1:5,000 | 1 h | |
| NCCpS89 | Loffing (Zurich) | 30 | Rb | 1:5,000 | 2 h | Invitrogen | GAR680 | 1:5,000 | 1 h | |
| NCCpT53 | Loffing (Zurich) | 60 | Rb | 1:5,000 | 2 h | Invitrogen | GAR680 | 1:5,000 | 1 h | |
| NHE3 | McDonough | 30 | 40 | Rb | 1:2,000 | O/N | Invitrogen | GAR680 | 1:5,000 | 1 h |
| NHE3pS552 | Santa Cruz | 30 | 40 | Mu | 1:1,000 | 2 h | LiCor | GAM800 | 1:5,000 | 1 h |
| NKAα1 | Kashgarian (Yale) | 2 | 2 | Mu | 1:200 | 2 h | Invitrogen | GAM680 | 1:5,000 | 1 h |
| NKAβ1 | McDonough | 20 | 2 | Rb | 1:500 | O/N | Invitrogen | GAR680 | 1:5,000 | 1 h |
| NKCC | C.Lytle (UCR) | 20 | 8 | Mu | 1:6,000 | O/N | Invitrogen | GAM680 | 1:5,000 | 1 h |
| NKCCpT96T101 | Forbush (Yale) | 20 | 8 | Rb | 1:2,000 | 2 h | LiCor | GAR800 | 1:5,000 | 1 h |
| OSR1 | DSTT, Dundee, UK | 80 | 40 | Sheep | 1:1,000 | O/N | Invitrogen | DAS680 | 1:5,000 | 1 h |
| SPAK | Delpire (Vanderbilt) | 20 | 4 | Rb | 1:3,000 | O/N | Invitrogen | GAR680 | 1:5,000 | 1 h |
| SPAKpS373/OSR1pS325 | DSTT, Dundee, UK | 80 | 40 | Sheep | 1:1,000 | 2 h | Invitrogen | DAS680 | 1:5,000 | 1 h |
| αENaC | Loffing (Zurich) | 80 | 37 | Rb | 1:5,000 | O/N | Invitrogen | GAR680 | 1:5,000 | 1 h |
| βENaC | Loffing (Zurich) | 80 | 37 | Rb | 1:15,000 | 2 h | Invitrogen | GAR680 | 1:5,000 | 1 h |
| γENaC | Palmer (Cornell) | 80 | 25 | Rb | 1:2,500 | O/N | Invitrogen | GAR680 | 1:500 | O/N |
| Immunochemistry | ||||||||||
| NCC | McDonough | Rb | 1:5,000 | 1.5 h | Mol. Probes | GAR488 | 1:500 | 1 h | ||
| NCCpT58 | DSTT, Dundee, UK | Sheep | 1:1,600 | 1.5 h | Mol. Probes | DAS568 | 1:500 | 1 h | ||
| NHE3 | McDonough | Rb | 1:100 | 1.5 h | Mol. Probes | GAR488 | 1:500 | 1 h | ||
| NKCC2 | C.Lytle (UCR) | Mu | 1:2,000 | 1.5 h | Mol. Probes | GAM555 | 1:500 | 1 h | ||
| NKCCpT96T101 | Forbush (Yale) | Rb | 1:500 | 1.5 h | Mol. Probes | GAR488 | 1:500 | 1 h | ||
Mu, mouse; Rb, rabbit; O/N, overnight; GAR, goat anti-rabbit; GAM, goat anti-mouse; DAS, donkey anti-sheep.
A new anti-NCC antibody was generated by immunizing rabbits with a peptide sequence from NH2-terminal amino acids 74–96 (PGEPRKVRPTLADLHSFLKQEG) of NCC (31) at Antibodies (Davis, CA). Serum was collected and tested for specificity by immunoblot against 40 μg of kidney homogenates from rat cortex (which contains NCC), medulla (which does not contain NCC but does contain NKCC to assess cross reactivity), and mouse whole kidney. Blots were probed with antibody diluted to 1:5,000. A frozen slice of renal cortex was probed with the new anti-NCC antibody as well as an antibody directed to NCCpT58.
Immunohistochemistry.
In a separate set of control vs. ANG II-infused rats (n = 4), kidneys were perfusion-fixed via the dorsal aorta as previously described (36). The fixed tissues were cryoprotected by overnight incubation with 30% sucrose in PBS, embedded in Tissue-Tek O.C.T. compound (Sakura Finetek, Torrance, CA), and frozen on dry ice. Cryosections (5 μm) from control vs. ANG II animals were sliced and transferred to the same Superfrost Plus-charged glass slide (Fisher) for direct side-by-side processing and viewing. For immunofluorescent labeling, the sections were rehydrated, washed, and blocked with 1% BSA/PBS before antibody incubation. All antibodies were diluted in 1% BSA/PBS (Table 1). The sections were mounted in Prolong Antifade containing DAPI (Invitrogen) and air dried overnight. Samples were viewed with a ZEISS LSM 510 confocal system. Fluorescence excitation and detector settings were the same for imaging sections from control and ANG II-treated kidneys.
Statistical analysis.
Differences in physiological parameters and in protein total abundance and phosphorylation were assessed by unpaired two-tailed Student's t-test. Difference in urinary angiotensinogen between control and ANG II groups before and after 2 wk of infusion was assessed by two-way ANOVA analysis. Data were expressed as means ± SE. Differences were regarded significant at P < 0.05.
RESULTS
Effects of ANG II-dependent hypertension on physiological parameters.
Baseline body weight, urine volume (V), [Na+], [K+], and osmolality were similar before treatment in both control and pre-ANG II groups (Table 2). As summarized in Table 3, ANG II infusion had the following effects: increased MAP measured from the carotid artery, increased heart weight, reduced rate of weight gain as reported previously (37), increased overnight urine volume >3-fold and increased urinary Na+, K+ and osmolar excretion indicative of chronic pressure-natriuresis and diuresis. Kaliuresis and increased solute excretion could be attributed to the reduced skeletal muscle mass (37) or increased food consumption (not measured). Interestingly, ANG II infusion significantly reduced plasma [Na+], likely associated with the dipsogenic effects of ANG II (39), which is also reflected in the threefold increase in urine volume. ANG II infusion increased urinary angiotensinogen levels more than 10 fold (Fig. 1), evidence for activation of intrarenal RAS (11).
Table 2.
Baseline physiological parameters
| Parameter | Control (n = 8) | Pre-ANG II (n = 8) |
|---|---|---|
| Initial body wt, g | 234 ± 3 | 239 ± 3 |
| UV, ml | 8.2 ± 0.6 | 9.5 ± 2.2 |
| UNaV, mmol | 1.5 ± 0.1 | 1.5 ± 0.1 |
| UKV, mmol | 2.6 ± 0.1 | 2.7 ± 0.1 |
| UK/UNa ratio | 1.9 ± 0.2 | 1.8 ± 0.1 |
| Uosm, mosmol/kgH2O | 1,768 ± 77 | 1,745 ± 157 |
| UosmV, mosmol | 14.4 ± 0.8 | 14.2 ± 0.8 |
Values represent means ± SE. Urine samples were collected over a 16-h period. UV, urine volume; UNaV, urinary Na+ excretion; UKV, urinary K+ excretion; Uosm, urine osmolality; UosmV, urine osmolality excretion. Urine was collected overnight (16 h) in metabolic cages.
Table 3.
Effects of chronic ANG II (400 ng·kg−1·min·1) infusion on physiological parameters
| Parameter | Control (n = 8) | ANG II (n = 8) |
|---|---|---|
| Final body wt, g | 306 ± 5 | 263 ± 9* |
| Body wt gain per day, g/day | 5.4 ± 0.3 | 1.7 ± 0.7* |
| MAP, mmHg | 119 ± 4 | 158 ± 5* |
| Heart wt, g/100 g body wt | 0.26 ± 0.002 | 0.35 ± 0.01* |
| PNa, mmol/l | 136.9 ± 1.5 | 131.1 ± 1.7* |
| PK, mmol/l | 4.6 ± 0.3 | 4.5 ± 0.3 |
| UV, ml | 9.1 ± 0.9 | 34.9 ± 4.0* |
| UNaV, mmol | 1.5 ± 0.1 | 2.5 ± 0.2* |
| UKV, mmol | 2.8 ± 0.1 | 3.4 ± 0.2* |
| Uosm, mosmol/kgH2O | 1,826 ± 115 | 687 ± 56* |
| UosmV, mosmol | 15.8 ± 0.5 | 22.6 ± 1.3* |
Values represent means ± SE. MAP, mean arterial pressure; P, plasma. Urine was collected overnight (16 h) in metabolic cages.
P < 0.05 vs. control.
Fig. 1.

Chronic ANG II (400 ng·kg−1·min−1) infusion increases urinary angiotensinogen. A: representative immunoblots of angiotensinogen in urine of rats infused with either vehicle (control) or ANG II (n = 8 each) assayed in 0.02% of overnight urine volume before (baseline) and after 2 wk of ANG II infusion (final). B: relative abundance displayed as individual records with means ± SE. Density values normalized to mean density of baseline values from their respective groups. *P < 0.05, final ANG II vs. final control. #P < 0.05, final ANG II vs. baseline ANG II, assessed by two-way ANOVA analysis.
Effects of ANG II-dependent hypertension on NHE3.
NHE3 is expressed in the apical membranes of epithelial cells along the entire proximal tubule (PT) as well as in the thick ascending loop of Henle (TALH); both tubule segments span cortex and medulla. ANG II infusion decreased NHE3 abundance in both regions (Fig. 2, A and B) to 0.78 ± 0.06 of control in cortex and to 0.54 ± 0.04 of control in medulla. NHE3 phosphorylation (NHE3pS552), a marker for its transit to the base of the microvilli (17), was unchanged in cortex and reduced in medulla. Kidney sections from cortex of control and ANG II-infused rats were placed on the same slide, costained with NHE3 (green) and the microvilli marker villin (red), and imaged at the same settings (Fig. 2C). In both control and ANG II-infused groups, NHE3 was resident in the microvilli overlapping with villin, and, as in the immunoblots, NHE3 staining was lower after ANG II infusion, evidence for a pressure-natriuresis response.
Fig. 2.
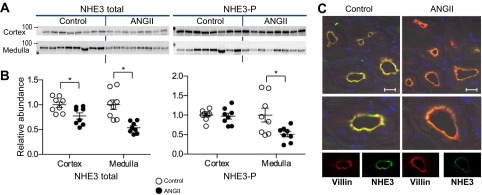
ANG II infusion decreases NHE3 abundance. A: immunoblots of total and phosphorylated NHE3 in renal cortex and medulla of rats infused with either vehicle (control) or ANG II (n = 8 each). Protein/lane in Table 1. B: relative abundance displayed as individual records with means ± SE. *P < 0.05. C: indirect immunofluorescence microscopy of NHE3 and villin in renal cortex. Sections were processed identically on same slide and imaged with the same settings. Antibody labeling specifics provided in Table 1. Bar, 20 μm.
Effects of ANG II-dependent hypertension on NKCC2.
NKCC2 is expressed in the apical membranes of TALH epithelial cells from medulla to cortex. The anti-NKCC antibody reagent used in this study recognizes not only NKCC2 but also the basolateral secretory NKCC1 isoform that has been detected along the collecting duct (21). The lack of detectable basolateral staining by immunohistochemistry with this anti-NKCC in cortex and medulla (Fig. 3) in this study indicates that the abundance of NKCC1 in kidney is quite low compared with NKCC2, and, with this caveat, we will refer to the signals detected as NKCC2. NKCC2 phosphorylation at threonine 96 and 101 (NKCC2-P) is indicative of transporter activation (9, 35). During ANG II infusion, total NKCC2 and NKCC2-P were differentially regulated in cortex vs. medulla (Fig. 3, A and B): total NKCC2 increased in cortex (to 1.71 ± 0.26 of control) and decreased in medulla (to 0.46 ± 0.06 of control); phosphorylated NKCC2 increased in cortex (to 1.74 ± 0.23 of control) and was unchanged in the medulla. Frozen sections from both groups were processed and analyzed on the same slide with same settings and colabeled with antibodies against NKCC2 (red) and NKCC2-P (green) (Fig. 3C). In cortex, location of NKCC2 was confirmed by apical localization and proximity to glomeruli. ANG II increased the fluorescent intensities of both NKCC2 and NKCC2-P in cortex, while in medulla ANG II decreased the intensities of both NKCC2 and NKCC-P compared with controls stained on the same slide. These findings suggest decreased NKCC2 transporter activity in the medullary TALH, consistent with a pressure-natriuresis effect, and increased NKCC2 activity in the cortical TALH, indicative of ANG II stimulation.
Fig. 3.

ANG II infusion provokes differential regulation of NKCC2 in cortex vs. medulla. A: immunoblots of total NKCC2 and NKCC2-P in cortex and medulla of rats infused with either vehicle (control) or ANG II (n = 8 each). Protein per lane in Table 1. B: relative abundance displayed as individual records with means ± SE. *P < 0.05. C: indirect immunofluorescence microscopy of NKCC2 and NKCC2-P. Kidneys from 2 rats were fixed for each condition. Sections were processed identically on same slide and imaged with the same settings. Two sections were fully examined and the representative images were chosen with glomeruli for identification of the cortex. Antibody labeling specifics provided in Table 1. Bar, 20 μm. G, glomerulus.
Effects of ANG II-dependent hypertension on sodium pump.
Na,K-ATPase (NKA) is expressed ubiquitously in the basolateral membranes of tubular cells. In cortex, the levels are high in both PT and DCT while in medulla NKA is primarily abundant in the TALH and low in other medullary segments (26). During ANG II infusion both NKAα1 and β1 decreased in medulla, to 0.77 ± 0.02 and 0.63 ± 0.05 of control, respectively (Fig. 4, A and B), suggesting that apical NKCC2 and basolateral NKA decrease in parallel in the medullary TALH during ANG II infusion, evidence of a pressure-natriuresis response. Cortical NKAα1 and β1 abundance, which reflects expression in both the proximal and distal tubules, was unchanged during ANG II infusion.
Fig. 4.

ANG II infusion decreases sodium pump subunits' abundance in the medulla. Immunoblots of NKAα1 (A) and NKAβ1 (B) in renal cortex and medulla of rats infused with either vehicle (control) or ANG II (n = 8). Protein per lane in Table 1. Relative abundance is displayed as individual records with means ± SE. *P < 0.05.
Effects of ANG II-dependent hypertension on NCC.
It has been established that ANG II infusion increases NCC abundance and phosphorylation (6, 11, 41). NCC-P is indicative of apical membrane localization (18) and activation (34). This study confirms that ANG II infusion increases NCC total (to 1.93 ± 0.16 of control) using a new polyclonal antibody (characterized in Fig. 5) as well as increases NCC phosphorylated at the following three sites: NCCpT53 by 5.2-fold, NCCpS71 by 2.3-fold, and NCCpS89 by 3.3-fold (Fig. 5).
Fig. 5.

Chronic ANG II (400 ng·kg−1·min−1) infusion increases NCC abundance and phosphorylation. A: immunoblots of NCC total, NCCpT53, NCCpS71, and NCCpS89 in renal cortex of rats infused with either vehicle (control) or ANG II (n = 8 each). Protein per lane in Table 1. B: relative abundance is displayed as individual records with means ± SE. Density values were normalized to mean density of control group. *P < 0.05. C: characterization of a new anti-NCC antibody produced in rabbits against NH2-terminal amino acids 74–96 (PGEPRKVRPTLADLHSFLKQEG). Forty micrograms of homogenate from untreated rat cortex (contains NCC and NKCC), medulla (contains NKCC), and mouse whole kidney were resolved and probed with the new anti-NCC diluted 1:5,000. The antibody detects a band around 150 kDa in cortex and very little in medulla, indicating that it detects NCC and not NKCC2. The band at 100 kDa in the medulla is nonspecific since there should be no NCC in the medulla, and the band at 70 kDa in mouse is unknown. D: frozen slice of renal cortex was probed with the new anti-NCC antibody as well as an antibody directed to NCCpT58. The antibodies both detect apical NCC, with little nonspecific labeling. Antibody labeling specifics provided in Table 1.
Effects of ANG II-dependent hypertension on kinases.
SPAK and the related kinase OSR1 colocalize with TALH NKCC2 and DCT NCC where they can phosphorylate the transporters and stimulate their transport activity; likewise, increased phosphorylations of SPAK and OSR1 are indicators of increased kinase activity (34, 35). Abundance and phosphorylation of SPAK and OSR1 were measured to assess whether they were differentially regulated by ANG II infusion in cortex vs. medulla. SPAK is expressed as three isoforms: full-length (FL-SPAK), SPAK2, and kidney-specific SPAK (KS-SPAK). KS-SPAK is reported to exert a dominant negative effect on both SPAK and OSR1 (24). We have previously identified FL- and KS-SPAK isoforms in rat kidney cortical and medullary homogenates and showed that the major isoform in the medulla is FL-SPAK (27). In this present study, ANG II infusion increased FL-SPAK in cortex to 2.07 ± 0.40-fold over control and decreased FL-SPAK in medulla to 0.61 ± 0.03 of control (Fig. 6). KS-SPAK was unchanged with ANG II infusion (1.03 ± 0.13) compared with control (1.00 ± 0.12); medullary KS-SPAK as well as SPAK-2 in cortex and medulla were too low to quantitate (data not shown). SPAK phosphorylated at Ser373 (SPAKpS373), an indicator of kinase activation, was increased in cortex during ANG II infusion to 1.82 ± 0.17 over control but was not significantly altered by ANG II in medulla, mimicking the pattern of NKCC2-P regulation (Fig. 3). Neither OSR1 total abundance nor OSR1 phosphorylated at Ser325 (detected with the same antibody that detects SPAKpS373) were altered by ANG II infusion (Fig. 7). Likewise, AMP-activated protein kinase (AMPK), reported to interact with and phosphorylate NKCC2 (10), was not regulated during ANG II infusion (not shown).
Fig. 6.

ANG II infusion differentially regulates SPAK and SPAK-P abundance in cortex vs. medulla. A: immunoblots of SPAK and SPAK-P in renal cortex and medulla of rats infused with either vehicle (control) or ANG II (n = 8 each). Protein per lane in Table 1. B: relative abundance displayed as individual records with means ± SE. *P < 0.05. FL, full-length SPAK; KS, kidney-specific SPAK.
Fig. 7.

Chronic ANG II (400 ng·kg−1·min−1) infusion does not change the abundance or phosphorylation of regulatory kinase oxidative stress response-1 (OSR1) in cortex or medulla. A: immunoblots of total OSR1 and OSR1-P in renal cortex and medulla of rats infused with either vehicle (control) or ANG II (n = 8). OSR1-P is detected by the same antibody used for SPAK-P. Protein per lane in Table 1. B: relative abundance displayed as individual records with means ± SE. *P < 0.05. #, Tubulin recognized by anti-OSR1 antibody (33).
Effects of ANG II-dependent hypertension on ENaC.
Epithelial Na+ channels, ENaC (made up of α, β and γ subunits), are located in the apical membranes of epithelia from the late DCT through CD. ANG II directly stimulates ENaC activity in the cortical collecting duct (CD) (23), increases αENaC protein abundance in kidney cortex (3), and increases Na+ reabsorption in the distal nephron (49). Proteolytic cleavage of α and γ has been shown to increase channel activity (15). Figure 8 demonstrates that ANG II infusion increases the abundance of the cleaved forms of α and γ in cortex (by 1.78 ± 0.23 and 1.67 ± 0.23 of control, respectively) and in medulla (by 2.23 ± 0.32 and 1.44 ± 0.13 of control, respectively). In addition, ANG II infusion increased full-length α (by 1.33 ± 0.11 in cortex and 1.83 ± 0.15 in medulla) and β subunit (by 1.24 ± 0.06 of control in cortex). These findings suggest activation of both cortical and medullary ENaC during ANG II infusion, confirming studies conducted previously in mouse (11).
Fig. 8.
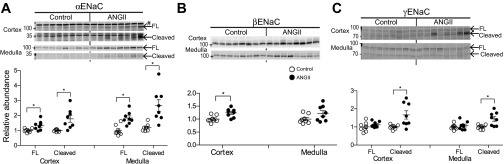
ANG II infusion increases proteolytic cleavage of the ENaC subunits in both cortex and medulla. Immunoblots of αENaC (A), βENaC (B), and γENaC (C) in renal cortex and medulla of rats infused with either vehicle (control) or ANG II (n = 8 each). Protein per lane in Table 1. Relative abundance displayed as individual records with means ± SE. *P < 0.05. FL, full-length; Cleaved, proteolytic cleavage product; #nonspecific band above FL-αENaC (40).
DISCUSSION
During ANG II-dependent hypertension, there is a homeostatic balance between ANG II stimulation of Na+ transporters (which may be direct or indirect via aldosterone stimulation), which raises ECFV and blood pressure, and hypertension-driven inhibition of Na+ transporters, which restores ECFV. If the pressure-natriuresis response was not appropriately activated, ECFV would be significantly expanded during ANG II infusion. Table 3 provides evidence for both hypertension (elevated mean arterial pressure and increased heart weight) and for pressure-natriuresis and diuresis (elevated urine volume and urine Na+ excretion) in this study. By separating cortex from medulla and analyzing all the sodium transporters along the nephron, this study was able to determine that ANG II infusion stimulates transporters (by increasing abundance, phosphorylation, and/or proteolytic cleavage) from the cortical TALH through the medullary CD, and that transporters are inhibited (presumably by the hypertension or ECFV signals) from the PT through the medullary TALH (Fig. 9).
Fig. 9.

Na+ transporter regulation along the nephron during ANG II-dependent hypertension. ANG II infusion activates distal nephron Na+ transporters (highlighted in red) consistent with increased Na+ reabsorption, elevated ECFV, and higher blood pressure. Hypertension provokes pressure-natriuresis to normalize ECFV by decreasing NHE3, medullary NKCC, and regulatory kinase SPAK (highlighted in green). See text for abbreviations.
Previous studies demonstrated stimulation of post-macula densa sodium transporters during ANG II hypertension, namely increased NCC abundance and phosphorylation and increased ENaC abundance and proteolytic cleavage (3, 6, 11, 41, 49). A component of the transporter stimulation may be secondary to stimulation of aldosterone secretion during ANG II infusion (28). However, recent studies (reviewed in 42) demonstrate ANG II stimulates NCC in adrenalectomized rats, mediated, at least in part, by the WNK4-SPAK-dependent cascade that is independent of aldosterone, while stimulation of ENaC is via a pathway that is additive to the effects of aldosterone. In this study, we confirmed that ANG II infusion increased abundance of the regulatory kinase SPAK and of phosphorylated SPAK (an indicator of SPAK activation) in renal cortex, increased abundance of NCC and NCC phosphorylated at multiple sites [a surrogate marker of NCC activation in apical membranes (16, 18)] in renal cortex (Figs. 5 and 6), and activated ENaC subunits, evidenced by their proteolytic cleavage (16, 18) (Fig. 8).
The TALH NKCC2 is also regulated by SPAK and, unlike NCC, is found in both cortex and medulla (Figs. 3 and 9). We recently reported that ANG II infusion in mice decreased total NKCC2 while increasing NKCC2-P, and increased SPAK-P without changing total SPAK, all measured in whole mouse kidney (11). This pattern of disparate regulation is clarified by the results of the present study. By analyzing kidney medulla separately from cortex, differential regulation along the TALH was revealed: cortical NKCC2 and NKCC2-P were stimulated by ANG II infusion while medullary NKCC2 abundance was decreased (NKCC2-P was not significantly decreased) (Fig. 3). Similarly, cortical SPAK and SPAK-P were stimulated by ANG II while medullary SPAK was decreased (SPAK-P was not significantly decreased) (Fig. 6). More than 90% of NKCC2-P is estimated to be localized to the plasma membrane (11), evidence of NKCC2 activation in the cortex. These findings indicate that the medullary TALH SPAK and NKCC2 are not stimulated by ANG II infusion when it is accompanied by hypertension; rather, the pool sizes of both are depressed and their phosphorylation is not increased, evidence for changes that facilitate the pressure-natriuresis response. Conversely, cortical TALH SPAK and NKCC2 and their phosphorylation are stimulated by ANG II. In contrast to SPAK, our analyses did not provide any evidence for regulation of OSR1 (Fig. 7) or AMPK, kinases reported to stimulate NKCC phosphorylation (10), in either cortex or medulla.
Recent studies in genetically modified mouse models provide some insight into the role of SPAK in regulating NKCC2: SPAKT243A/T243A knockin mice (with inactive SPAK, intact KS-SPAK) exhibit less NKCC2 phosphorylation than wild-type mice (33), and SPAK KO (also lack KS-SPAK) exhibit increased NKCC2 total abundance (45) and phosphorylation (12, 24, 45) as well as increased OSR1 phosphorylation (24, 45) consistent with the lack of the dominant negative influence of KS-SPAK on SPAK and OSR1 activation. Together with our in vivo observations in rats in this study, we postulate that as long as SPAK is present, it can play a significant role in regulating NKCC2.
At the basolateral side of the TALH cell the sodium pump drives salt reabsorption; it accounts for most medullary NKA (26). The decrease in NKAα1 and NKAβ1 abundance in medulla (Fig. 4), whether primary or secondary to decreased apical NKCC2 activity, is another likely molecular mechanism contributing to the pressure-natriuretic response in the face of ANG II infusion with hypertension. The design of this study does not allow determination of NKA regulation in nephron-specific regions because of the heterogeneity of tubules with different levels of NKA. However, previous studies have reported that ANG II stimulation alone rapidly increases PT apical membrane Na,K-ATPase (46), and that acute hypertension alone decreases Na,K-ATPase activity (48) indicating that cortical Na,K-ATPase is likely regulated in a region specific manner during ANG II-dependent hypertension.
ANG II stimulation (without accompanying hypertension) has been reported to stimulate pre-macula densa NHE3 and NKCC2 (2, 36, 38). Specifically, blocking ANG II production with an ACE inhibitor redistributes PT NHE3 from body to the base of the microvilli and decreases PT Na+ reabsorption (19, 44), and ANG II infusion (without hypertension) redistributes NHE3 into the microvilli and activates transport (36). In contrast, NHE3 redistributes to the base of the microvilli in response to acute hypertension without ANG II stimulation (25). In the present study the PT NHE3 is subjected to the simultaneous opposing forces of ANG II and hypertension. Interestingly, the ANG II stimulation appears to retain NHE3 in the body of the microvilli, yet there is a compensatory decrease in NHE3 abundance (by both immunoblot and immunohistochemistry) (Fig. 2). Nonetheless, the persistent localization of NHE3 in the microvilli during ANG II infusion may blunt the magnitude of the natriuresis for a given increase in blood pressure and contribute to the rise in blood pressure. Whether the decreased pool size of NHE3 during ANG II hypertension is due to depressed synthesis or elevated degradation remains to be determined. It will also be interesting to learn whether the NHE3 abundance increases initially in response to ANG II alone prior to the development of hypertension and subsequently decreases after the blood pressure goes up. In an earlier study with Gurley et al. (13), we reported that elimination of the ANG II-receptor type 1A from the mouse PT improved the pressure-natriuresis response during ANG II-dependent hypertension, evident as larger decreases in NHE3 abundance and lower blood pressure, thus illustrating the important role of the PT in blood pressure regulation.
Differential regulation of NKCC2 in medullary vs. cortical TALH may be a function of ion transport characteristics of NKCC2 isoforms. Isoform NKCC2F is expressed mainly in medullary TALH while NKCC2B is expressed mainly in cortical TALH (7, 30). Both furosemide administration and chronic water loading alter expression of NKCC2 in an isoform-specific manner (4), suggesting that splicing machinery may be influenced by the filtered load and/or the intracellular [Cl−]. During ANG II hypertension, the decrease in PT NHE3 would contribute to increased flow into the TALH, which could alter the expression of NKCC2 splice variants along the TALH and activate parallel changes in NKA. However, we have previously established that increased flow out of the PT (during acute hypertension or in response to a PT diuretic) actually increases NKA activity in the medulla (22). Since the upstream kinase SPAK, localized to the TALH and DCT, also responds to ANG II infusion with a differential pattern (increase in cortical SPAK, SPAK-P, and decrease in medullary SPAK), SPAK is more likely a target for effecting differential regulation of NKCC2.
Potential candidates, regulated by ANG II and/or by tubular flow, that could suppress SPAK, NKCC2, and NKA in medullary TALH would include changes in WNKs (14), nitric oxide, and reactive oxygen species (5). High blood pressure has been shown to stimulate release of the cytochrome P-450 metabolite 20-hydroxyeicosatetraenoic acid, 20-HETE (43), which inhibits Na+ transport and NKA activity in the PT and TALH (32, 47) and NKCC transport activity in isolated medullary TALH (8). ANG II hypertension stimulates release of 20-HETE in rat kidney (1) suggesting the possibility that 20-HETE could counteract the antinatriuretic influence of ANG II from the PT through the medullary TALH by suppressing NHE3, NKA, and NKCC2.
What is the significance of these findings? Evidence indicates that ANG II can stimulate transporter abundance and/or activity all along the nephron. However, if ANG II stimulation is accompanied by hypertension, compensatory natriuretic responses override the antinatriuresis of ANG II. This study determined that ANG II hypertension increases transporters' abundance and activation from the cortical TALH to the medullary CD (NKCC2, NCC, ENaC, and regulatory kinase SPAK) and that this stimulation is balanced by a compensatory inhibition of transporters (NHE3 and medullary: NKCC2, NKA, SPAK) from PT through medullary TALH, presumably driven by elevated blood pressure. That is, hypertension overrides the effects of ANG II from PT through medullary TALH. Futures studies should recognize and consider that the ANG II-dependent hypertension model generates opposing signals to maintain ECFV homeostasis. On a practical note, region-specific regulation of NKCC and SPAK will not be evident in analyses of whole kidney homogenates: it is important to dissect cortex from medulla to discriminate regions of antinatriuretic response vs. natriuretic response. Understanding where ANG II stimulates Na+ transporters along the nephron is crucial for targeting with antihypertensive therapies. Natriuretic mediators that counteract the effects of ANG II warrant further investigation.
GRANTS
This study was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-083785 (to A. A. McDonough); and by National Institutes of Health (NIH) Grants GM-074771 and DK-093501 (to E. Delpire). Microscopy was performed by the Cell and Tissue Imaging Core of the USC Research Center for Liver Diseases (NIH Grants P30-DK-048522 and S10-RR-022508).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.T.X.N., E.D., and A.A.M. conception and design of research; M.T.X.N. and D.H.L. performed experiments; M.T.X.N. and A.A.M. analyzed data; M.T.X.N., D.H.L., E.D., and A.A.M. interpreted results of experiments; M.T.X.N. prepared figures; M.T.X.N. drafted manuscript; M.T.X.N., D.H.L., E.D., and A.A.M. edited and revised manuscript; M.T.X.N., D.H.L., E.D., and A.A.M. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Michelle MacVeigh-Aloni for expertise in confocal microscopy and Kristen Gadel for excellent technical assistance.
REFERENCES
- 1. Alonso-Galicia M, Maier KG, Greene AS, Cowley AW, Jr, Roman RJ. Role of 20-hydroxyeicosatetraenoic acid in the renal and vasoconstrictor actions of angiotensin II. Am J Physiol Regul Integr Comp Physiol 283: R60–R68, 2002 [DOI] [PubMed] [Google Scholar]
- 2. Banday AA, Lokhandwala MF. Oxidative stress causes renal angiotensin II type 1 receptor upregulation, Na+/H+ exchanger 3 overstimulation, and hypertension. Hypertension 57: 452–459, 2011 [DOI] [PubMed] [Google Scholar]
- 3. Beutler KT, Masilamani S, Turban S, Nielsen J, Brooks HL, Ageloff S, Fenton RA, Packer RK, Knepper MA. Long-term regulation of ENaC expression in kidney by angiotensin II. Hypertension 41: 1143–1150, 2003 [DOI] [PubMed] [Google Scholar]
- 4. Brunet GM, Gagnon E, Simard CF, Daigle ND, Caron L, Noel M, Lefoll MH, Bergeron MJ, Isenring P. Novel insights regarding the operational characteristics and teleological purpose of the renal Na+-K+-Cl2 cotransporter (NKCC2s) splice variants. J Gen Physiol 126: 325–337, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cabral PD, Garvin JL. Luminal flow regulates NO and O2− along the nephron. Am J Physiol Renal Physiol 300: F1047–F1053, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Castaneda-Bueno M, Cervantes-Perez LG, Vazquez N, Uribe N, Kantesaria S, Morla L, Bobadilla NA, Doucet A, Alessi DR, Gamba G. Activation of the renal Na+:Cl− cotransporter by angiotensin II is a WNK4-dependent process. Proc Natl Acad Sci USA 109: 7929–7934, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Castrop H, Schnermann J. Isoforms of renal Na-K-2Cl cotransporter NKCC2: expression and functional significance. Am J Physiol Renal Physiol 295: F859–F866, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Escalante B, Erlij D, Falck JR, McGiff JC. Effect of cytochrome P450 arachidonate metabolites on ion transport in rabbit kidney loop of Henle. Science 251: 799–802, 1991 [DOI] [PubMed] [Google Scholar]
- 9. Flemmer AW, Gimenez I, Dowd BF, Darman RB, Forbush B. Activation of the Na-K-Cl cotransporter NKCC1 detected with a phospho-specific antibody. J Biol Chem 277: 37551–37558, 2002 [DOI] [PubMed] [Google Scholar]
- 10. Fraser SA, Gimenez I, Cook N, Jennings I, Katerelos M, Katsis F, Levidiotis V, Kemp BE, Power DA. Regulation of the renal-specific Na+-K+-2Cl− co-transporter NKCC2 by AMP-activated protein kinase (AMPK). Biochem J 405: 85–93, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gonzalez-Villalobos RA, Janjoulia T, Fletcher NK, Giani JF, Nguyen MT, Riquier-Brison AD, Seth DM, Fuchs S, Eladari D, Picard N, Bachmann S, Delpire E, Peti-Peterdi J, Navar LG, Bernstein KE, McDonough AA. The absence of intrarenal ACE protects against hypertension. J Clin Invest 123: 2011–2023, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grimm PR, Taneja TK, Liu J, Coleman R, Chen YY, Delpire E, Wade JB, Welling PA. SPAK isoforms and OSR1 regulate sodium-chloride co-transporters in a nephron-specific manner. J Biol Chem 287: 37673–37690, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gurley SB, Riquier-Brison AD, Schnermann J, Sparks MA, Allen AM, Haase VH, Snouwaert JN, Le TH, McDonough AA, Koller BH, Coffman TM. AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab 13: 469–475, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hoorn EJ, Nelson JH, McCormick JA, Ellison DH. The WNK kinase network regulating sodium, potassium, and blood pressure. J Am Soc Nephrol 22: 605–614, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hughey RP, Mueller GM, Bruns JB, Kinlough CL, Poland PA, Harkleroad KL, Carattino MD, Kleyman TR. Maturation of the epithelial Na+ channel involves proteolytic processing of the alpha- and gamma-subunits. J Biol Chem 278: 37073–37082, 2003 [DOI] [PubMed] [Google Scholar]
- 16. Kleyman TR, Carattino MD, Hughey RP. ENaC at the cutting edge: regulation of epithelial sodium channels by proteases. J Biol Chem 284: 20447–20451, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kocinsky HS, Dynia DW, Wang T, Aronson PS. NHE3 phosphorylation at serines 552 and 605 does not directly affect NHE3 activity. Am J Physiol Renal Physiol 293: F212–F218, 2007 [DOI] [PubMed] [Google Scholar]
- 18. Lee DH, Maunsbach AB, Riquier-Brison AD, Nguyen MT, Fenton RA, Bachmann S, Yu AS, McDonough AA. Effects of ACE inhibition and ANG II stimulation on renal Na-Cl cotransporter distribution, phosphorylation, and membrane complex properties. Am J Physiol Cell Physiol 304: C147–C163, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Leong PK, Devillez A, Sandberg MB, Yang LE, Yip DK, Klein JB, McDonough AA. Effects of ACE inhibition on proximal tubule sodium transport. Am J Physiol Renal Physiol 290: F854–F863, 2006 [DOI] [PubMed] [Google Scholar]
- 20. Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H, Amann M, Anderson HR, Andrews KG, Aryee M, Atkinson C, Bacchus LJ, Bahalim AN, Balakrishnan K, Balmes J, Barker-Collo S, Baxter A, Bell ML, Blore JD, Blyth F, Bonner C, Borges G, Bourne R, Boussinesq M, Brauer M, Brooks P, Bruce NG, Brunekreef B, Bryan-Hancock C, Bucello C, Buchbinder R, Bull F, Burnett RT, Byers TE, Calabria B, Carapetis J, Carnahan E, Chafe Z, Charlson F, Chen H, Chen JS, Cheng AT, Child JC, Cohen A, Colson KE, Cowie BC, Darby S, Darling S, Davis A, Degenhardt L, Dentener F, Des Jarlais DC, Devries K, Dherani M, Ding EL, Dorsey ER, Driscoll T, Edmond K, Ali SE, Engell RE, Erwin PJ, Fahimi S, Falder G, Farzadfar F, Ferrari A, Finucane MM, Flaxman S, Fowkes FG, Freedman G, Freeman MK, Gakidou E, Ghosh S, Giovannucci E, Gmel G, Graham K, Grainger R, Grant B, Gunnell D, Gutierrez HR, Hall W, Hoek HW, Hogan A, Hosgood HD, 3rd, Hoy D, Hu H, Hubbell BJ, Hutchings SJ, Ibeanusi SE, Jacklyn GL, Jasrasaria R, Jonas JB, Kan H, Kanis JA, Kassebaum N, Kawakami N, Khang YH, Khatibzadeh S, Khoo JP, Kok C, Laden F, Lalloo R, Lan Q, Lathlean T, Leasher JL, Leigh J, Li Y, Lin JK, Lipshultz SE, London S, Lozano R, Lu Y, Mak J, Malekzadeh R, Mallinger L, Marcenes W, March L, Marks R, Martin R, McGale P, McGrath J, Mehta S, Mensah GA, Merriman TR, Micha R, Michaud C, Mishra V, Hanafiah KM, Mokdad AA, Morawska L, Mozaffarian D, Murphy T, Naghavi M, Neal B, Nelson PK, Nolla JM, Norman R, Olives C, Omer SB, Orchard J, Osborne R, Ostro B, Page A, Pandey KD, Parry CD, Passmore E, Patra J, Pearce N, Pelizzari PM, Petzold M, Phillips MR, Pope D, Pope CA, 3rd, Powles J, Rao M, Razavi H, Rehfuess EA, Rehm JT, Ritz B, Rivara FP, Roberts T, Robinson C, Rodriguez-Portales JA, Romieu I, Room R, Rosenfeld LC, Roy A, Rushton L, Salomon JA, Sampson U, Sanchez-Riera L, Sanman E, Sapkota A, Seedat S, Shi P, Shield K, Shivakoti R, Singh GM, Sleet DA, Smith E, Smith KR, Stapelberg NJ, Steenland K, Stockl H, Stovner LJ, Straif K, Straney L, Thurston GD, Tran JH, Van Dingenen R, van Donkelaar A, Veerman JL, Vijayakumar L, Weintraub R, Weissman MM, White RA, Whiteford H, Wiersma ST, Wilkinson JD, Williams HC, Williams W, Wilson N, Woolf AD, Yip P, Zielinski JM, Lopez AD, Murray CJ, Ezzati M, AlMazroa MA, Memish ZA. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380: 2224–2260, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu W, Schreck C, Coleman RA, Wade JB, Hernandez Y, Zavilowitz B, Warth R, Kleyman TR, Satlin LM. Role of NKCC in BK channel-mediated net K+ secretion in the CCD. Am J Physiol Renal Physiol 301: F1088–F1097, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Magyar CE, Zhang Y, Holstein-Rathlou NH, McDonough AA. Downstream shift in sodium pump activity along the nephron during acute hypertension. J Am Soc Nephrol 12: 2231–2240, 2001 [DOI] [PubMed] [Google Scholar]
- 23. Mamenko M, Zaika O, Ilatovskaya DV, Staruschenko A, Pochynyuk O. Angiotensin II increases activity of the epithelial Na+ channel (ENaC) in distal nephron additively to aldosterone. J Biol Chem 287: 660–671, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McCormick JA, Mutig K, Nelson JH, Saritas T, Hoorn EJ, Yang CL, Rogers S, Curry J, Delpire E, Bachmann S, Ellison DH. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab 14: 352–364, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McDonough AA. Mechanisms of proximal tubule sodium transport regulation that link extracellular fluid volume and blood pressure. Am J Physiol Regul Integr Comp Physiol 298: R851–R861, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McDonough AA, Magyar CE, Komatsu Y. Expression of Na+-K+-ATPase alpha- and beta-subunits along rat nephron: isoform specificity and response to hypokalemia. Am J Physiol Cell Physiol 267: C901–C908, 1994 [DOI] [PubMed] [Google Scholar]
- 27. Nguyen MT, Yang LE, Fletcher NK, Lee DH, Kocinsky H, Bachmann S, Delpire E, McDonough AA. Effects of K+-deficient diets with and without NaCl supplementation on Na+, K+, and H2O transporters' abundance along the nephron. Am J Physiol Renal Physiol 303: F92–F104, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ortiz RM, Graciano ML, Seth D, Awayda MS, Navar LG. Aldosterone receptor antagonism exacerbates intrarenal angiotensin II augmentation in ANG II-dependent hypertension. Am J Physiol Renal Physiol 293: F139–F147, 2007 [DOI] [PubMed] [Google Scholar]
- 29. Palmer LG, Patel A, Frindt G. Regulation and dysregulation of epithelial Na+ channels. Clin Exp Nephrol 16: 35–43, 2012 [DOI] [PubMed] [Google Scholar]
- 30. Payne JA, Forbush B., 3rd Alternatively spliced isoforms of the putative renal Na-K-Cl cotransporter are differentially distributed within the rabbit kidney. Proc Natl Acad Sci USA 91: 4544–4548, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pisitkun T, Hoffert JD, Saeed F, Knepper MA. NHLBI-AbDesigner: an online tool for design of peptide-directed antibodies. Am J Physiol Cell Physiol 302: C154–C164, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Quigley R, Baum M, Reddy KM, Griener JC, Falck JR. Effects of 20-HETE and 19(S)-HETE on rabbit proximal straight tubule volume transport. Am J Physiol Renal Physiol 278: F949–F953, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rafiqi FH, Zuber AM, Glover M, Richardson C, Fleming S, Jovanovic S, Jovanovic A, O'Shaughnessy KM, Alessi DR. Role of the WNK-activated SPAK kinase in regulating blood pressure. EMBO Mol Med 2: 63–75, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Richardson C, Rafiqi FH, Karlsson HK, Moleleki N, Vandewalle A, Campbell DG, Morrice NA, Alessi DR. Activation of the thiazide-sensitive Na+-Cl− cotransporter by the WNK-regulated kinases SPAK and OSR1. J Cell Sci 121: 675–684, 2008 [DOI] [PubMed] [Google Scholar]
- 35. Richardson C, Sakamoto K, de Los Heros P, Deak M, Campbell DG, Prescott AR, Alessi DR. Regulation of the NKCC2 ion cotransporter by SPAK-OSR1-dependent and -independent pathways. J Cell Sci 124: 789–800, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Riquier-Brison AD, Leong PK, Pihakaski-Maunsbach K, McDonough AA. Angiotensin II stimulates trafficking of NHE3, NaPi2, and associated proteins into the proximal tubule microvilli. Am J Physiol Renal Physiol 298: F177–F186, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Semprun-Prieto LC, Sukhanov S, Yoshida T, Rezk BM, Gonzalez-Villalobos RA, Vaughn C, Michael Tabony A, Delafontaine P. Angiotensin II induced catabolic effect and muscle atrophy are redox dependent. Biochem Biophys Res Commun 409: 217–221, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Silva GB, Garvin JL. Angiotensin II-dependent hypertension increases Na transport-related oxygen consumption by the thick ascending limb. Hypertension 52: 1091–1098, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Skott O. Body sodium and volume homeostasis. Am J Physiol Regul Integr Comp Physiol 285: R14–R18, 2003 [DOI] [PubMed] [Google Scholar]
- 40. Sorensen MV, Grossmann S, Roesinger M, Gresko N, Todkar AP, Barmettler G, Ziegler U, Odermatt A, Loffing-Cueni D, Loffing J. Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int 83: 811–824, 2013 [DOI] [PubMed] [Google Scholar]
- 41. van der Lubbe N, Lim CH, Fenton RA, Meima ME, Jan Danser AH, Zietse R, Hoorn EJ. Angiotensin II induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter independent of aldosterone. Kidney Int 79: 66–76, 2011 [DOI] [PubMed] [Google Scholar]
- 42. van der Lubbe N, Zietse R, Hoorn EJ. Effects of angiotensin II on kinase-mediated sodium and potassium transport in the distal nephron. Curr Opin Nephrol Hypertens 22: 120–126, 2013 [DOI] [PubMed] [Google Scholar]
- 43. Williams JM, Murphy S, Burke M, Roman RJ. 20-Hydroxyeicosatetraeonic acid: a new target for the treatment of hypertension. J Cardiovasc Pharmacol 56: 336–344, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang LE, Leong PK, McDonough AA. Reducing blood pressure in SHR with enalapril provokes redistribution of NHE3, NaPi2, and NCC and decreases NaPi2 and ACE abundance. Am J Physiol Renal Physiol 293: F1197–F1208, 2007 [DOI] [PubMed] [Google Scholar]
- 45. Yang SS, Lo YF, Wu CC, Lin SW, Yeh CJ, Chu P, Sytwu HK, Uchida S, Sasaki S, Lin SH. SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J Am Soc Nephrol 21: 1868–1877, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yingst DR, Araghi A, Doci TM, Mattingly R, Beierwaltes WH. Decreased renal perfusion rapidly increases plasma membrane Na-K-ATPase in rat cortex by an angiotensin II-dependent mechanism. Am J Physiol Renal Physiol 297: F1324–F1329, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yu M, Lopez B, Dos Santos EA, Falck JR, Roman RJ. Effects of 20-HETE on Na+ transport and Na+-K+-ATPase activity in the thick ascending loop of Henle. Am J Physiol Regul Integr Comp Physiol 292: R2400–R2405, 2007 [DOI] [PubMed] [Google Scholar]
- 48. Zhang Y, Magyar CE, Norian JM, Holstein-Rathlou NH, Mircheff AK, McDonough AA. Reversible effects of acute hypertension on proximal tubule sodium transporters. Am J Physiol Cell Physiol 274: C1090–C1100, 1998 [DOI] [PubMed] [Google Scholar]
- 49. Zhao D, Seth DM, Navar LG. Enhanced distal nephron sodium reabsorption in chronic angiotensin II-infused mice. Hypertension 54: 120–126, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]