

The nitrogenase molybdenum–iron (MoFe)–protein contains two types of metalloclusters, the [8Fe:7S] P–cluster and the [7Fe:9S:Mo:C:homocitrate] FeMo–cofactor, that have been the focus of considerable interest to define their roles in the mechanism of biological nitrogen fixation.[1] In addition to these two metalloclusters, a third metal binding site is present in the MoFe–protein that has attracted much less attention. The metal at this site has been assigned to either Ca2+ or Mg2+ in different MoFe–protein structures and is positioned at the interface between the two β–subunits, approximately 25 Å and 21 Å away from the P–cluster and FeMo–cofactor, respectively.[2] Six ligands coordinate the metal in approximate octahedral geometry: the side chain oxygens from β–Glu109, β’–Asp353, and β’–Asp357, the backbone carbonyl oxygen of β–Arg108, and two water molecules (β and β’ denote the two distinct β–subunits in an α2β2 MoFe–protein tetramer). The assignments as either Ca2+ or Mg2+ were inferred from the value of the electron density at this position, the coordination environment at the metal binding site and/or the presence of Mg2+ in the crystallization solution.[2a,b,d] As the identified species are not redox–active metals, it was speculated that this metal site most likely serves a structural rather than a mechanistic role. However, as shown below, our multiple–wavelength anomalous diffraction (MAD) data around the Fe K–edge unambiguously demonstrate the presence of Fe at this binding site, which we will subsequently refer to as the mononuclear metal binding site (MMB–site) and the metal at this site as Fe16.
MoFe–protein crystals were grown under anaerobic conditions as described previously[1b] using wildtype protein isolated from Azotobacter vinelandii. Two sets of anomalous diffraction data were collected at 7080 eV and 7130 eV (below and above the Fe K–edge, respectively (Table S1)) to 2.1 Å resolution. As shown in Figure 1, well–defined density is clearly present at the MMB–site in the double difference (Δanom7130eV – Δanom7080eV) anomalous density map, and the peak density is about half of that observed at the Fe sites in the FeMo–cofactor and P–cluster. In contrast, the density around non–Fe heavy atoms (e.g. S and Mo) is comparable to the noise level. The double difference anomalous density maps clearly indicate the presence of Fe at this site and the comparison of the anomalous density to the Fe sites in the two metalloclusters suggests a partial occupancy.
Figure 1.
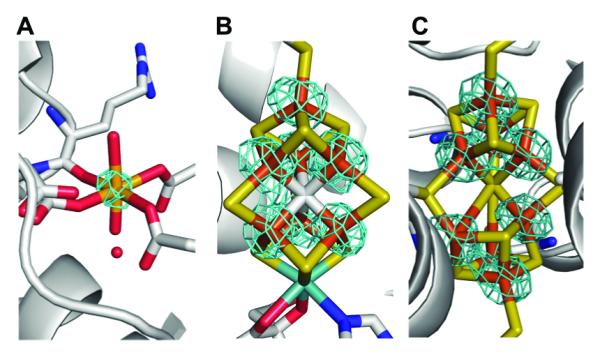
The double difference (Δanom7130eV – Δanom7080eV) anomalous density maps (highlighted in cyan) at the MMB–site (A), the FeMo–cofactor (B) and the P–cluster (C). The Fe atoms are shown in orange, Mo in cyan, S in yellow, C in gray, N in blue and O in red. These maps are contoured at the level of 10 sigma.
To determine the oxidation state and chemical environment of Fe16, site–specific Fe K–edge absorption spectra were extracted from anomalous differences present in a 10° wedge of diffraction data (plus inverse beam) measured from a MoFe–protein crystal at 19 different energies across the Fe K–edge (Table S2). The Δf” for heavy atoms (Fe, Mo and S) in the MoFe–protein structure were then refined as described previously.[3] As shown in the top panel of Figure 2, the well–defined absorption edge feature of Fe16 confirms unambiguously the presence of an iron at this site, consistent with the anomalous density analysis shown above. Significantly, the absorption edge of the Δf” spectrum for Fe16 is about 5 eV higher relative to the averaged Δf” spectra for the iron atoms in the two metalloclusters. Because the cluster Fe are mainly four–coordinated ferrous irons, the 5 eV shift suggests either a different coordination environment and/or a different oxidation state. Indeed, the edge energy and the fine structure around the absorption edge of Fe16 are similar to that of a hexa–coordinated ferrous model compound (ferrous sulfate, bottom panel of Figure 2), and differ markedly from the corresponding ferric model compound (ferric sulfate, bottom panel of Figure 2). The average Fe–O distance for Fe16 is 2.23 Å, which matches the average Fe–O distances of hexa–coordinated oxygen–only ferrous compounds (2.16 Å ± 0.08 for 61 ferrous FeO6 compounds and 2.00 Å ± 0.02 for 14 ferric FeO6 compounds with R factor ≤ 0.05) in the Cambridge Crystallographic Structure Database.[4] Combining these observations, we conclude that Fe16 has properties consistent with a hexa–coordinated ferrous iron.
Figure 2.
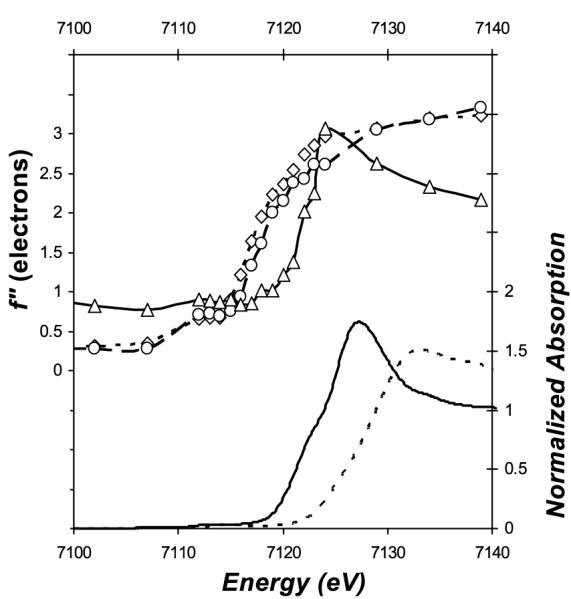
The refined Δf” spectra for Fe sites in the MoFe–protein with comparison to that of Fe models. In the top panel are the refined Δf” for Fe16 (solid line with Δ symbol) and the averaged Δf” for all Fe in the P–cluster (dash line with ◇ symbol) and FeMo–cofactor (broken line with ○ symbol), respectively; in the bottom panel are the Δf” converted from the X–ray absorption spectra of ferrous sulfate heptahydrate (FeSO4•7H2O, solid line) and ferric sulfate hydrate (Fe2(SO4)3 •xH2O, broken line), respectively. No background removal or normalization were applied to the refined Δf” spectra for Fe16 and other Fe in the MoFe–protein. The absorption edge of Δf” curve for Fe16 is ~5 eV higher relative to the metalloclusters; the shift towards higher energy for the FeMo–cofactor relative to the P–cluster is consistent with a more reduced state for the Fe in the latter.
As a further check, we examined the recent 1 Å resolution structure of the MoFe–protein (PDB code 3U7Q)[1f] for the feasibility of fitting a partially occupied Fe at the MMB–site. Indeed, the refinement results show the electron density at the MMB–sites is approximately equivalent to either 0.5 Fe, 0.7 Ca or 1 Mg/Na per site. The shape of the electron density in the MMB–sites, however, indicates it may be described as a superposition of two distinct arrangements (Figure 3A). Currently, the non-spherical electron density around Fe16 is modelled with two mutually exclusive arrangements that contain either the Fe16 (Figure 3B) or Na1 (Figure 3C), offset by 1.2 Å. The electron density also indicates that the side chain of Glu109 has two distinct conformations. Surrounding these peaks are two half–occupied water molecules (designated HOH–2A and HOH–2B) that could be coordinated to Fe16 and Na1, respectively (Figure 3). The average Na–O distance of 2.37 Å in the pentahedral coordination environment around Na1 is consistent with that of Na+, but not Mg2+ or H2O binding sites reported in the Cambridge Crystallographic Structure Database and the Protein Data Bank (Table S3).[5] The M–O distances around Na1 are also compatible with Ca2+, but the electron density does not account for a half–occupied Ca2+.
Figure 3.

The refined crystallographic structure at the MMB site in the Av MoFe–protein superimposed on the electron density map contoured at 2 sigma (A). The observed density of the MMB–site is interpreted as a superposition of partially occupied Fe16–bound (B) and Fe16–free (C) forms, with the latter modeled as a half–occupied Na+ (Na1). The iron atom is highlighted in orange, oxygen in red, nitrogen in blue, carbon in silver, and sodium in purple blue color.
The presence of any transition metals other than Fe and Mo in the MoFe-protein has been ruled out by hard X–ray emission spectroscopy (XES) studies and inductively coupled plasma mass spectrometry (ICP–MS) analysis of the metal composition of the MoFe–protein. As shown in Figure S1, Fe, Zn, Cu and V are the only transition metals with atomic number Z ≤ 30 observed in the X–ray emission spectra of the MoFe–protein crystal and the crystallization solution. The Cu and V Kα peak intensities in the spectrum of the MoFe–protein crystal are present in both the crystallization solution and metal–free water (data not shown), suggesting that they are primarily contributed by the emission from the beamline components. From the ICP–MS analysis, the Fe/Mo ratio averages 15.6 ± 0.3 (Table S4), an excess of 0.6 over the iron present in the FeMo–cofactor and P–cluster, but this excess is within the experimental error of ICP-MS analysis on Fe. In addition to Fe and Mo, Mg and Ca are present at 1.7 – 2.6 and 0.8 – 1.5 atoms per MoFe–protein tetramer, respectively. The only other metal detected at a level of ~ 0.1 atom per molecule by ICP–MS analysis is Zn.
To quantify the Fe16 occupancy from the MAD data, we compared the edge jump for Fe16 to the average of that observed for all the Fe in the P–cluster and FeMo–cofactor, with the assumption that the latter corresponds to unit occupancy. The edge jump here refers to the difference between the Δf” values refined above and below the Fe K–edge (at 7130 and 7080 eV, respectively). Eight sets of the MAD data were collected for this study, which were acquired from different MoFe–protein crystals and with resolutions ranging from 2.1 Å to 1.39 Å (Table S5). When all 8 data sets are analyzed, the Fe16 occupancies average 0.5, ranging from 0.1 to 1.1. The ratios of the electron density at the Fe16 site to the average of all the Fe in the metalloclusters average 0.7, with a range of 0.4 to 1.1; the higher value relative to the anomalous density–derived quantity reflects the contribution of all scatterers at this site to the electron density, while only iron contributes to the absorption edge jump. The observed variation could reflect changes in the iron content during protein purification or crystallization; as Fe16 is in an approximate octahedral geometry with O–only ligands, the observed large variation in the edge jumps cannot be explained by the polarization effect.[6] Substoichiometric occupancies of metal binding sites are not unprecedented and have been noted in several metalloenzymes, such as the copper containing particulate methane monooxygenase,[7] the tungsten–iron–sulfur enzyme acetylene hydratase,[8] acetyl–CoA synthase/carbon monoxide dehydrogenase,[9] and in the ferrous form of superoxide reductase,[10] as well as for copper–bound metallochaperones.[11]
A critical question concerns the possible roles of Fe16 at the MMB–site. Protein sequence comparisons of nitrogenase homologues indicate that the residues comprising the MMB–site may be important (Table S6); the identities of the three β–subunit residues (Glu109, Asp353 and Asp357) involved in Fe16 binding are highly conserved in almost all the nitrogenase homologues within and across species (Figure S2). Arg108 is the only Fe16-binding residue not strictly conserved, but as it coordinates through its backbone carbonyl oxygen, it is therefore not restricted to any specific side chain. Intriguingly, one of the Fe16–bound ligands, Asp357, is conserved in the nitrogenase–like, light–independent protochlorophyllide reductase BchNB.[12] This residue in BchNB is close to the substrate binding site and has been shown to be involved in proton transfer during the reduction of protochlorophyllide (Figure S2F).[13] The possibility of inferring potential functional roles for Fe16 from the functions of comparable sites in other proteins is precluded by the apparent absence of mononuclear iron sites with similar coordination environments in the Protein Data Bank, as searched with version 10 of the MESPEUS database.[14] Indeed, the best match for Fe16 is one of the two iron sites present in di–iron centers of rubrerythrins (Figure S3), although the correspondence is incomplete and there is no evidence for a second iron site in our electron density maps. While the conservation of the residues at the MMB–site emphasizes the potential importance of the site in nitrogenase, the precise function has yet to be defined.
The identification of Fe16 in the MMB–site, particularly at partial occupancy, is a remarkable demonstration of the analytical capability of MAD to site-specifically identify elements, even in complex systems such as nitrogenase that have been extensively studied for over 40 years. In the case of the MoFe–protein, Fe16 at full occupancy would account for ~6% of the Fe, a value not reliably measurable by conventional metal analysis while its presence has potential implications for the interpretation of other biophysical studies. A review of past literature suggests that Fe16 may have been “ghosting” around in spectroscopic investigations of nitrogenase. A minor ferrous “impurity”, varied from 3% to 8% of total Fe in nitrogenase, has been reported in Mössbauer studies.[15]
The characteristics of this ferrous “impurity” are typical for a hexa–coordinated, high–spin ferrous species with an oxygen and/or nitrogen ligand sphere,[15b] matching the chemical environment around Fe16. Because a high–spin ferrous iron cannot be easily characterized by conventional electronic paramagnetic resonance spectroscopy studies typically conducted on nitrogenase,[16] the presence of this iron species may have been overlooked in past studies. It is important to keep the possible presence of a 16th Fe in nitrogenase in mind when interpreting results from structural and crystallographic and spectroscopic studies on nitrogenase.
Supplementary Material
Footnotes
The work was supported by NIH (grant GM45162 to D.C.R.), NSERC PDF award (to L.M.Z.), and Deutsche Forschungsgemeinschaft (grants Ei–520/7 to O.E., An–676/1 to S.L.A.A., and IRTG 1478). G.M. is a Marie Curie International Outgoing Fellow (European Commission, grant no. 252961). We thank staff at Beamline 12–2, Stanford Synchrotron Radiation Lightsource (SSRL) and staff at HXMA Beamline, Canada Light Source (CLS).SSRL is operated for the DOE and supported by its OBER and by the NIH, NIGMS (P41GM103393) and the NCRR (P41RR001209). CLS is supported by NSERC, NRC, CIHR and the University of Saskatchewan. We acknowledge the Gordon and Betty Moore Foundation, the Beckman Institute, and the Sanofi–Aventis Bioengineering Research Program at Caltech for their generous support of the Molecular Observatory at Caltech. The assistance of Nathan Dalleska and the resources of the Caltech Environmental Analysis Center are greatly appreciated.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Dr. Limei Zhang, Division of Chemistry and Chemical Engineering, Howard Hughes Medical Institute, California Institute of Technology, Pasadena, CA 91125, USA
Dr. Jens T. Kaiser, Division of Chemistry and Chemical Engineering, Howard Hughes Medical Institute, California Institute of Technology, Pasadena, CA 91125, USA
Dr. Gabriele Meloni, Division of Chemistry and Chemical Engineering, Howard Hughes Medical Institute, California Institute of Technology, Pasadena, CA 91125, USA
Dr. Kun–Yun Yang, Division of Chemistry and Chemical Engineering, Howard Hughes Medical Institute, California Institute of Technology, Pasadena, CA 91125, USA
Dr. Thomas Spatzal, Division of Chemistry and Chemical Engineering, Howard Hughes Medical Institute, California Institute of Technology, Pasadena, CA 91125, USA
Dr. Susana L. A. Andrade, Institut für Biochemie and BIOSS Centre for Biological Signaling Studies, Albert–Ludwigs–Universität Freiburg, 79104 Freiburg; Germany
Dr. Oliver Einsle, Institut für Biochemie and BIOSS Centre for Biological Signaling Studies, Albert–Ludwigs–Universität Freiburg, 79104 Freiburg; Germany
Dr. James B. Howard, Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, Minneapolis, MN 55455, USA
Dr. Douglas C. Rees, Division of Chemistry and Chemical Engineering, Howard Hughes Medical Institute, California Institute of Technology, Pasadena, CA 91125, USA.
References
- [1] a).Burgess BK, Lowe DJ. Chem. Rev. 1996;96:2983–3011. doi: 10.1021/cr950055x. [DOI] [PubMed] [Google Scholar]; b) Einsle O, Tezcan FA, Andrade SLA, Schmid B, Yoshida M, Howard JB, Rees DC. Science. 2002;297:1696–1700. doi: 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]; c) Howard JB, Rees DC. Proc. Natl. Acad. Sci. USA. 2006;103:17088–17093. doi: 10.1073/pnas.0603978103. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Seefeldt LC, Hoffman BM, Dean DR. Annu. Rev. Biochem. 2009;78:701–722. doi: 10.1146/annurev.biochem.78.070907.103812. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Hu Y, Ribbe MW. Acc. Chem. Res. 2010;43:475–484. doi: 10.1021/ar900254x. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Spatzal T, Aksoyoglu M, Zhang LM, Andrade SLA, Schleicher E, Weber S, Rees DC, Einsle O. Science. 2011;334:940–940. doi: 10.1126/science.1214025. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Lancaster KM, Roemelt M, Ettenhuber P, Hu Y, Ribbe MW, Neese F, Bergmann U, DeBeer S. Science. 2011;334:974–977. doi: 10.1126/science.1206445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2] a).Kim JS, Rees DC. Science. 1992;257:1677–1682. doi: 10.1126/science.1529354. [DOI] [PubMed] [Google Scholar]; b) Kim JS, Rees DC. Nature. 1992;360:553–560. doi: 10.1038/360553a0. [DOI] [PubMed] [Google Scholar]; c) Kim J, Woo D, Rees DC. Biochemistry. 1993;32:7104–7115. doi: 10.1021/bi00079a006. [DOI] [PubMed] [Google Scholar]; d) Lawson DM, Mayer SM, Gormal CA, Roe SM, Smith BE. J. Mol. Biol. 1999;292:871–891. doi: 10.1006/jmbi.1999.3107. [DOI] [PubMed] [Google Scholar]
- [3].Einsle O, Andrade SLA, Dobbek H, Meyer J, Rees DC. J. Am. Chem. Soc. 2007;129:2210–2211. doi: 10.1021/ja067562o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Allen FH. Acta Crystallogr. Sect. B. 2002;58:380–388. doi: 10.1107/s0108768102003890. [DOI] [PubMed] [Google Scholar]
- [5] a).Harding MM. Acta Crystallogr. Sect. D. 2002;58:872–874. doi: 10.1107/s0907444902003712. [DOI] [PubMed] [Google Scholar]; b) Harding MM. Acta Crystallogr. Sect. D. 2006;62:678–682. doi: 10.1107/S0907444906014594. [DOI] [PubMed] [Google Scholar]
- [6] a).Templeton DH, Templeton LK. Acta Crystallogr. Sect. A. 1980;36:237–241. [Google Scholar]; b) Smith TA, Pennerhahn JE, Berding MA, Doniach S, Hodgson KO. J. Am. Chem. Soc. 1985;107:5945–5955. [Google Scholar]
- [7].Smith SM, Rawat S, Telser J, Hoffman BM, Stemmler TL, Rosenzweig AC. Biochemistry. 2011;50:10231–10240. doi: 10.1021/bi200801z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Einsle O, Niessen H, Abt DJ, Seiffert G, Schink B, Huber R, Messerschmidt A, Kroneck PM. Acta Crystallogr. Sect. F. 2005;61:299–301. doi: 10.1107/S174430910500374X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Darnault C, Volbeda A, Kim EJ, Legrand P, Vernede X, Lindahl PA, Fontecilla-Camps JC. Nat. Struct. Biol. 2003;10:271–279. doi: 10.1038/nsb912. [DOI] [PubMed] [Google Scholar]
- [10].Yeh AP, Hu YL, Jenney FE, Adams MWW, Rees DC. Biochemistry. 2000;39:2499–2508. doi: 10.1021/bi992428k. [DOI] [PubMed] [Google Scholar]
- [11].Wernimont AK, Huffman DL, Lamb AL, O’Halloran TV, Rosenzweig AC. Nat. Struct. Biol. 2000;7:766–771. doi: 10.1038/78999. [DOI] [PubMed] [Google Scholar]
- [12].Broecker MJ, Schomburg S, Heinz DW, Jahn D, Schubert WD, Moser J. J. Biol. Chem. 2010;285:27336–27345. doi: 10.1074/jbc.M110.126698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Muraki N, Nomata J, Ebata K, Mizoguchi T, Shiba T, Tamiaki H, Kurisu G, Fujita Y. Nature. 2010;465:110–114. doi: 10.1038/nature08950. [DOI] [PubMed] [Google Scholar]
- [14].Hsin K, Sheng Y, Harding MM, Taylor P, Walkinshaw MD. J. Appl. Crystallogr. 2008;41:963–968. [Google Scholar]
- [15] a).Ravi N, Moore V, Lloyd SG, Hales BJ, Huynh BH. J. Biol. Chem. 1994;269:20920–20924. [PubMed] [Google Scholar]; b) Krahn E, Weiss BJR, Krockel M, Groppe J, Henkel G, Cramer SP, Trautwein AX, Schneider K, Müller A. J. Biol. Inorg. Chem. 2002;7:37–45. doi: 10.1007/s007750100263. [DOI] [PubMed] [Google Scholar]; c) Mclean PA, Papaefthymiou V, Orme-Johnson WH, Münck E. J. Biol. Chem. 1987;262:12900–12903. [PubMed] [Google Scholar]
- [16] a).Hendrich MP, Debrunner PG. J. Magn. Reson. 1988;78:133–141. [Google Scholar]; b) Hendrich MP, Debrunner PG. Biophys. J. 1989;56:489–506. doi: 10.1016/S0006-3495(89)82696-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.