

Abstract
Modulation of protein–protein interactions (PPIs) with small molecules has been hampered by a lack of lucid methods capable of reliably identifying high-quality hits. In fragment screening, the low ligand efficiencies associated with PPI target sites pose significant challenges to fragment binding detection. Here, we investigate the requirements for ligand-based NMR techniques to detect rule-of-three compliant fragments that form part of known high-affinity inhibitors of the PPI between the von Hippel–Lindau protein and the alpha subunit of hypoxia-inducible factor 1 (pVHL:HIF-1α). Careful triaging allowed rescuing weak but specific binding of fragments that would otherwise escape detection at this PPI. Further structural information provided by saturation transfer difference (STD) group epitope mapping, protein-based NMR, competitive isothermal titration calorimetry (ITC), and X-ray crystallography confirmed the binding mode of the rescued fragments. Our findings have important implications for PPI druggability assessment by fragment screening as they reveal an accessible threshold for fragment detection and validation.
Keywords: NMR fragment screening, protein−protein interactions, binding affinity, druggability
Protein–protein interactions (PPIs) and interfaces are preponderant in nearly every facet of cellular function, exhibiting a wide range of topologically complex and diverse interactions.1−4 The recognized value of modulating PPIs for therapeutic intervention have led to recent efforts, both in academic and pharmaceutical research, to target them using drug-like small molecules.5−8 However, to date, this has remained a frustratingly slow and difficult goal to achieve, in part due to challenges in identifying high-quality starting hits.1,9 Nevertheless, several small molecules have been developed to bind PPI sites with desired levels of affinity, yet they tend to be much larger, on average, than compounds targeting more classical binding sites and, as a result, exhibit much lower ligand efficiency (LE), i.e., binding free energy per heavy atoms.10
To aid more successful targeting of PPIs, it is important to focus efforts onto those PPIs that are most likely to yield high-quality, high-LE small molecule binders, i.e., exhibit highest druggability.11,12 Several approaches are being used to assess target propensity to ligand binding, including computational methods based on three-dimensional structures of the target.13−15 A method that is being increasingly used for this purpose is biophysical fragment screening.11 On the one hand, fragment screening can be performed on any protein targets that are readily expressed recombinantly without requiring in principle prior structural information on the target. On the other hand, the lower LEs typically associated with small molecule binding at PPIs raise the barrier to the detection of bona fide low-affinity fragment hits. One way to approach this challenge would be to screen larger compounds.16,17 The problem associated with this strategy is that larger compounds will inevitably explore chemical space less efficiently than the smaller fragments, and as a result, larger libraries would be required. However, a more general and simpler approach would be to increase the sensitivity of binding detection in a fragment screen. Here, we interrogate for the first time the low limits of fragment binding detection using a successfully targeted PPI as a model system.
Our recent discovery of high-affinity, lead-like small molecule inhibitors of the interaction between the von Hippel–Lindau protein (pVHL) and the alpha subunit of hypoxia-inducible factor 1 (HIF-1α) has revealed this PPI to be druggable.17−19 The compounds were designed using a crystal structure of HIF-1α peptide bound to the stable multiprotein complex pVHL-ElonginC:Elongin B (VCB) (Figure 1). Surprisingly, however, we could not detect binding of the constitutive Rule-of-three (Ro3)20 compliant fragments of our inhibitors (molecular weight < 300, ClogP < 3, the number of hydrogen bond donors and acceptors each should be <3, and the number of rotatable bonds should be <3).17 Moreover, a screen of ∼1300 Ro3 obeying fragments library proved unsuccessful for targeting the pVHL:HIF-1α interface.17 We were intrigued by these observations, in part because there is a growing belief that target druggability correlates with hit rates from fragment screening.11 To resolve this conundrum, we decided to investigate the ability to triage bona fide weak binding fragments using ligand-based NMR spectroscopy, arguably one of the most sensitive biophysical techniques that is widely applied to hit generation in drug discovery.21
Figure 1.
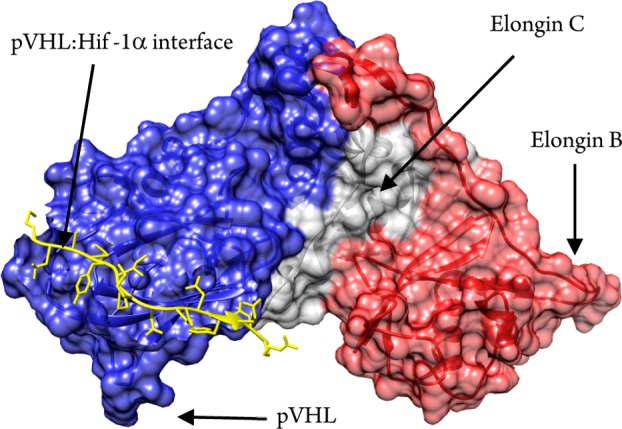
Structural representation of the VCB multiprotein complex and the pVHL:HIF-1α interface.
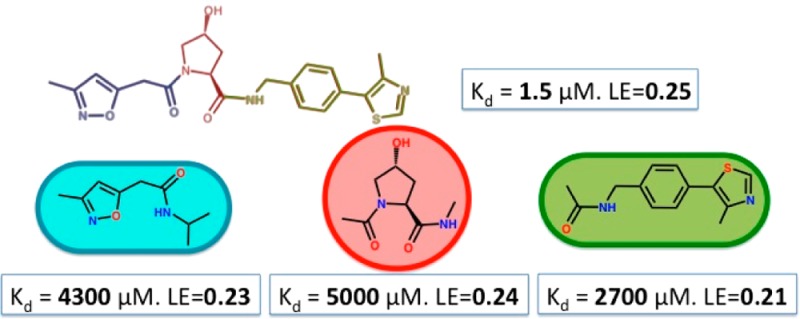
A library of 12 compounds was designed by defragmenting known inhibitors 1 and 2 (see Figures S1 and S3, Supporting Information),18,19 and screened against the target protein using three distinct ligand-based 1D 1H NMR experiments (Table 1): first, saturation transfer difference (STD)22 experiments apply a selective pulse to saturate protein resonances. Only ligands that bind to the protein will receive saturation transfer, resulting in their signals to appear as positive in a difference spectrum between the unsaturated and saturated spectra; second, Carr–Purcell–Meiboom–Gill (CPMG)23 experiments exploit the faster T2 relaxation times of macromolecules relative to small molecules. Upon binding to the protein, ligands relaxation time will decrease, causing a line broadening and a consequent decrease in intensity on their signals; finally, the water-ligand observed via gradient spectroscopy (WaterLOGSY)24 experiments apply a selective pulse to saturate resonances of water molecules. In the absence of protein, cross-relaxation from water will yield control ligand signals that are phased downward; in the presence of protein, small molecule binders will receive an NOE contribution from water that is of opposite sign relative to control, resulting in their signals either showing a reduction in intensity, or even pointing upward.
Table 1. Biophysical Characterization of Small Molecules Binding to VCBa.
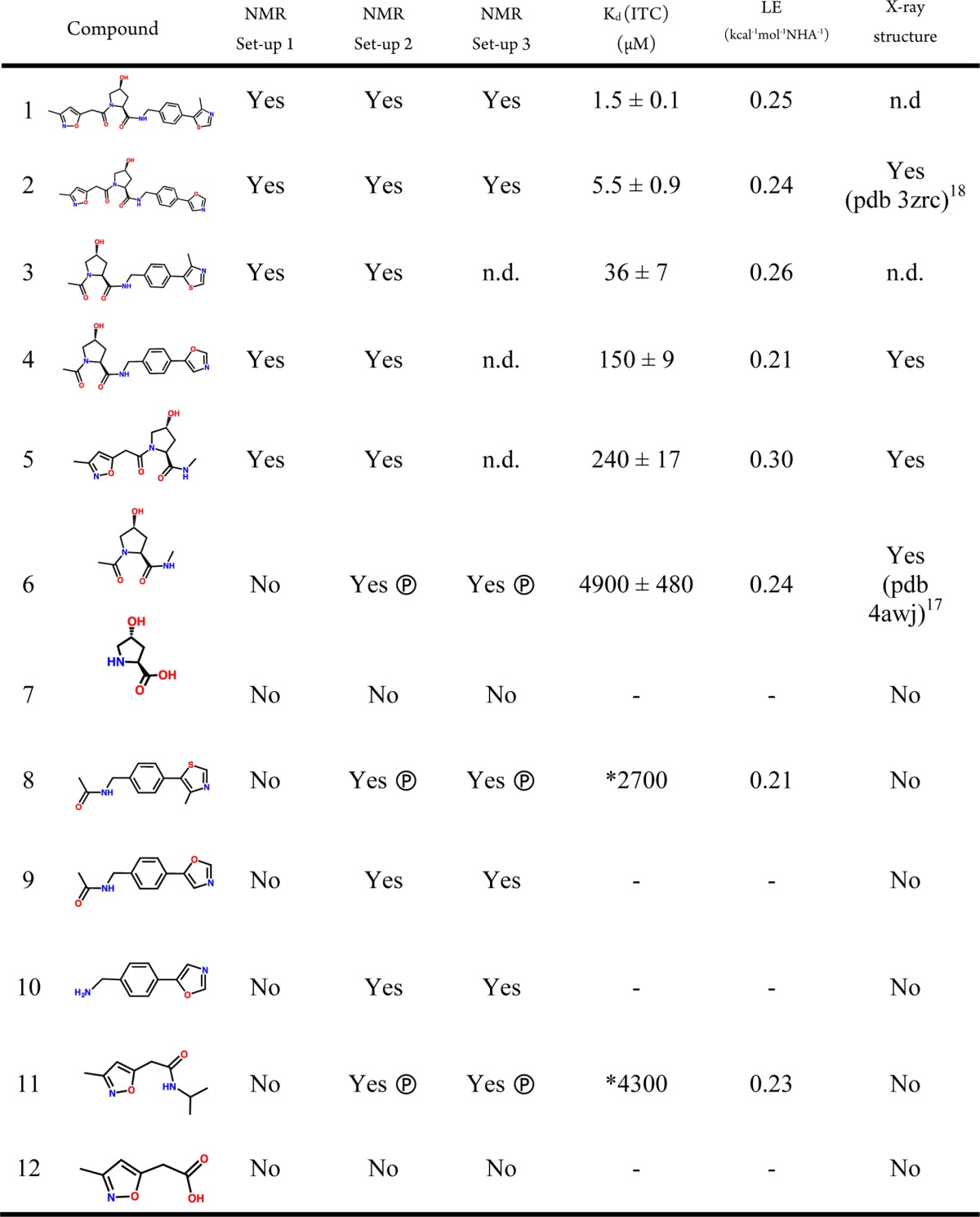
NMR binding detection was assessed by all three experiments (STD, CPMG, and WaterLOGSY). In all cases, the three experiments were found to be in agreement with each other; therefore, a single Yes/No answer is tabulated under each set-up. Ⓟ, displaced by 19-mer HIF-1α peptide; *, obtained by competitive ITC assay; n.d., not determined.
Experimental conditions of 1 mM ligand and 10 μM protein, which are typically used in NMR fragment screening, enabled unambiguous detection in each of the three NMR experiments of the larger, Ro3-breaking compounds 3–5 (set-up 1, Table 1). We were also able to characterize binding thermodynamics by isothermal titration calorimetry (ITC) and determine bound X-ray structures by crystallographic soaks, confirming the expected binding modes at the HIF-1α site (Table 1, Figures 2 and S4–S5, Supporting Information). In contrast, binding detection was unfruitful for Ro3-compliant fragments 6–12 under set-up 1 (Table 1 and Figures S6–12a–c, Supporting Information). This observation is consistent with our previous results,17 raising the question whether this is due to the absence of binding (true negative) or limitations of the screening setup at detecting weak binding (false negative).
Figure 2.
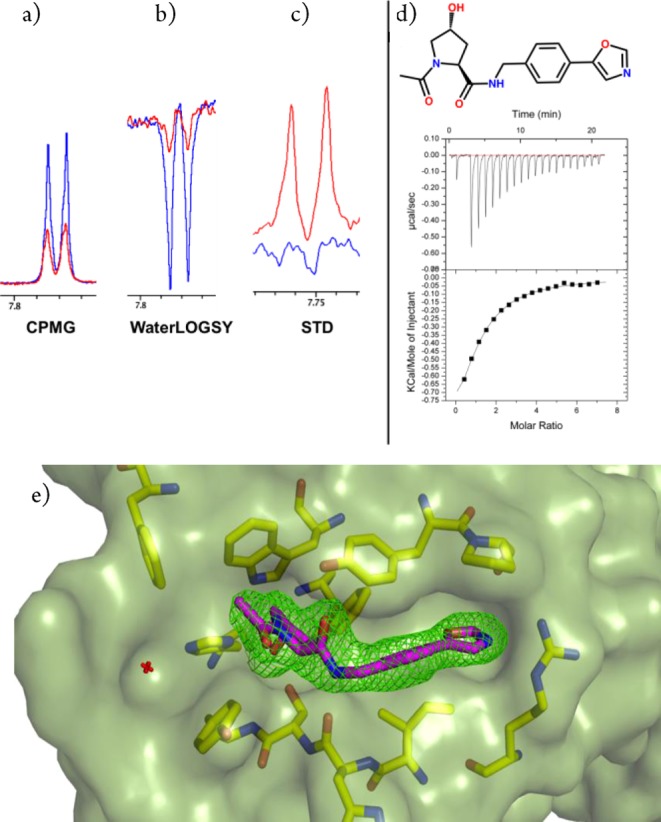
Binding detection at the pVHL:HIF-1α interface for 4. (a–c) NMR spectra for VCB + 4 using set-up 1 (red) and the compound alone (blue). (d) Direct ITC titration for 4 (3 mM compound and 100 μM VCB). Data was fitted with Ka = 6.7 × 103 ± 400 M–1; ΔH = −1450 ± 100 cal/mol; and ΔS = 12.6 cal/mol/degree. (e) Crystal structure of VCB in complex with 4 (purple carbon sticks). The omit electron density maps (Fo – Fc) are shown in green contoured at 2.5σ around the ligand. The protein surface is shown in green at 40% transparency.
To extend the binding detection range, we set to modify the NMR experimental conditions by first increasing the protein concentration to 40 μM while maintaining ligand concentrations fixed at 1 mM (setup 2); and second by increasing concentrations of protein and ligand to 30 μM and 3 mM (set-up 3), respectively, thereby maintaining a protein/ligand excess of 100-fold as in set-up 1.
We first applied the revised set-ups to the capped hydroxyproline (Hyp) core fragment 6, that had successfully yielded an X-ray bound structure17 but had otherwise proven elusive to biophysical detection. Each NMR experiment distinctly detected binding of 6 under both set-ups 2 and 3 but not set-up 1, placing this compound as a true positive hit (Figures 3 and S6, Supporting Information).
Figure 3.
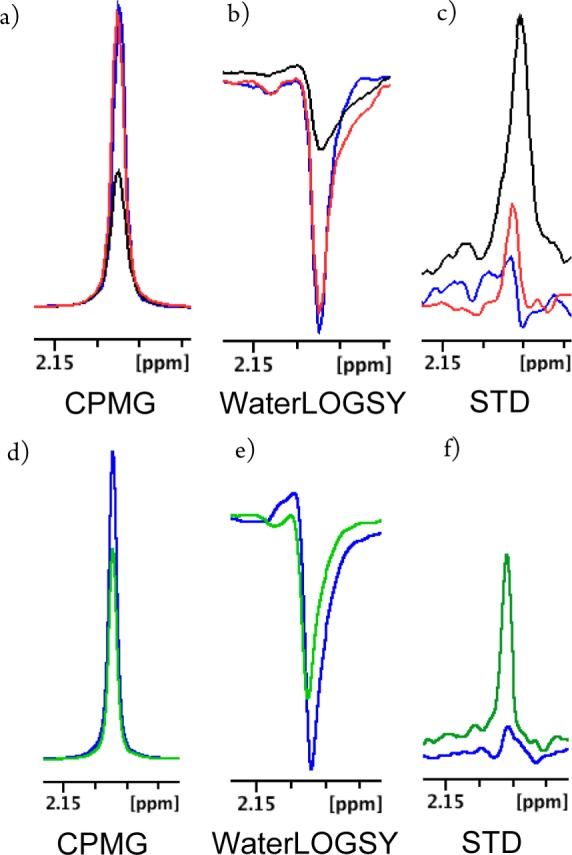
Detection of 6 binding to VCB using ligand-based NMR spectroscopy. (a–c) NMR spectra for VCB + 6 using set-ups 1 and 2, in red and black respectively, and the compound alone (blue). (d– f) Spectra for VCB + 6 using set-up 3 (green) and compound alone (blue).
Set-up 2 provided the most reliable detection profile for 6 in CPMG and WaterLOGSY, a direct result of increasing the fractional bound ligand, while maintaining total ligand concentration constant at 1 mM. In contrast, set-up 3 gave the best result in STD, as this technique is unaffected by increasing free ligand concentration. Similarly, we were able to unambiguously detect binding of compounds 8–11 to VCB, whereas binding of compounds 7 and 12 remained undetected under each revised setup (Table 1 and Figures S7 and S12, Supporting Information). This highlighted that the newly adopted set-ups enable robust discrimination between true binders and nonbinders, which is a critical requisite in biophysical fragment screening.
Aiming further characterization of the rescued binders, we asked if they targeted specifically the pVHL-HIF-1α interface and whether they would recapitulate the binding mode shown as part of the intact parent compounds 1 and 2. To address this, we first attempted to compete binding of 6 and 8–11 using a high-affinity 19-mer HIF-1α peptide. Compounds 6, 8, and 11 were displaced by the peptide, placing them at this PPI (Table 1 and Figures S6, S8, and S11, panels a, b and c, Supporting Information). To assess binding affinity for the displaced compounds, competitive ITC experiments were carried out using inhibitor 2 as the titrant in the presence of fragments 8 and 11, yielding apparent Kd of 2.7 and 4.3 mM for 8 and 11, respectively (Table 1 and Figure S8 and S11, panel g, Supporting Information). A matching Kd of ∼5 mM was obtained for 6 under both direct and competitive conditions, thereby validating the approach (Figure S6, panels g and h, Supporting Information). These fragments maintained similar LE values (Table 1) of the parent inhibitors, which notably fell around the value of 0.24 kcal mol–1 NHA–1 obeying LE’s generally observed for PPI-targeting small molecules.1
To gain information about the fragments binding mode, we first turned to group epitope mapping (GEM) characterization of their STD-NMR spectra.25 Relative degrees of saturation of the individual protons were normalized to the highest saturated proton of the compounds, yielding some information of the proximity of each proton and the interacting protein surface. The NMR GEM data for compound 6 (Table S2, Supporting Information, and Figure 4) suggests the protons adjacent to the hydroxyl group to be in closer contact with the protein, in perfect agreement with the binding mode observed in the X-ray structure (Figure S6, panel i, Supporting Information). In contrast, the GEM data for 8 and 11 (Tables S3–4, Supporting Information) could not conclusively inform about their binding modes as different saturation build up rates are often observed between aliphatic and aromatic protons.
Figure 4.
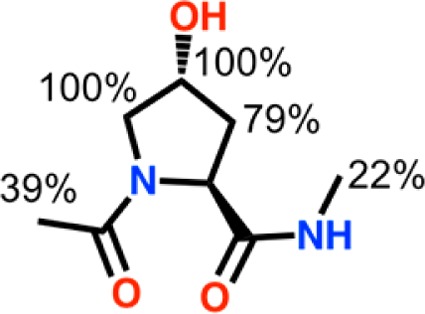
Group epitope mapping (GEM) obtained from STD-NMR for 6.
As no crystal structure could be obtained for compounds 8 and 11, further structural validation of the compounds binding mode was achieved using 1H–15N heteronuclear single quantum coherence (HSQC) chemical shift perturbation (CSP). The backbone assignments for the VCB complex are available,26 and we experimentally validated our residue-specific screening using compound 1 (Figure S2, Supporting Information)
Being the VCB complex a 42 kDa system and intrinsically challenging for the majority of protein NMR techniques, an alternative labeling scheme was adopted. Apart from 15N, the VCB complex was also perdeuterated. When using this labeling scheme and for this molecular weight, the yielded spectra (Figure S8, panel h, Supporting Information) allowed improved signal-to-noise and less resonance overlap. The expected binding site of compound 8 was successfully confirmed by demonstrating chemical shifts in the 1H–15N HSQC spectra for residues at the pVHL-HIF-1α interface as a result of compound titration (Figure 5a,b).
Figure 5.

Residue specific mapping using 1H–15N HSQC. (a) 1H–15N HSQC of 0.3 mM perdeuterated VCB (black contours) showing illustrative residues at the pVHL-HIF-1α interface when titrated with 3 mM (blue contours) and 5 mM (red contours) of 8 (see Figure S8h, Supporting Information, for full spectra). Lower panels show the mapped region of residues exhibiting chemical shift perturbation onto the pVHL structure (green) (c) and the modeled fragment bound to pVHL (d).
The recapitulation of the binding site occupied by 8 when it is part of the Hyp-containing inhibitors confidently reinforces it as a true fragment hit for this PPI (Figure 5c,d) and is consistent with the previously observed displacement by the 19-mer HIF-1α peptide. Taken together our data suggest that Ro3 compliant native subsite binders (namely, 6, 8, and 11) can be found by carefully triaged fragment based screening. These hits would have retrospectively provided key starting points to the design of 1 and 2 even in the absence of information from the natural peptide ligand.
In summary, this work has addressed for the first time the problem of detecting and structurally validating the weak affinities and low ligand efficiencies that are expected for fragments effectively targeting PPIs. Standard biophysical fragment screening approaches have been historically optimized against classical drug targets, such as enzymes and receptors that typically exhibit LE values well beyond a threshold of 0.3 kcal–1 mol–1 NHA–1. Using a model druggable PPI, we show that a revision on the experimental conditions of ligand-based NMR techniques opens the possibility of rescuing genuine binders, thus delivering more sensitive and robust set-ups for NMR screening. Screening at higher receptor concentrations could be particularly beneficial in the case of less stable or more transient multisubunit complexes, as this would also increase the relative population of the functional target site. Provided requirements for compound and protein solubilities are met, our fine-tuned NMR conditions should yield more accurate hit rates of true positives. This in turn will enable a more reliable assessment of PPI target druggability and tractability by fragment screening and better inform the prioritization of PPI targets for drug discovery programs.
Acknowledgments
We thank Dr. Mark Bycroft (University of Cambridge) for the 1H–15N assignments of the pVHL-ElonginC-ElonginB complex and the staff of the Diamond Light Source I02 line for help with data collection.
Supporting Information Available
Chemical and biochemical procedures, characterization of novel compounds, all NMR spectra, ITC data, and details of the X-ray diffraction data collection and analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The crystal structures of pVHL-ElonginB-ElonginC in complex with 4 and 5 described in this letter have been deposited in the Protein Data Bank (PDB codes 4bks and 4bkt, respectively)
Author Present Address
§ (I.V.M.) VIB Department of Structural Biology, Structural Biology Brussels, Vrije Universiteit, Brussels, Belgium
Author Present Address
∥ (M.G.J.B., C.G., and A.C.) College of Life Sciences, Division of Biological Chemistry and Drug Discovery, University of Dundee, Dow Street, DD1 5EH Dundee, U.K.
This work was supported by the Fundação para a Ciência e a Tecnologia (FCT, SFRH/BD/81735/2011 Studentship to D.M.D.), the U.K. BBSRC (BB/G023123/1, David Phillips Fellowship to A.C.), the European Research Council ERC-2012-StG-311460 DrugE3CRLs (Starting Grant to A.C.), the EC PIEF-GA-2010-275683 (Marie-Curie Intra European Fellowship to I.V.M.), and the EMBO ASTF 165-2012 (Short-Term Fellowship to C.G.).
The authors declare no competing financial interest.
Supplementary Material
References
- Wells J. A.; McClendon C. L. Reaching for High-Hanging Fruit in Drug Discovery at Protein–Protein Interfaces. Nature 2007, 450, 1001–1009. [DOI] [PubMed] [Google Scholar]
- Thompson A. D.; Dugan A.; Gestwicki J. E.; Mapp A. K. Fine-Tuning Multiprotein Complexes Using Small Molecules. ACS Chem. Biol. 2012, 7, 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M. C.; Gestwicki J. E. Features of Protein–Protein Interactions That Translate Into Potent Inhibitors: Topology, Surface Area and Affinity. Expert Rev. Mol. Med. 2012, 14, e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy E. D.; Pereira-Leal J. B. Evolution and Dynamics of Protein Interactions and Networks. Curr. Opin. Struct. Biol. 2008, 18, 349–357. [DOI] [PubMed] [Google Scholar]
- Mullard A. Protein–Protein Interaction Inhibitors Get into the Groove. Nat. Rev. Drug Discovery 2012, 11, 173–175. [DOI] [PubMed] [Google Scholar]
- Thiel P.; Kaiser M.; Ottmann C. Small-Molecule Stabilization of Protein–Protein Interactions: an Underestimated Concept in Drug Discovery?. Angew. Chem., Int. Ed. 2012, 51, 2012–2018. [DOI] [PubMed] [Google Scholar]
- Fry D. C. Protein–Protein Interactions as Targets for Small Molecule Drug Discovery. Biopolymers 2006, 84, 535–552. [DOI] [PubMed] [Google Scholar]
- Immekus F.; Barandun L. J.; Betz M.; Debaene F.; Petiot S.; Sanglier-Cianferani S.; Reuter K.; Diederich F.; Klebe G. Launching Spiking Ligands Into a Protein–Protein Interface: a Promising Strategy to Destabilize and Break Interface Formation in a tRNA Modifying Enzyme. ACS Chem. Biol. 2013, 8, 1163–1178. [DOI] [PubMed] [Google Scholar]
- Fuller J. C.; Burgoyne N. J.; Jackson R. M. Predicting Druggable Binding Sites at the Protein–Protein Interface. Drug Discovery Today 2009, 14, 155–161. [DOI] [PubMed] [Google Scholar]
- Higueruelo A. P.; Schreyer A.; Bickerton G. R. J.; Pitt W. R.; Groom C. R.; Blundell T. L. Atomic Interactions and Profile of Small Molecules Disrupting Protein–Protein Interfaces: the TIMBAL Database. Chem. Biol. Drug Des. 2009, 74, 457–467. [DOI] [PubMed] [Google Scholar]
- Edfeldt F. N. B.; Folmer R. H. A.; Breeze A. L. Fragment Screening to Predict Druggability (Ligandability) and Lead Discovery Success. Drug Discovery Today 2011, 16, 284–287. [DOI] [PubMed] [Google Scholar]
- Surade S.; Blundell T. L. Structural Biology and Drug Discovery of Difficult Targets: the Limits of Ligandability. Chem. Biol. 2012, 19, 42–50. [DOI] [PubMed] [Google Scholar]
- Schmidtke P.; Le Guilloux V.; Maupetit J.; Tufféry P. Fpocket: Online Tools for Protein Ensemble Pocket Detection and Tracking. Nucleic Acids Res. 2010, 38, W582–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger D. M.; Gohlke H. DrugScorePPI Webserver: Fast and Accurate in Silico Alanine Scanning for Scoring Protein-Protein Interactions. Nucleic Acids Res. 2010, 38, W480–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne N. J.; Jackson R. M. Predicting Protein Interaction Sites: Binding Hot-Spots in Protein–Protein and Protein–Ligand Interfaces. Bioinformatics 2006, 22, 1335–1342. [DOI] [PubMed] [Google Scholar]
- Fry D. C.; Wartchow C.; Graves B.; Janson C.; Lukacs C.; Kammlott U.; Belunis C.; Palme S.; Klein C.; Vu B. Deconstruction of a Nutlin: Dissecting the Binding Determinants of a Potent Protein–Protein Interaction Inhibitor. ACS Med. Chem. Lett. 2013, 4, 660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Molle I.; Thomann A.; Buckley D. L.; So E. C.; Lang S.; M Crews C.; Ciulli A. Dissecting Fragment-Based Lead Discovery at the Von Hippel-Lindau Protein:Hypoxia Inducible Factor 1α Protein-Protein Interface. Chem. Biol. 2012, 19, 1300–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley D. L.; Van Molle I.; Gareiss P. C.; Tae H. S.; Michel J.; Noblin D. J.; Jorgensen W. L.; Ciulli A.; Crews C. M. Targeting the Von Hippel-Lindau E3 Ubiquitin Ligase Using Small Molecules to Disrupt the VHL/HIF-1α Interaction. J. Am. Chem. Soc. 2012, 134, 4465–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley D. L.; Gustafson J. L.; Van Molle I.; Roth A. G.; Tae H. S.; Gareiss P. C.; Jorgensen W. L.; Ciulli A.; Crews C. M. Small-Molecule Inhibitors of the Interaction Between the E3 Ligase VHL and HIF1α. Angew. Chem., Int. Ed. 2012, 51, 11463–11467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congreve M.; Carr R.; Murray C.; Jhoti H. A “Rule of Three” for Fragment-Based Lead Discovery?. Drug Discovery Today 2003, 8, 876–877. [DOI] [PubMed] [Google Scholar]
- Pellecchia M.; Bertini I.; Cowburn D.; Dalvit C.; Giralt E.; Jahnke W.; James T. L.; Homans S. W.; Kessler H.; Luchinat C.; Meyer B.; Oschkinat H.; Peng J.; Schwalbe H.; Siegal G. Perspectives on NMR in Drug Discovery: a Technique Comes of Age. Nat. Rev. Drug Discovery 2008, 7, 738–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer M.; Meyer B. Characterization of Ligand Binding by Saturation Transfer Difference NMR Spectroscopy. Angew. Chem., Int. Ed. 1999, 38, 1784–1788. [DOI] [PubMed] [Google Scholar]
- Wang C.; Grey M.; Palmer A. CPMG Sequences with Enhanced Sensitivity to Chemical Exchange. J. Biomol. NMR 2001, 21, 361–366. [DOI] [PubMed] [Google Scholar]
- Dalvit C.; Pevarello P.; Tatò M.; Veronesi M.; Vulpetti A.; Sundström M. Identification of Compounds with Binding Affinity to Proteins via Magnetization Transfer From Bulk Water. J. Biomol. NMR 2000, 18, 65–68. [DOI] [PubMed] [Google Scholar]
- Mayer M.; Meyer B. Group Epitope Mapping by Saturation Transfer Difference NMR to Identify Segments of a Ligand in Direct Contact with a Protein Receptor. J. Am. Chem. Soc. 2001, 123, 6108–6117. [DOI] [PubMed] [Google Scholar]
- Knauth K.; Cartwright E.; Freund S.; Bycroft M.; Buchberger A. VHL Mutations Linked to Type 2C Von Hippel-Lindau Disease Cause Extensive Structural Perturbations in pVHL. J. Biol. Chem. 2009, 284, 10514–10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.