

Abstract
Novel photolabile amphiphiles containing thioxanthone-based fluorogenic caging groups are developed. Photoinduced fragmentation in dithiane-thioxanthone adducts was demonstrated to occur with 100% quantum efficiency at λ ~ 320 nm and more than 50% at λ ~ 360 nm. A plausible mechanism involves homolytic fission of a carbon-carbon single bond in the excited thioxanthone followed by disproportionation via hydrogen transfer. The critical feature of the system is that fluorescence of a substituted thioxanthone is recovered as a result of photofragmentation, making dithiane-thioxanthone adducts efficient fluorogenic caging groups. Photolabile amphiphiles containing these fluorogens are synthesized and their photoinduced disassembly is probed while following the fluorescence recovery. This methodology allows for destabilizing supramolecular assemblies of amphiphiles and at the same time offers a feedback mechanism for monitoring the process by fluorescence.
Keywords: thioxanthone, dithiane, fluorogenic group, amphiphile, photoinduced fragmentation, pulsed field gradients
1. Introduction
Photoinduced fragmentation has been playing an increasingly important role in chemical and biochemical applications, especially in cases when a photochemical event triggers a fast reaction, which otherwise is difficult to follow. Photoinduced uncaging of protected functionalities found numerous uses for triggered release of neurotransmitters and similar applications [1]. Primary utility of photoinduced uncaging lies with its ability to nearly instantaneously release an active form of a biological effector, which allows for subsequent monitoring of the resulting fast kinetics by spectroscopic or electrophysiological means. Laser pulses can be timed and focused in a very precise fashion, so the method offers an unprecedented spatial and temporal control. However, it is the physiological response which is measured as a physical observable resulting from the initial photochemical event.
Direct monitoring of the actual photochemical event is difficult, especially in biological systems. At some point it was therefore recognized that fluorogenic caging groups can fill this void by providing an independent physical observable, i.e. fluorescence, to independently confirm that the expected release actually occurred.
Existing fluorogenic caging systems are based on two general approaches. One approach involves tethering a fluorescent marker to a quencher via a photolabile linker, so that in the armed state most of fluorescence is quenched. Photoinduced fragmentation separates the two moieties, with fluorescence recovering [2]. The other approach involves covalent caging of a peripheral hydroxy group of a generic fluorescence marker by a generic photoprotecting group – such caging normally causes decrease in fluorescence [3]. Besides the fact that this solution is a somewhat artificial hybrid of old technologies, the caveat is that the UV-Vis absorption of the caged fluorophore is often higher than the photoremovable group, which inevitably reduces the quantum efficiency of release and may cause other complications. Nevertheless, compact fluorogenic caging groups show great promise as demonstrated by numerous accounts, including an elegant solution by Lawrence [4] for probing intracellular protein kinase activity with a light activated probe. The probe has the enzyme substrate, outfitted with a fluorophore and, in the immediate vicinity of this fluorophore – a strategically placed serine residue, decorated with the nitroveratryl. Photoinduced fragmentation deprotects the serine residue which leads to recovery of the function concomitant with fluorescence recovery.
Our approach to fluorogenic caging groups, which we term “release and report”, based on dithiane adducts of substituted thioxanthones [5]. In this paper we demonstrate how these groups can be incorporated in photolabile amphiphiles for monitoring of photoinduced morphological changes in amphiphilic supramolecular assemblies.
2. Results and Discussion
2-Alkoxy and 2-aminothioxanthones have strong emission in 400–500 nm range, which fluorescence quantum yields as high as 80%. Our caging methodology is based on disrupting conjugation in these aromatic ketones via addition of lithiated dithianes, which results in a blue shift of the absorption maxima and dramatic decrease in fluorescence intensity, Scheme 1.
Scheme 1.
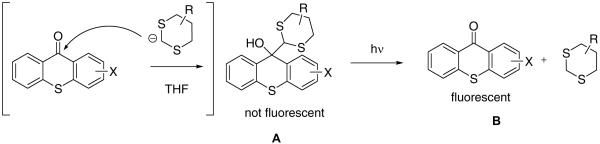
For example, in the case of 2-amidothioxanthones such disruption of conjugation reduces their fluorescence quantum yield in aprotic solvents from 60–70% to nearly zero. However, the remaining p-amidodiphenyl sulfide moiety has considerable residual absorption tailing beyond 400 nm. Excitation of the dithiane-thioxanthone adduct leads to expulsion of dithiane radical followed by disproportionation in the initially formed radical pair D regenerating emissive thioxanthone B and dithiane, Scheme 2. While direct homolytic cleavage in the singlet excited state of the adduct is the most plausible mechanism, we cannot completely rule out a three step process in which initial intramolecular electron transfer produces charge-separated species C, which undergoes fragmentation to furnish the two disproportionating radicals D.
Scheme 2.

Release of the fluorophore, thioxanthone can be accurately followed by bulk fluorescence measurements as shown in Figure 1.
Figure 1.
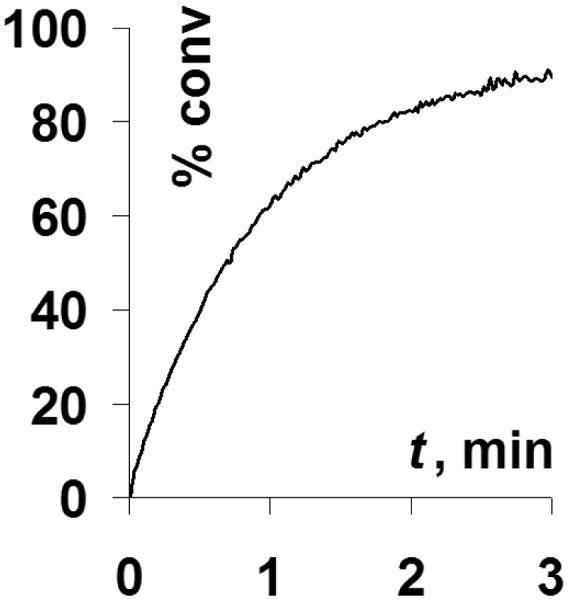
Fluorescence recovery as a function of irradiation time of 2 × 10−5 M acetonitrile solution of 2-butanoylamino-thioxanthone – methyl dithiane adduct at 320 nm.
We have synthesized two types of fluorogenic amphiphiles: one, in which the thioxanthone moiety carries a phosphocholine pendant (7, Scheme 3) and the other equipped with polyethyleneglycol brushes (12, Scheme 4).
Scheme 3.

Scheme 4.

2-Aminothioxanthone 3, synthesized from 2-chloro-5-nitrobenzoic acid 1 and thiophenol, was outfitted with a carboxylate “handle” via ring opening of glutaric anhydride. 2-Undecyl-1,3-dithaine was deprotonated with BuLi in THF and reacted with 4 to afford carboxylate 5, which was converted into N-hydroxysuccinimide ester and coupled with aminoethyl phosphocholine 6 to furnish amphiphile 7 (85%).
Alternatively, bis-hydroxythioxanthone 9 was obtained by condensation of thiosalicylic acid and catechol in sulfuric acid, bisalkylated by methyl triethyleneglycol tosylate and reacted with lithiated 5,5-bis(hexadecyl)-2-methyl-1,3-dithiane 11 to furnish PEGylated fluorogenic amphiphile 12 in 55% yield.
One important property of any novel caging functionality is its dark stability. To ensure high quantum yield of photoinduced fragmentation we designed a system with a weakened C-C bond, prone to homolytic cleavage (both thioxanthyl- and dithiane radical are very stable radicals). Naturally, these weakening can go only so far, before the dark instability, i.e. spontaneous thermal fragmentation in the absence of UV photons, renders the caging group useless. In order to assess the dark stability of dithiane-thioxanthone adducts the following experiments were carried out. Small amount of 2-aminothioxanthone 3 was converted into butyramide derivative 14 as shown in Scheme 5 and the adduct 14 was heated at 50°C for two weeks. No changes were detected by NMR after this treatment. The temperature dependence of the rate of the thermal fragmentation for the same adduct 14, plotted as ln(k/T) vs 1/T (Figure 2), gave activation enthalpy ΔH≠ = 27.5 kcal/mol, which is arguably the best optimal value. Our DFT computations for the bond dissociation energy with B3LYP functional, corrected by vibrational analysis (zpe), produced very similar value of 28.9 kcal/mol. The experimental graph in Figure 2 extrapolates to the lifetime of almost half a thousand years at 20°C attesting to more than satisfactory stability of the caging group. On the other hand, weakening the bond even further is problematic: for example, for a slightly smaller (20 kcal/mol) activation barrier the lifetimes are reduced to minutes at 20° C. Another important advantage of the new caging systems is that unlike in the o-nitrobenzyl group, the carbon-carbon bond between the thioxanthone and dithiane moieties is not a subject to hydrolytic cleavage.
Scheme 5.
Figure 2.

Determination of activation enthalpy, ΔH≠, for thermal (“dark”) fragmentation in 2-butanoylamino-thioxanthone – methyl dithiane adduct in DMSO.
Dithianes are readily outfitted with hydrophobic alkyl chains either at position 2 or 5 of the dithiane rings. This modification does not affect the ability of lithiated dithianes to add to ketones, which we used to synthesize photoactive fluorogenic amphiphiles carrying thioxanthone modified with hydrophilic groups, either phosphocholine or polyethylene glycol (PEG) brushes. 2-Alkyldithianes are most readily accessible from 1,3-propanedithiol and long chain aliphatic aldehydes. The synthesis of 5,5-dialkyl substituted dithianes is more involved as outlined in Scheme 6 for 5,5-bis(hexadecyl)-2-methyl-1,3-dithiane.
Scheme 6.
In all cases we studied, the bulk photolysis of the thioxanthone-dithiane based amphiphiles dissolved in an organic solvent (acetonitrile or dichloromethane) was accompanied by clearly observable fluorescence recovery ranging from 1 to 2 orders of magnitude.
Quantum yields of photoinduced fragmentation were measured for the model compound 14 at two wavelengths: 320 nm and 360 nm. At the shorter wavelength the quantum yield of the fragmentation was unity within the error of our measurements. At 360 nm, the quantum yield was measured to be 56%, which is comparable to that of the o-nitrobenzyl protecting groups.
As stated in the introduction, our design of fluorogenic amphiphiles offers an independent physical observable (= fluorescence) for monitoring the photochemical changes (= photoinduced fragmentation) which is expected to change morphology of amphiphilic self-assemblies. This initial hypothesis was tested in a series of NMR Pulse Field Gradient (PFG) experiments in an aqueous micellar solution.
Fluorogenic amphiphile 7, mixed with dodecyl phosphocholine (DPC) 2:1 produces stable micellar solution, which is virtually non-fluorescent. Irradiation of these solutions led to full fluorescence recovery, which can be fitted to a pseudo-first order kinetics as long as one uses a stable UV source. The PFG experiments (a typical experiment is shown in Figure 3) had reveled that, concomitant to the fluorescence recovery, the diffusion coefficient measured using several different dithiane protons was increasing.
Figure 3.

A typical PFG experiment with the logarithm of signal intensity plotted against the squared value of the gradient strength, (Gauss/cm)2
This indicates that the micelles housing these dithianes were shrinking in size as the photolysis was continued. The initial diffusion coefficient was measured to be 2.6 ± 0.7 × 10−7 cm2 s−1, which corresponds to rather large micellar assemblies having a hydrodynamic radius of more than 8 nm. In 5 minutes of photolysis the diffusion coefficient increased to 4.1 ± 1.2 × 10−7 cm2 s−1 (~ 5 nm hydrodynamic radius). We interpret these results in terms of considerable loss of micellar volume due to departing hydrophilic fluorophore.
In conclusion, we have described here a novel general approach to fluorogenic amphiphiles based on thioxanthone-dithiane adducts and demonstrated its applicability for monitoring of the actual photochemical events with fluorescence measurements. Work is in progress in our laboratory to implement this methodology in light-sensitive fluorogenic bilayers and liposomes.
3. Experiment
Common reagents were purchased from Sigma-Aldrich and used without further purification. THF was refluxed over and distilled from potassium benzophenone ketyl prior to use. The 1H and 13C NMR spectra were recorded at 25°C on a Varian Mercury 400 MHz instrument, CDCl3, DMSO-d6, CD3OD and CD3CN as solvents, and TMS was used as internal standard. The diffusion coefficients were measured with Varian Mercury's Performa I pulse field gradient module and 4 nucleus autoswitchable PFG probe. Column chromatography was performed on Silica Gel, 70–230 mesh ASTM. UV-Vis spectra were recorded on a Beckman DU-640 Spectrophotometer, and fluorescence spectra were recorded on Varian Cary Eclipse instrument. The irradiations were carried out in a carousel Rayonet photo reactor (RPR-3500, RPR-3000 lamps), or using the Oriel Photomax housing with a 200W medium pressure ozone free mercury lamp and a grating monochromator or different wavelength bandpass filters, such as a U-360 broad bandpass (360 nm ± 45 nm) and 405 nm ± 5 nm narrow bandpass interference filter.
3.1 Synthetic Procedures
3.1.1. N-(9-Oxo-9H-thioxanthen-2-yl)-butyramide (13)
2-Aminothioxanthen-9-one (1g, 4.4 mmol) was dissolved in 15mL of dichloromethane, and butyryl chloride (0.7g. 6.6 mmol) and catalytic amount of triethyl amine was added to this solution under stirring. The reaction mixture was stirred at room temperature overnight. The resulting solid was filtered, washed with water, dried to furnish yellow powder (1.18 g, 90%). 1H NMR (DMSO-d6, 400 MHz): δ 10.25 (s, 1H), 8.71 (d, J=2.35Hz,1H), 8.45 (d, J=8.17Hz,1H), 8.04 (dd, J1=2.36Hz, J2=8.76Hz,1H), 7.83–7.73 (m, 3H), 7.58–7.54 (m, 1H), 2.30 (t, J=7.28Hz, 2H), 1.65–1.60 (m, 2H), 0.90 (t, J=7.35Hz, 2H). 13C NMR (DMSO-d6, 400 MHz): δ 179.31, 172.22, 138.91, 137.35, 133.49, 129.81, 129.47, 128.65, 127.75, 127.29, 127.22, 125.27, 118.65, 118.63, 39.02, 19.16, 14.31.
UV-Vis: λmax: 395 nm
Fluorescence: λex: 395 nm, λem: 460 nm, Φf = 0.646 (10−5 M, Acetonitrile)
3.1.2. N-[9-Hydroxy-9-(2-methyl-1, 3-dithian-2-yl)-9H-thioxanthen-2-yl]-butyramide (14)
Methyldithiane (902 mg, 6.73 mmol) was dissolved in 15 mL of freshly distilled THF under nitrogen. To this solution 2.52 mL of butyl lithium (1.6M solution in hexanes, 4 mmol) was added dropwise upon stirring at room temperature. The resulting mixture was stirred for 10 min at this temperature to generate the anion. Thioxanthone 13 (400mg, 1.35 mmol) in 10 mL of THF was added dropwise to the vigorously stirred solution of the methyldithianyl anion. The reaction mixture was stirred overnight at room temperature. The subsequent aqueous workup included quenching the reaction mixture with a 30 mL 1 M solution of ammonium chloride, extracting twice with ether, and drying the organic layer over sodium sulfate. The solvent was removed under vacuum, the crude reaction mixture was purified by silica gel column chromatography, hexane:EtOAc (9:1 & 3:2) as an eluent to afford pale yellow solid (380 mg, 65.2%). 1H NMR (CDCl3, 400 MHz): δ 8.03–8.02 (m, 2H), 7.72 (d, J=8.49Hz, 1H), 7.35–7.26 (m, 4H), 4.03 (s, 1H), 2.91–2.86 (m, 2H), 2.73–2.66 (m, 2H), 2.27 (t, J=7.48Hz, 2H), 1.91–1.86 (m, 4H), 1.74–1.64 (m, 2H), 1.49 (s, 3H), 0.95 (t, J=7.35Hz, 3H). 13C NMR (CDCl3, 400 MHz): δ 171.5, 135.51, 134.47, 133.34, 132.42, 130.67, 128.21, 127.43, 126.45, 125.91, 125.219, 121.84, 120.24, 80.20, 76.92, 62.42, 39.86, 27.86, 25.77, 24.58, 19.20, 13.99. Calcd. for C22H25NO2S3,%: C, 61.22; H, 5.84. Found: C, 61.18; H, 5.86.
3.1.3. 2,3-Dihydroxythioxanthone (9)
2,3-Dihydroxythioxanthone was prepared via a modified procedure of Roberts and Smiles [6]. Catechol (36.3 g 0.33 mol) and thiosalicylic acid 8 (10.3 g, 0.067mol) were heated in 100 mL of H2SO4 for 4 h at 80°C. The mixture was poured into 1 L of boiling water. The suspension was cooled and the formed precipitate was filtered and dried. The crude product was refluxed in isopropanol for 1 h and the suspension was filtered while hot. The precipitate was collected, dried, and used without further purification.
3.1.4. 2,3-Bis(methoxyethyloxyethyloxyethyloxy)thioxanthone (10)
2,3-Dihydroxythioxantone (0.25 g, 1.02 mmol) was added to a suspension of NaH (0.09 g (60%) ~ 2.3 mmol) in 50 mL of DMF. The reaction was stirred 15 min and methyl triethyleneglycol tosylate (0.65 g, 2.04 mmol) was added. The mixture was stirred at 20°C overnight, after that the solvent was evaporated, and the residue was passed through alumina (CH2Cl2/THF 10: 1) to furnish the product as a yellow oil, 0.29 g (53%). 1H NMR (CDCl3, 400 MHz): δ 8.61 (ddd, 1H, J1 = 8.1 Hz, J2 = 1.4 Hz, J3 = 0.7 Hz), 8.06 (s, 1H), 7.61–7.55 m (2H), 7.47 (ddd, J1 = 8.2 Hz, J2 = 6.4 Hz, J3 = 2.0 Hz), 6.99 (s, 1H), 4.32–4.27 (m, 4H), 3.96–3.94 (m, 4H), 3.79–3.75 m (4H), 3.70–3.64 (m, 8H), 3.55–3.53 (m, 4H), 3.372 (s, 3H), 3.369 (s, 3H). MALDI-TOF (M+Na), 559.2, calcd for C27H36NaO9S 559.1972.
3.1.5. 2,2-Bis(hexadecyl)-1,3-propanediol (16)
LiAlH4 (10 g, 0.26 mol) was suspended in 500 mL of THF. Diethyl 2,2-bis(hexadecyl)malonate 15 (70 g, 0.115 mol), obtained by alkylation of diethyl malonate via previously described procedures, was dissolved in 100 mL of THF and added to the suspension at 20°C. The reaction mixture was refluxed for 1 hr while stirring, quenched with 5% HCl and extracted with ether (3 × 500 mL). The organic layer was dried over Na2SO4, and the solvent was evaporated to furnish the crude product as a white solid. The diol was recrystallized from a minimal amount of heptane: 49.5 g, (82%). 1H NMR (CDCl3, 400 MHz): δ 3.55 (s, 4H), 2.54 (s, 2H), 1.34–1.18 (m, 60H), 0.88 (t, 6H, J = 7.0 Hz).
3.1.6. 2,2-Bis(hexadecyl)-1,3-diiodopropane (17)
Triphenylphosphine (55 g, 0.21 mol) was dissolved in 500 mL of benzene and 50 mL of pyridine. To this solution iodine (55.9 g, 0.22 mol) was slowly added and the mixture was stirred at 20°C for 15 min. Diol 16 (50 g, 0.095 mol) was added to the mixture and the reaction was refluxed for 2 hr while stirring. The precipitate was filtered off and the solvent was evaporated. The product was purified on silica gel using hexane/EtOAc (10: 1) as an eluent: 70g, (84%).
3.1.7. 2,2-Bis(hexadecyl)-1,3-bis(acetylthio)propane (18)
To a DMF/THF (1:1) solution of diiodide 17 (50 g, 0.067 mol) potassium tioacetate (16.9g 0.147 mol) was added and the reaction was refluxed for 2 hr while stirring. The reaction mixture was poured into water, extracted with ether (3 × 150 ml), and the organic layer was dried. The solvent was evaporated and the product was used without further purification (95%). NMR showed no impurities. 1H NMR (CDCl3, 400 MHz): δ 2.92 (s, 4H), 2.33 (s, 6H), 1.28–1.22 (m, 60H), 0.88 (t, 6H, J = 7.0 Hz).
3.1.8. 2,2-Bis(hexadecyl)-1,3-propanedithiol (19)
Method A
To a suspension of LiAlH4 (6.4 g, 0.17 mol) in 500 mL of THF solution of bis-thioacetate 18 (50 g, 0.078 mol) in 100 mL of THF was added at 20°C. The reaction was refluxed for 1h while stirring under nitrogen, quenched with HCl (5%), the product was extracted with ether (3 × 300 ml) dried over Na2SO4, and the solvent was evaporated. The resulting colorless oil was cyclized into dithiane without further purification.
Method B
Bis-thioacetate 18 (5 g, 7.81 mmol) was dissolved in 20 mL of THF and 5 drops of water. To this solution 3g of crushed NaOH pellets were added and the reaction was stirred overnight at 20°C under nitrogen. The reaction was quenched with 10% HCl to pH 1, the product was extracted with ether, the organic extract was dried, and the solvent was evaporated. The product was cyclized into dithiane without further purification.
3.1.9. 5,5-Bis(hexadecyl)-2-methyl-1,3-dithiane (20)
Dithiol (19) (5.57 g, 10 mmol) and acetaldehyde (0.84 mL, 15 mmol) were dissolved in 100 mL CH2Cl2. Boron trifluoride etherate (5.6 mL, 50 mmol) was slowly added to the solution and the reaction was stirred at 20°C overnight. The reaction was washed with 10% aqueous NaOH (2 × 100 mL), water and the organic layer was separated. The aqueous layer was extracted with 100 mL CH2Cl2, the extracts were combined and dried over Na2SO4. The solvent was evaporated and the product was purified by column chromatography on silica gel (hexane/EtOAc 10: 1). (4.26 g, 73%). 1H NMR (CDCl3, 400 MHz): δ 3.99 (q, 1H, J = 6.9 Hz), 2.70 (d, 2H, J = 14.1 Hz), 2.52 (d, 2H, J = 14.1 Hz), 1.48 (d, 3H, J = 7.0 Hz), 1.32–1.20 (m, 60H), 0.88 (t, 6H, j = 7.0 Hz).
3.1.10. Fluorogenic amphiphile (12)
5,5-Bis(hexadecyl)-2-methyl-1,3-dithiane 11 (0.41 g, 0.7 mmol) was dissolved in 15 mL of freshly distilled THF under nitrogen atmosphere. To this solution 0.5 mL of butyl lithium (1.6M solution in hexanes) was added dropwise upon stirring at room temperature. The resulting mixture was stirred for 10 min at this temperature to generate the anion. PEG-Thioxanthone 10, (0.150 g, 0.26 mmol) in 10 mL of THF was added dropwise to the vigorously stirred solution of the dithianyl anion. The reaction mixture was stirred overnight at room temperature. The subsequent aqueous workup included quenching the reaction mixture with a 30 mL 1 M solution of ammonium chloride, extracting with ether (2 × 50mL), and drying the organic layer over Na2SO4. The solvent was removed under vacuum, the crude reaction mixture was purified by silica gel column chromatography, hexane:EtOAc (10:1 to pure EtOAc) as an eluent to afford pale yellow solid (183 mg, 55%). 1H NMR (CDCl3, 400 MHz): δ 8.08 (d, 1H, J = 7.0 Hz), 7.69 (s, 1H), 7.34–7.26 (m, 3H), 6.84 (s, 1H), 4.22 (ddd, 2H, J1 = 4.97 Hz, J2 = 4.9 Hz, J3 = 1.0 Hz), 4.17 (t, 2H, J = 5.1 Hz), 4.01 (s, 1H), 3.87 (q, 4H, J = 4.6 Hz), 3.77–3.72 (m, 4H), 3.69–3.61 (m, 8H), 3.56–3.52 (m, 2H), 3.51–3.48 (m, 2H), 3.37 (s, 3H), 3.32 (s, 3H), 2.48 (t, 2H, J = 14 Hz), 2.37 (dd, 2H, J1 = 14.0 Hz, J2 = 4.8 Hz), 1.4–1.2 (m, 60H), 0.879 (t, 3H, J = 6.9 Hz), 0.873 (t, 3H, J = 6.9 Hz). MALDI-TOF, (M+Na) 1141.7, Calcd for C64H110NaO9S3: 1141.7210.
3.1.11. N-(tert-butoxycarbonyl)-O-phosphorylcholine ethanolamine
To a stirred mixture of 2.0 g of N-tert-butoxycarbonylethanolamine (12 mmol), 2.2 mL of triethylamine (14 mmol) and 100 mL of benzene in 250 mL round bottom flask 1.2 mL of 2-chloro-1,3,2-dioxaphospholane-2-oxide (13 mmol) was added at 10°C. The stirring was continued overnight at room temperature. Then 200 mL of diethyl ether were added to the reaction mixture, triethylamine hydrochloride was removed by filtration and the solvent was removed. The residue was dissolved in 100 mL of acetonitrile in a 250 mL glass bomb, cooled to −20°C and trimethylamine was condensed into the stirred reaction mixture at that temperature. Upon accumulating 10 g of trimetylamine the reaction mixture was heated in the sealed bomb at 60°C for 24h, the solvent was removed to afford pure product as a colorless solid. Yield 4.0g (quantitative). 1H NMR (DMSO-d6, 400 MHz): δ 7.08 (m, 1H), 4.04 (m, 2H), 3.63 (q, 2H, J = 6.1 Hz), 3.51 (t, 2H, J=4.9 Hz), 3.12 (s, 9H), 3.04 (m, 2H), 1.35 (s, 9H).
3.1.12. O-phosphorylcholine ethanolamine (6)
To rapidly stirred mixture of 1.0 g of Boc-protected amine (3.1 mmol) in a 50 mL round bottom flask the solution of 1.55 g of 95–98% sulfuric acid (~15.5 mmol) in 10 mL of water was added at room temperature and stirring was continued overnight. Barium hydroxide octahydrate (4.8 g, 15.5 mmol) was added and stirred for 24h, precipitated barium sulfate was filtered off and the solution was evaporated to give 0.7 g of 6, (quantitative). 1H NMR (D2O, 400 MHz): δ 4.15 (m, 2H), 3.80 (q, 2H, J = 5.5Hz), 3.51 (m, 2H,), 2.81 (m, 2H).
3.1.13. Undecyldithiane adduct 5
To rapidly stirred solution of 9.7 g of dithiane (35 mmol) in 150 mL of THF 22 mL of 1.6M solution of butyl lithium (35 mmol) was added at 0°C and stirred for 10 min. Then the solution of 2.0 g of the acid (5.9 mmol) in 100 mL of THF was added at 0°C and stirred overnight at ambient temperature. The reaction mixture was poured in saturated solution of ammonium chloride, acidified by 5% solution of hydrochloric acid and extracted with ethyl acetate. The organic layer was dried over Na2SO4, and the solvent was evaporated. The residue was subjected to gel filtration on silica gel in a sintered glass funnel using ethyl acetate – methanol mixture 10:1 as the eluent, to afford 2.6g (72%). 1H NMR (CDCl3, 400 MHz): δ 8.07 (m, 2H), 7.61 (m, 2H), 7.28 (m, 3H), 7.23 (d, 1H, J = 8.5 Hz), 3.73 (s, 1H), 3.12 (m, 2H), 2.58 (m, 2H), 2.42 (q, 4H, J = 6.8Hz), 2.01 (m, 2H), 1.94 (m, 2H), 1.81 (m, 2H), 1.50 (m, 2H), 1.04–1.41 (m, 18H), 0.87 (t, 3H, J = 6.8Hz).
3.1.14. NHS ester of 5
To the solution of 2.6 g of the adduct 5 (4.2 mmol) in 150 mL of DCM 0.73 g of N-hydroxysuccinimide (6.3 mmol) and 0.97g of EDC (5.1 mmol) was added and stirred overnight. The reaction mixture was washed with water, brine, organic layer was separated, dried, the solvent was removed to give the product. Yield 2.8g, 93%. 1H NMR (CDCl3, 400 MHz): δ 8.07 (m, 2H), 7.62 (m, 2H), 7.30 (m, 3H), 7.25 (d, 1H, J = 8.5 Hz), 3.63 (s, 1H), 3.09 (m, 2H), 2.87 (m, 4H), 2.69 (t, 2H, J = 6.8Hz), 2.55–2.60 (m, 2H), 2.45 (t, 2H, J = 6.8Hz), 2.15 (m, 2H), 1.90–1.98 (m, 2H), 1.75–1.85 (m, 2H), 1.52 (m, 2H), 1.04–1.41 (m, 18H), 0.87 (t, 3H, J = 6.8Hz).
3.1.15. Fluorogenic phosphocholine 7
The mixture of 0.43 g of O-phosphorylcholine ethanolamine (1.9mmol), 1.13 g of 5-NHS (1.6 mmol) and 50 mg of DMAP in 30 ml of DMSO was stirred for 48h in a round bottom flask. The solvent was removed on a high vacuum pump, the residue was dissolved in DCM, washed with 5% solution of hydrochloric acid and brine. The organic layer was separated, dried, the solvent was removed to afford 1.11g (85%) of pure 7. 1H NMR (DMSO-d6; 400 MHz): δ 8.37 (m, 1H), 8.25 (m, 1H), 8.09 (m, 1H), 7.77 (m, 1H), 7.25 (m, 3H), 7.17 (m, 1H), 6.32 (s, 1H), 4.06 (m, 2H), 3.69 (m, 2H), 3.53 (m, 2H), 3.45 (m, 2H), 3.18 (m, 2H), 3.13 (s, 4H), 2.53 (m, 2H), 2.34 (m, 2H), 2.09 (m, 2H), 2.15 (m, 2H), 1.97–2.09 (m, 2H), 1.75–1.83 (m, 2H), 1.60 (m, 2H), 0.94–1.32 (m, 18H), 0.83 (t, 3H, J = 6.8Hz).
3.2. Quantum Yields of Fragmentation
Determination of quantum yields of fragmentation was carried out by using the onitrobenzaldehyde actinometer as a reference (Φstd = 0.5). Monochromated output of the Oriel PhotoMax UV source (200 W, ozone free mercury-xenon lamp) was directed at the rotating carousel of quartz tubes containing the solution of the standard actinometer in alternation with quartz tubes containing the adduct 14. At 365 nm the quantum yield was determined to be 56%. At 320 nm the quantum yield was determined to be unity within the accuracy of our measurements.
3.3. Quantum Yield of Fluorescence
Thioxanthone was used as a reference compound for determining the quantum yield of fluorescence (Φf=0.12 in methanol). The quantum yield of fluorescence was determined using the following equation.
where As = Absorbance of the standard, Au = Absorbance of the unknown, Fs = Emission intensity of the standard, Fu = Emission intensity of the unknown, Φs = quantum yield of the standard (0.12)
3.4. Preparation of micelles
6.47 mg of phosphocholine amphiphile 7 (20 mmol) and 3.51mg of DPC (10 mmol) were shaken in 2ml of D2O for 0.5h to produce a clear solution of 10mM : 5mM 7 : DPC. The diffusion coefficient of the produced micelles was measured by pulse-field gradient NMR experiments to be 2.6 ± 0.7 × 10−7 cm2 s−1, which corresponds to approximately 8.3nm hydrodynamic radius.
3.5. Pulse Field Gradient (PFG) diffusion measurements
A typical experiment included a series of gradient field increases from 0 to approximately 20 G/cm. The natural logarithm of signal intensities was then plotted against G2 and the diffusion coefficient was obtained from the slope of this linear plot according to the following equation:
where γ - magnetogyric ratio, 2.6752 × 10 4 1/G s, δ - length of pulsed field gradient (s), G - gradient strength (G), Δ - diffusion time = δ + gstab1 + pulse width + diff, Ds - diffusion coefficient.
Supplementary Material
Acknowledgment
Support of this research by the NIH, GM067655, is gratefully acknowledged.
References
- [1].Goeldner M, Givens R, editors. Dynamic Studies in Biology: Phototriggers, Photoswitches and Caged Biomolecules. Wiley; 2005. [Google Scholar]
- [2].For a representative example, see Pellois J-P, Hahn ME, Muir TW. J. Am. Chem. Soc. 2004;126:7170. doi: 10.1021/ja0499142.
- [3].For a representative example, see Marriott G. Bioconjugate Chem. 1998;9:143. doi: 10.1021/bc970147o.
- [4].Veldhuyzen WF, Nguyen Q, McMaster G, Lawrence DS. J. Am. Chem. Soc. 2003;125:13358. doi: 10.1021/ja037801x. [DOI] [PubMed] [Google Scholar]
- [5].For initial communication see Majjigapu JRR, Kurchan AN, Kottani R, Gustafson TP, Kutateladze AG. J. Am. Chem. Soc. 2005;127:12458. doi: 10.1021/ja053654m.
- [6].Roberts KC, Smiles S. J. Chem. Soc. 1929:863. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.