

Abstract
The importance of thyroid hormone signaling in the acute regulation of metabolic activity has been recognized for decades. Slowly, the underlying mechanisms responsible for this activity are being elucidated. A prominent characteristic of thyroid signaling is rapid increases in oxygen consumption and ATP production. This discovery implicated a non-genomic regulation of mitochondrial metabolism by thyroid hormones. Another important clue came from the discovery that thyroid hormones stimulated fatty acid oxidation (FAO) in a variety of tissues in a receptor-dependent, but transcriptional-independent manner. Recently, key linkages between thyroid hormone signaling and specific mitochondrial-targeted pathways have been discovered. This review focuses on the molecular mechanisms by which mitochondrial FAO can be increased through thyroid hormone signaling. The roles of both the full-length and shortened mitochondrial isoforms of thyroid hormone receptor will be discussed. Additionally, the impact of thyroid hormone signaling on dyslipidemias such as obesity, type II diabetes, and fatty liver disease will be considered.
Keywords: thyroid hormone; thyroid hormone receptor; TRα; TRβ; 3,3,5 triiodothyronine; T3,3,5 diiodothyroinine; T2; fatty acid; fatty acid oxidation; energy homeostasis; mitochondria; type II diabetes; insulin resistance; obesity; metabolic syndrome; non-alcoholic fatty liver disease; steatosis; fatty acid trafficking; dyslipidemia; non-genomic; c-erbA; p28; p30; p43; adenosine monophosphate activated protein-kinase; AMPK; acetyl-coA carboxylase; ACC; carnitine palmitoyl transferase; CPT; mitochondrial trifunctional protein; MTP; long-chain acyl CoA dehydrogenase; LCHAD; thyrotoxicity; thyroid hormone analogs; GC-1; CO23; MB07344
INTRODUCTION
Energy homeostasis is strongly regulated by thyroid hormones and activated thyroid hormone receptors. Although they are traditionally better known for transcriptional regulation, this review focuses on non-genomic cellular responses of thyroid hormone receptor signaling. In particular, we have focused on the long established relationship between thyroid hormones and the regulation of mitochondrial metabolism. Recent work has linked this signaling pathway to fatty acid oxidation (FAO), which is capable of metabolizing significant amounts of lipids independently of gene expression. We begin with a brief review of thyroid hormone receptor subtypes and their cellular localization. This is followed by an assessment of the various non-genomic roles of thyroid hormone signaling in fatty acid trafficking and oxidation. We end with a discussion of the potential impact of this metabolic signaling pathway on pathophysiology.
Thyroid hormone receptor subtypes, shortened isoforms and localization
Two genes encode all subtypes of the thyroid hormone receptor. The first gene, thyroid hormone receptor α (TRα), was mapped to the c-erbA gene (also known as Thra) on chromosome 17 based on its homology to steroid hormone receptors [1]. Transcription of c-erbA results in several mRNAs - the full-length TRα1, and two variants that encode proteins which do not bind to thyroid hormone [2, 3]; reviewed in [4]. The second thyroid hormone receptor gene, Thrb, is located on chromosome 3 and has high sequence homology to c-erbA. The protein it encodes, TRβ, exhibits its greatest differences in the amino terminal domains compared to TRα. Both proteins have isoform-specific functions, but can also act redundantly, for example, when one isoform is genetically deleted (reviewed in [4, 5]). The remainder of this review will focus primarily on the TRα, which has been better characterized for its ability to stimulate FAO.
Shortened TRα1 isoforms: Expression and localization
The full-length mRNA for TRα1 contains several alternative start sites that result in translation of at least 3 isoforms of decreasing length [1, 6, 7]. The shortened isoforms were first described by Wienberger and colleagues after in vitro translation of the c-erbA coding region [1]. Later, Bigler and Eisenman described and further characterized these multiple translation products in chicken erythroid cells, and showed that mutation of the alternative start sites eliminated translation of the shortened isoforms [6, 7]. Today these isoforms are named according to their sizes in kilodaltons: p43, p30, and p28 (Figure 1). Of these isoforms, the p43 variant has been most characterized, whereas relatively little has been described about the function of the p30 variant.
Figure 1. Thyroid hormone receptor alpha shortened isoforms.
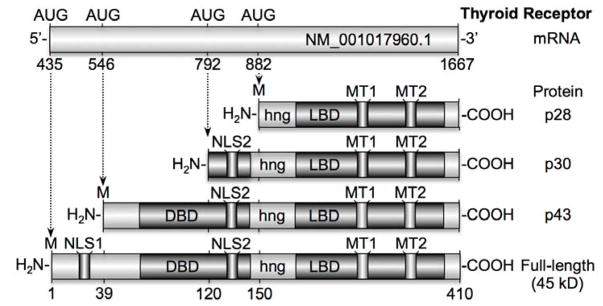
TRα mRNA consists of 1232 base-pairs encoded by c-erbA. Three alternative start sites on the mRNA are translated into 3 shortened isoforms (43, 30, and 28 kD) and 1 full-length isoform (45 kD). The full-length isoform contains 2 separate nuclear localization signals (NLS) as well as 2 mitochondrial targeting signals (MT), a DNA-binding domain (DBD), a hinge region (hng) and a ligand binding domain (LBD). The full-length TRα and p30 localize predominantly in the nucleus, whereas p28 and p43 localize to mitochondria.
Full-length TRα1 mRNA encodes a 45–50 kDa protein with structural similarity to other nuclear hormone receptors - namely, it encodes an N-terminal domain, a DNA binding domain, a regulatory hinge region, and a ligand binding domain. Full-length TRα1 is normally shuttled between the nucleus and cytoplasm by virtue of the two nuclear localization signals (NLS; [8–10]). The first NLS is found in the N-terminal domain of the full-length protein only [11]. The second NLS is near the hinge region and is also found in p43 and p30, but not p28 [10]. Fusion of each NLS to non-nuclear proteins is capable of targeting them to the nucleus [10, 11], and so either would be expected to be sufficient for nuclear targeting. However, only p30 appears to localize to the nucleus [6], while p43 localizes to mitochondria [12–14]. This difference in localization may be due in part to the increased length of the N-terminus in p43 compared to p30. This longer N-terminus was proposed to obscure the second NLS and render it inaccessible to the requisite targeting proteins [15]. The second NLS is not translated in p28, making its targeting similar to p43. Lacking a strong NLS, p43 and p28 are instead trafficked to mitochondria by virtue of two mitochondrial targeting sequences located in the ligand binding domain [15]. Trafficking of p43 has been studied in greater detail, and takes advantage of two separate pathways into mitochondria. One route utilizes classical import mechanisms and is directed by mitochondrial targeting sequences within helices 10 and 11 of the ligand binding domain. Fusion of this portion of p43 to enhanced green fluorescence protein (eGFP) produces an ATP-dependent and membrane potential-driven import of the fusion protein into mitochondria [15]. This classic pathway is characteristic of the proteins imported into mitochondria via the translocase of the outer membrane (TOM) and the translocase of the inner membrane (TIM) complexes. TOM and TIM import linearized proteins into the mitochondria with the aid of chaperone proteins in the mitochondrial spaces [16]. The second pathway of p43 import into mitochondria does not follow well-characterized routes, in that it does not require ATP or a high membrane potential in order to enter the organelle. This alternative import pathway is directed via targeting sequences found in helices 5 and 6 of the ligand binding domain [15]. The exact mechanism by which ATP and potential-independent mitochondrial targeting occurs remains unclear. Of additional interest is the fact that while p43 localizes to the matrix [12–14], p28 associates with the inner membrane [14]. The mechanism of this differential targeting is not currently understood.
Full-length TRα and TRβ are expressed in a majority of tissues [4]. However, expression of full-length TRα is predominant in cardiac tissue while expression in the liver is dominated by full-length TRβ. The shortened isoform p43 is expressed in white adipose, brown adipose, liver [14], skeletal muscle [17, 18], heart, brain, kidneys, spleen [18], and fibroblasts [19]. Expression of p28 and p30 in various tissues has not yet been fully investigated, and it remains to be determined whether mechanisms exist to control expression of the shortened TRα isoforms in different tissues. In addition, physiological data obtained by our group shows that thyroid hormones acutely regulate mitochondrial metabolism, even in the absence of the full-length TRα gene [19]. These results will be discussed in greater detail below, but they suggest that a functional redundancy exists in the TRα and TRβ isoforms.
Given the differential localization and expression of shortened TRα isoforms, it is logical that these receptors exert a variety of effects within the cell and in different tissues. In the following section, we emphasize the non-genomic impact that these receptors and their hormone have on fatty acid metabolism via two important pathways. First, thyroid hormone and full length TRα increase the subcellular trafficking of fatty acids to the primary site of oxidation, the mitochondria. Second, thyroid hormone and p43 cause greater activity of enzymes that catalyze the conversion of acyl-CoA into acetyl-CoA.
Thyroid hormone receptor signaling increases trafficking of fatty acids into the mitochondria
Triacylglycerol (TAG) is the main storage form of fatty acids in the body [20]. The largest storage depot of TAG is in adipose tissue; however TAG is also stored in the lipid droplets of individual cells. TAG can be hydrolyzed by lipases that release free-fatty acids from glycerol. These free-fatty acids (FFAs) are then conveyed to the site of metabolism [21]. Adipose tissue releases FFAs to albumin in the circulatory system for dispersal to the rest of the body. Long-chain FFAs are transferred into individual cells via fatty acid transport proteins [20, 21].
Once inside the cell, FFAs are converted into acyl-CoAs [21] and used in the cell for a variety of purposes ranging from the synthesis of new membrane to fuel for mitochondrial metabolism via fatty acid oxidation (FAO). For the latter process, acyl-CoA must be transported to the site of oxidation on the inner mitochondrial membrane. Because the outer mitochondrial membrane is impermeable to acyl-CoA, an acyl-carnitine shuttle is used to traffic fatty acids into mitochondria. Acyl-CoAs are converted into acyl-carnitine via the action of carnitine palmitoyl transferase 1 (CPT1), an integral, outer-mitochondrial membrane protein. Carnitine acylcarnitine translocase (CACT) is then able to exchange acyl-carnitines outside of the mitochondria for free carnitine inside mitochondria, thereby trafficking acyl-carnitine inside. Once inside mitochondria, CPT2 exchanges CoA for carnitine on the fatty acid. The fatty acyl-CoA is then available for oxidation by mitochondrial enzymes [21, 22] (Figure 2).
Figure 2. Thyroid hormone and non-genomic upregulation of fatty acid trafficking.
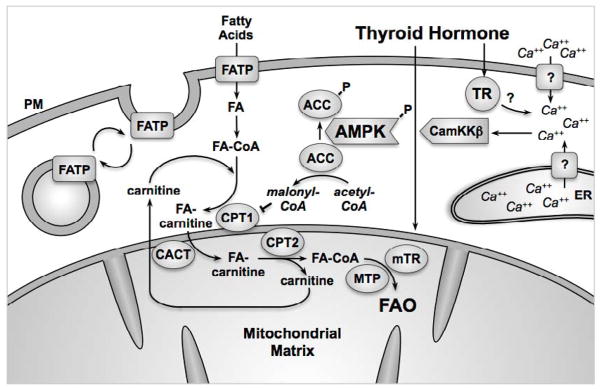
In cytosol, TR binds to TH. Activated TR immediately elicits an increase in cytosolic Ca++ from intracellular and extracellular pools (see text). Increased Ca++ causes phosphorylation and activation of CamKKβ, which then phosphorylates and activates AMPK. Phospho-AMPK in turn increases localization of FATP to the plasma membrane, thereby increasing fatty acid import and modification into fatty acyl-CoA. Similarly, phospho-AMPK phosphorylates and so inactivates ACC. Inactive phospho-ACC does not synthesize malonyl-CoA. Malonyl-CoA normally inhibits CPT1, thus reducing fatty acid trafficking into mitochondria. When the inhibition is relieved by the actions of phospho-AMPK, CPT1 exchanges the CoA for carnitine on the fatty acid. Fatty-acyl carnitine is translocated into the mitochondria by CACT while carnitine is translocated out. CPT2 then exchanges carnitine for CoA on the fatty acid inside the motochondria. The fatty acyl-CoA then enters FAO to be catabolized into 2-carbon acetyl-CoA molecules. Long-chain fatty acyl-CoA FAO involves MTP, which is also regulated by mitochondrial TRs.
Whether the acyl-CoAs are used for energy or are stored depends on the activity of AMP-activated protein kinase (AMPK), which acts as a master sensor of the energy status of the cell [23]. When cells need energy, the amounts of AMP and ADP are high relative to the amount of ATP. This, in turn, causes AMPK to activate via phosphorylation, driving the regulation of multiple processes that ultimately lead to increased energy production [23].
AMPK plays a central role in the regulation of FAO; it is perhaps not surprising then that AMPK is itself a target of thyroid hormone action. Thyroid hormone treatment increases phosphorylation of AMPK in muscle both in vitro [24] and in vivo [25–28]. Of particular interest, AMPK phosphorylation occurs rapidly after thyroid hormone treatment, within 30 minutes in vitro [24], and within 1–2 hours in vivo [25, 29]. Because these changes were observed so quickly after treatment, it appeared unlikely that thyroid hormone was acting in the classical manner, i.e. via transcriptional activation. Indeed, Yamauchi and co-workers demonstrated that knock-down of Ca++/calmodulin-dependent protein kinase-β (CaMKKβ) decreased thyroid hormone-stimulated FAO. They suggested that thyroid hormone could activate AMPK in muscle by causing thyroid hormone-dependent Ca++ movement and activation of CaMKKβ. Activated CaMKKβ, in turn, mediated phosphorylation of AMPK [24]. The mechanism of thyroid hormone-dependent mobilization of Ca++ is less clear. In Xenopus oocytes, we found that thyroid hormone binding to a shortened, transcriptionally inactive TR isoform increased the periodicity of IP3-induced Ca++ waves in oocytes, although thyroid hormone by itself did not stimulate Ca++ release [30]. Removal of Ca++ from media does not eliminate the response of cells to TH; this indicates that at least some of the increased cytosolic Ca++ is due to movement from intracellular stores to the cytosol [31, 32]. Nevertheless, use of membrane impermeant TH was able to elicit Ca++ increases [31], indicating the plasma membrane does play some role in eliciting Ca++ increases. Some intracellular release of Ca++ might occur through activation of IP3 [31, 33–35]. However, Del Viscovo and colleagues showed that depletion of IP3 Ca++ stores failed to decrease cytoplasmic Ca++ after TH, even though inhibition of downstream IP3 signaling molecules did reduce cytoplasmic Ca++ [32].
Regardless of the precise mechanism, AMPK is clearly activated via phosphorylation after TH treatment. An important downstream target of phosphorylated AMPK (phospho-AMPK) is fatty acid transport protein (FATP). Phospho-AMPK causes FATP to translocate from endosomal vesicles to the plasma membrane in hyperthyroid rat hearts, probably through activation of Rab proteins, which are essential for vesicular transport [28, 36]. Similarly, phospho-AMPK increases membrane localization of the fatty acid translocase CD36 via activation of Rab8a in cardiomyocytes after insulin treatment [37]. Altogether, phospho-AMPK-induced trafficking of either FATP or CD36 to the plasma membrane serves to increase fatty acid import from the periphery into the cell.
Another target of phospho-AMPK is acetyl-CoA carboxylase (ACC), which catalyzes the production of malonyl-CoA from acetyl-CoA and is one of the initial steps of fatty acid synthesis [22, 38]. When ACC activity is high, a greater concentration of malonyl-CoA is produced. Malonyl-CoA binds and inhibits CPT1, thereby reducing the amount of fatty acyl-CoA entering mitochondria, and consequently, FAO. Conversely, when ACC is phosphorylated by phospho-AMPK, its activity is inhibited. This lack of malonyl-CoA production, in turn, restores CPT1 function and allows FAO in the mitochondria to proceed [38]. Concomitant to AMPK phosphorylation (and thus ACC phosphorylation), increased CPT1 activity is observed in muscles treated with thyroid hormone [26, 27], consistent with a mechanism of action increasing fatty acid trafficking into mitochondria (Figure 2).
While AMPK phosphorylation clearly increases with thyroid hormone treatment in HeLa cells [24], heart [28], and skeletal muscle [25–27], the opposite result occurs within the hypothalamus, which is a crucial regulator of whole-body metabolism and feeding [39]. Lopez et al. observed that hyperthyroid rats have significantly lower levels of phospho-AMPK and phospho-ACC within the hypothalamus, whereas hypothyroidism increased phosphorylation of both proteins. Similarly, intracerebroventricular treatment with thyroid hormone lowered AMPK phosphorylation within an hour. The decreased phospho-AMPK, in turn, increased malonyl-CoA and therefore reduced CPT1 activity. Remarkably, the net effect of thyroid hormone in the hypothalamus was to increase burning of fat for heat in brown adipose tissue. Thus, although thyroid hormone decreased phospho-AMPK, energy expenditure was increased in the whole body [39]. These results underscore the need for caution when extending mechanisms of action for thyroid hormone across different cell types and tissues.
In addition to cytoplasmic AMPK, it appears that targets within mitochondria can drive thyroid hormone-induced increases in FAO. Lombardi et al. demonstrated that thyroid hormone is capable of increasing fatty acid import, β-oxidation and oxygen consumption in isolated mitochondria [40]. These authors found that thyroid hormone (T2)-induced increases in flavine adenine dinucleotide (FADH2)-linked respiratory pathways, but not nicotinamide adenine dinucleotide (NADH)-linked pathways [27, 40]. They suggested that CPT1 itself could be a target of T2, because FAO was highest when palmitoyl-CoA was used as a substrate compared to palmitoyl-carnitine. This observation was confirmed by direct measurements of CPT activity. They also reported that activity of mitochondrial thioesterase 1 (MTE-1), an enzyme that cleaves acyl-CoA to fatty acid and CoA, was increased by thyroid hormone treatment of isolated mitochondria. Complimentary to these observations, our laboratory found that thyroid hormone could stimulate FAO, oxygen consumption, and increase mitochondrial membrane potentials in isolated mitochondria [19, 30]. In addition, we showed that thyroid hormone could stimulate FAO inCV-1 cells that lacked endogeneous TR receptors, but only when these cells were expressing an exogenous, mitochondrial-targeted TRα isoform, p43 [19]. We also showed that a mutant full-length TRα without a start site for p43 could not3 be stimulated by thyroid hormone to increase FAO [19]. Together, these data suggest that thyroid hormones can increase FAO independently of the phospho-AMPK mediated upregulation of fatty acid trafficking.
The impact of thyroid hormones on enzymatic activity in FAO
FAO within mitochondria occurs via a series of reactions that progressively shorten acyl-CoA by 2 carbons during each reaction cycle, producing acetyl-CoA. Four separate enzymatic activities are responsible for the catabolism of fatty acids, which occurs via oxidation of the β carbon on the fatty acid and consequently, is referred to as β oxidation. First, the acyl-CoA undergoes dehydrogenation to form a double bond on the β carbon, yielding a trans-2 enoyl-CoA and reducing equivalents in the form of FADH2. Second, the new carbon-carbon double bond is hydrated by a hydratase to form hydroxyacyl-CoA. This new hydroxy-group is then oxidated by dehydrogenase, creating a β-ketoacyl-CoA and more reducing equivalents in the form of NADH. Finally, the β-ketone is cleaved from the rest of the molecule by β-ketothiolase, yielding a shortened acyl-CoA and acetyl-CoA [21]. Acetyl-CoA can then be used for the tricarboxylic acid (TCA) cycle, ketogenesis, or cholesterol/steroid synthesis (Figure 3).
Figure 3. Thyroid hormone and mitochondrial TR increase the amount of assembled MTP, thereby increasing FAO activity.

Long-chain fatty acyl-CoA is reduced by LCHAD in the first reaction of FAO. MTP catalyzes the remaining 3 reactions. The α-subunit has enoyl-CoA hydratase and hydroxyacyl-CoA dehydrogenase activity, whereas the β-subunit has β-ketothiolase activity. At the end of the series of 4 reactions, acetyl-CoA is created along with a shortened fatty-acyl CoA that can reenter the FAO cycle. Acetyl-CoA can be used as a substrate for the TCA cycle, for cholesterol biosynthesis, and for ketone body synthesis. MTP activity is dependent on full assembly of the protein, and mitochondrial TR can influence the amount of assembled MTP. It remains unclear whether this influence is exerted via decreased MTP degradation or increased assembly.
The specific enzymes that perform FAO are highly dependent on the number of carbons in the acyl group. Long chain fatty acids (>12 carbons) are recognized by long-chain acyl-CoA dehydrogenase (LCHAD) and by very long chain acyl-CoA dehydrogenase (VLCHAD). Medium chain fatty acids (6–12 carbons) are also recognized by LCHAD, whereas short-chain fatty acids (<6 carbons) are recognized by short-chain acyl-CoA dehydrogenase (SCHAD) [21].
To identify potential mitochondrial mediators of the thyroid hormone-stimulated FAO, mitochondrial lysates were screened for potential binding partners to a shortened TR isoform, and our laboratory discovered that mitochondrial trifunctional protein (MTP) was an associated protein [19]. MTP catalyzes the last three reactions of FAO (hydration, dehydrogenation, and cleavage) on long-chain fatty acids. MTP has two subunits, α and β, each of which form tetramers within the assembled, functional protein [41]. Indeed, the assembly of α and β subunits is crucial for the activity and stability of the protein [42]. We found that thyroid hormone treatment increased the levels of assembled MTP within 15 minutes of treatment, suggesting that some of the increased FAO observed after thyroid hormone treatment might be due to increased MTP activity rather than increased fatty acid trafficking into mitochondria. In support of this mechanism of action, blocking fatty acid entry into mitochondria with the CPT1 inhibitor etomoxir did not impair thyroid hormone-induced MTP subunit assembly. In addition, when AMPK phosphorylation was maximized using the AMPK activator 5-amino-1-β-D-ribofuranosyl-imidazole-4-carboxamide (AICAr), thyroid hormone treatment still increased FAO, further indicating that the flux of fatty acids that normally increases in response to AMPK phosphorylation was not contributing to this mitochondrial mechanism of action. Not surprisingly, elimination of fatty acid trafficking into mitochondria using either the AMPK inhibitor compound C or the CPT1 inhibitor etomoxir eliminated thyroid hormone-mediated increases in FAO, simply due to the absence of substrate [19]. Future experiments will be required to determine a precise mechanism by which p43 interacts with and regulates MTP activity. A model for this mechanism is presented in Figure 3.
Pathobiology of fatty acid metabolism and the effect of thyroid hormones on dyslipidemia
Obesity and metabolic syndrome
Metabolic syndrome refers to the panoply of pathological symptoms that arise concurrently with the excess storage of fatty acids in organs rather than in adipose tissue [43–45]; it is most often associated with obesity. Symptoms include steatohepatitis (fatty liver), cardiovascular disease, insulin resistance and type II diabetes [45]. In 2009–2010, nearly 36% of adults and 17% of children in the United States were considered obese with a body mass index greater that 30 [46]. Dietary and lifestyle intervention will be the key to reducing the epidemic of obesity. Unfortunately, lifestyle itself is very difficult to change, and past studies indicate that they succeed in less than 35% of those that try to lose weight [47]. Consequently, it is more likely that significant reduction in the population’s rate of obesity will require a combinatorial approach of both lifestyle changes and pharmacological intervention. In this light, it has long been hoped that thyroid hormone receptor signaling could be used as pharmacological targets to reduce obesity and its associated syndromes. As presented in the following sections, encouraging insights and significant progress are being made in this direction.
Insulin resistance and type II diabetes
Approximately 25% of obese individuals are insulin resistant [48]. Patients compensate for this resistance by secreting more insulin, but eventually become unable to secrete sufficient insulin to control blood sugar, resulting in type II diabetes [45]. In addition to regulating glucose uptake, insulin normally reduces gluconeogenesis and increases lipogenesis within the liver. In patients with type II diabetes, insulin does not reduce gluconeogenesis even though lipogenesis is increased (for reasons not well understood) [49]. The net result is the release of significant amounts of glucose, free fatty acids, and other lipids into circulation by the liver. This is compounded by the absence of insulin inhibition of lipolysis in adipose tissue, which releases even greater amounts of free fatty acids into the circulatory system of these patients. Altogether, insulin resistance in type II diabetes patients contributes to dyslipidemia initiated by obesity, and thereby increases cardiovascular risk [45, 48].
Diabetes has been associated with hypothyroidism in 11–13% of diabetes patients [50, 51], while hyperthyroidism has been associated with insulin resistance [52, 53], with hyperthyroid patients having 2.5 times greater chance of having diabetes [54]. Consistent with these data, mice lacking TRα do not become insulin resistant in response to a high fat diet, unlike wild-type mice [55]. On the other hand, thyroid hormones are known to increase insulin sensitivity in skeletal muscle by upregulating expression of uncoupling protein 3 (UCP3). Loss of UCP3 expression is associated with insulin resistance and decreased AMPK signaling [56–59]. The function of UCP3 is not well defined, but appears to result in less energy from the same amount of fatty acid fuel, presumably due to uncoupling of catabolism from mitochondrial energy production [60, 61].
Non-alcoholic steatohepatitis (NASH) and non-alcoholic fatty liver disease (NAFLD)
The liver plays a central role in regulating the lipid homeostasis of the entire body. In NASH and NAFLD, an abnormal amount of lipids accumulate in the liver, such that at least 5% of its weight is due to excess fat [62]. In the “two-hit” hypothesis, this excess accumulation of lipid (the first hit) causes an inflammatory response, which serves to further damage the liver tissue (second hit), thereby resulting in steatohepatitis and liver disease [63]. Estimates for the prevalence of NAFLD vary, but on average effects 24–45% of obese adults and 38–53% of obese children within the United States [62]. Many have recognized that liver-specific activation of thyroid receptors has the potential to effectively treat many liver diseases. For example, Lanni and colleagues demonstrated that 3,5 diiodo-L-thyronine (T2) could specifically increase hepatic FAO in the liver mitochondria without causing thyrotoxicity [64]. Similarly, steatosis and plasma lipids were markedly reduced in NAFLD mouse and rat models when liver TRβ was selectively activated with MB07344 [65]. Specific targeting of thyroid hormone signaling to liver or liver mitochondria is important for successful treatment, because thyroid hormones can also stimulate adipose tissue to release fatty acid, which then circulates to the liver and exacerbates steatosis [66–68].
Treating disorders of FAO using TH analogs
Prolonged hyperthyroidism has significant detrimental effects, most severely within the heart (reviewed in [69], [70]). Cardiac symptoms from excess thyroid hormone include tachycardia [71, 72], atrial fibrillation [73–75], and increased left ventricular thickness [72].
It is possible that certain thyroid hormone analogs could be used to acutely stimulate specific FAO pathways without the side effects that long-term hyperthyroidism elicit. For instance, TR activity of different isoforms within specific tissues could be targeted. Baxter and colleagues discovered that TRα and TRβ have a single amino acid difference in the ligand binding pockets (serine 277 in TRα versus asparagine 331 in TRβ) that could be exploited to create selective agonists for either TRα or TRβ. They designed a TRβ selective agonist GC-1 and showed that the affinity for TRβ was 9 times greater than for TRα [76]. TRα is the major functional isoform that affects heart function [77], and impressively, GC-1 was able to decrease plasma lipids without affecting the heart [76]. Similarly, others showed that GC-1 administration to rat models of NAFLD was able to reverse steatosis [78]. As noted above, another TRβ-selective agonist, MB07344, also reversed steatosis in rats [65], indicating that selective activation of TRβ in the liver is a viable strategy to treat disease.
The differences in ligand binding domain were also exploited to create a potential TRα specific agonist, CO23, which activated TRα in cultured cells and promoted TRα-selective growth patterns in Xenopus tadpoles [79]. Unfortunately, testing of CO23 in vivo showed that CO23 also activates TRβ in mice [80].
New therapeutic targets
Ideally, new drugs that target thyroid hormone stimulated FAO without affecting transcriptional activity need to be identified. Most of the cardiac pathologies attributed to thyroid hormones can be explained by changes in cardiac gene expression [81, 82] [83–85]. A promising new target revealed by our recent work suggests that this may be possible. Specifically, shortened, mitochondrial-targeted TR isoforms appear to increase FAO without adversely affecting gene expression, because DNA binding domains are not present (Figure 1). To date, no selective agonists to these shortened isoforms have been described, although di-iodothyronine (T2), along with its analogs, are potential candidates. T2 rapidly increases respiration in isolated mitochondria, enhances CPT1 activity (and therefore fatty acid trafficking into mitochondria), and finally, stimulates FAO [27]. It is not yet clear whether these observed changes are dependent on a TR receptor. The rapidity with which the T2-induced changes occur suggested that gene expression changes were not involved. However, T2 itself appeared to indirectly affect gene expression in the liver. Grasselli and colleagues found that expression of peroxisome proliferator-activated receptors (PPAR), which are essential transcriptional regulators of lipid metabolism, were elevated after 30 days of T2 treatment in rats [86]. Modulation of PPAR expression did not seem dependent on TR, because T2 treatment of hepatoma cells lacking TRs still caused lipid-lowering effects consistent with activation of PPARs [87]. Nevertheless, T2 treatment successfully prevented weight gain, steatosis, and insulin resistance associated with high fat diet without affecting the heart [88]. Together, these data are very encouraging and suggest that appropriately designed small-molecule screens against mitochondrial-targeted TRs may be a fruitful avenue to identify new therapeutic compounds that acutely regulate FAO with minimal long-term detrimental effects.
References
- 1.Weinberger C, Thompson CC, Ong ES, Lebo R, Gruol DJ, Evans RM. Nature. 1986;324:641. doi: 10.1038/324641a0. [DOI] [PubMed] [Google Scholar]
- 2.Mitsuhashi T, Tennyson GE, Nikodem VM. Proc Natl Acad Sci USA. 1988;85:5804. doi: 10.1073/pnas.85.16.5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mitsuhashi T, Nikodem VM. J Biol Chem. 1989;264:8900. [PubMed] [Google Scholar]
- 4.Cheng SY, Leonard JL, Davis PJ. Endocr Rev. 2010;31:139. doi: 10.1210/er.2009-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng SY. Rev Endocr Metab Disord. 2000;1:9. doi: 10.1023/a:1010052101214. [DOI] [PubMed] [Google Scholar]
- 6.Bigler J, Eisenman RN. Mol Cell Biol. 1988;8:4155. doi: 10.1128/mcb.8.10.4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bigler J, Hokanson W, Eisenman RN. Mol Cell Biol. 1992;12:2406. doi: 10.1128/mcb.12.5.2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bunn CF, Neidig JA, Freidinger KE, Stankiewicz TA, Weaver BS, McGrew J, Allison LA. Molecular Endocrinology. 2001;15:512. doi: 10.1210/mend.15.4.0619. [DOI] [PubMed] [Google Scholar]
- 9.Maruvada P, Baumann CT, Hager GL, Yen PM. Journal of Biological Chemistry. 2003;278:12425. doi: 10.1074/jbc.M202752200. [DOI] [PubMed] [Google Scholar]
- 10.Dang CV, Lee WM. Journal of Biological Chemistry. 1989;264:18019. [PubMed] [Google Scholar]
- 11.Andersson ML, Vennstrom B. FEBS Lett. 1997;416:291. doi: 10.1016/s0014-5793(97)01223-4. [DOI] [PubMed] [Google Scholar]
- 12.Sterling K, Lazarus J, Milch P, Sakurada T, Brenner M. Science. 1978;201:1126. doi: 10.1126/science.210507. [DOI] [PubMed] [Google Scholar]
- 13.Sterling K, Campbell GA, Brenner MA. Acta Endocrinol (Copenh) 1984;105:391. doi: 10.1530/acta.0.1050391. [DOI] [PubMed] [Google Scholar]
- 14.Wrutniak C, Cassar-Malek I, Marchal S, Rascle A, Heusser S, Keller JM, Fléchon J, Daua M, Samarut J, Ghysdael J, Cabello G. Journal of Biological Chemistry. 1995;270:16347. doi: 10.1074/jbc.270.27.16347. [DOI] [PubMed] [Google Scholar]
- 15.Carazo A, Levin J, Casas F, Seyer P, Grandemange S, Busson M, Pessemesse L, Wrutniak-Cabello C, Cabello G. J Cell Physiol. 2012;227:3768. doi: 10.1002/jcp.24085. [DOI] [PubMed] [Google Scholar]
- 16.Schmidt O, Pfanner N, Meisinger C. Nat Rev Mol Cell Biol. 2010;11:655. doi: 10.1038/nrm2959. [DOI] [PubMed] [Google Scholar]
- 17.Casas F, Pessemesse L, Grandemange S, Seyer P, Baris O, Gueguen N, Ramonatxo C, Perrin F, Fouret G, Lepourry L, Cabello G, Wrutniak-Cabello C. PLoS One. 2009;4:e5631. doi: 10.1371/journal.pone.0005631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morrish F, Buroker NE, Ge M, Ning XH, Lopez-Guisa J, Hockenbery D, Portman MA. Mitochondrion. 2006;6:143. doi: 10.1016/j.mito.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 19.Chocron ES, Sayre NL, Holstein D, Saelim N, Ibdah JA, Dong LQ, Zhu X, Cheng SY, Lechleiter JD. Molecular Endocrinology. 2012;26:1117. doi: 10.1210/me.2011-1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stremmel W, Pohl L, Ring A, Herrmann T. Lipids. 2001;36:981. doi: 10.1007/s11745-001-0809-2. [DOI] [PubMed] [Google Scholar]
- 21.Houten SM, Wanders RJ. J Inherit Metab Dis. 2010;33:469. doi: 10.1007/s10545-010-9061-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wakil SJ, Abu-Elheiga LA. J Lipid Res. 2009;50(Suppl):S138. doi: 10.1194/jlr.R800079-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hardie DG, Ross FA, Hawley SA. Nat Rev Mol Cell Biol. 2012;13:251. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamauchi M, Kambe F, Cao X, Lu X, Kozaki Y, Oiso Y, Seo H. Mol Endocrinol. 2008;22:893. doi: 10.1210/me.2007-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Irrcher I, Walkinshaw DR, Sheehan TE, Hood DA. J Appl Physiol. 2008;104:178. doi: 10.1152/japplphysiol.00643.2007. [DOI] [PubMed] [Google Scholar]
- 26.de Lange P, Senese R, Cioffi F, Moreno M, Lombardi A, Silvestri E, Goglia F, Lanni A. Endocrinology. 2008;149:6462. doi: 10.1210/en.2008-0202. [DOI] [PubMed] [Google Scholar]
- 27.Lombardi A, de Lange P, Silvestri E, Busiello RA, Lanni A, Goglia F, Moreno M. Am Journal of Physiol Endocrinol Metab. 2009;296:E497. doi: 10.1152/ajpendo.90642.2008. [DOI] [PubMed] [Google Scholar]
- 28.Heather LC, Cole MA, Atherton HJ, Coumans WA, Evans RD, Tyler DJ, Glatz JF, Luiken JJ, Clarke K. Endocrinology. 2010;151:422. doi: 10.1210/en.2009-0593. [DOI] [PubMed] [Google Scholar]
- 29.Lombardi A, de Lange P, Silvestri E, Busiello RA, Lanni A, Goglia F, Moreno M. Am J Physiol Endocrinol Metab. 2009;296:E497. doi: 10.1152/ajpendo.90642.2008. [DOI] [PubMed] [Google Scholar]
- 30.Saelim N, John LM, Wu J, Park JS, Bai Y, Camacho P, Lechleiter JD. J Cell Biol. 2004;167:915. doi: 10.1083/jcb.200409011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.D’Arezzo S, Incerpi S, Davis FB, Acconcia F, Marino M, Farias RN, Davis PJ. Endocrinology. 2004;145:5694. doi: 10.1210/en.2004-0890. [DOI] [PubMed] [Google Scholar]
- 32.Del Viscovo A, Secondo A, Esposito A, Goglia F, Moreno M, Canzoniero LM. American journal of physiology Endocrinology and metabolism. 2012;302:E1419. doi: 10.1152/ajpendo.00389.2011. [DOI] [PubMed] [Google Scholar]
- 33.Furuya F, Hanover JA, Cheng SY. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:1780. doi: 10.1073/pnas.0510849103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Storey NM, Gentile S, Ullah H, Russo A, Muessel M, Erxleben C, Armstrong DL. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:5197. doi: 10.1073/pnas.0600089103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hiroi Y, Kim HH, Ying H, Furuya F, Huang Z, Simoncini T, Noma K, Ueki K, Nguyen NH, Scanlan TS, Moskowitz MA, Cheng SY, Liao JK. Proc Natl Acad Sci USA. 2006;103:14104. doi: 10.1073/pnas.0601600103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luiken JJ, Coort SL, Koonen DP, van der Horst DJ, Bonen A, Zorzano A, Glatz JF. Pflugers Arch. 2004;448:1. doi: 10.1007/s00424-003-1199-4. [DOI] [PubMed] [Google Scholar]
- 37.Samovski D, Su X, Xu Y, Abumrad NA, Stahl PD. Journal of Lipid Research. 2012;53:709. doi: 10.1194/jlr.M023424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saggerson D. Annual Review of Nutrition. 2008;28:253. doi: 10.1146/annurev.nutr.28.061807.155434. [DOI] [PubMed] [Google Scholar]
- 39.Lopez M, Varela L, Vazquez MJ, Rodriguez-Cuenca S, Gonzalez CR, Velagapudi VR, Morgan DA, Schoenmakers E, Agassandian K, Lage R, Martinez de Morentin PB, Tovar S, Nogueiras R, Carling D, Lelliott C, Gallego R, Oresic M, Chatterjee K, Saha AK, Rahmouni K, Dieguez C, Vidal-Puig A. Nat Med. 2010;16:1001. doi: 10.1038/nm.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lombardi A, de Matteis R, Moreno M, Napolitano L, Busiello RA, Senese R, de Lange P, Lanni A, Goglia F. Am Journal of Physiol Endocrinol Metab. 2012;303:E1222. doi: 10.1152/ajpendo.00037.2012. [DOI] [PubMed] [Google Scholar]
- 41.Rector RS, Payne RM, Ibdah JA. Adv Drug Deliv Rev. 2008;60:1488. doi: 10.1016/j.addr.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ushikubo S, Aoyama T, Kamijo T, Wanders RJ, Rinaldo P, Vockley J, Hashimoto T. Am J Hum Genet. 1996;58:979. [PMC free article] [PubMed] [Google Scholar]
- 43.Fabbrini E, Magkos F, Mohammed BS, Pietka T, Abumrad NA, Patterson BW, Okunade A, Klein S. Proceedings of the National Academy of Sciences. 2009;106:15430. doi: 10.1073/pnas.0904944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cali AMG, De Oliveira AM, Kim H, Chen S, Reyes-Mugica M, Escalera S, Dziura J, Taksali SE, Kursawe R, Shaw M, Savoye M, Pierpont B, Constable RT, Caprio S. Hepatology. 2009;49:1896. doi: 10.1002/hep.22858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bremer AA, Mietus-Snyder M, Lustig RH. Pediatrics. 2012;129:557. doi: 10.1542/peds.2011-2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ogden CL, Carroll ME, Kit BK, Flegal KM. NCHS Data Brief. 2012;82:1. [PubMed] [Google Scholar]
- 47.Wadden TA, Volger S, Sarwer DB, Vetter ML, Tsai AG, Berkowitz RI, Kumanyika S, Schmitz KH, Diewald LK, Barg R, Chittams J, Moore RH. New England Journal of Medicine. 2011;365:1969. doi: 10.1056/NEJMoa1109220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abbasi F, Brown BW, Jr, Lamendola C, McLaughlin T, Reaven GM. J Am Coll Cardiol. 2002;40:937. doi: 10.1016/s0735-1097(02)02051-x. [DOI] [PubMed] [Google Scholar]
- 49.Brown MS, Goldstein JL. Cell Metab. 2008;7:95. doi: 10.1016/j.cmet.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 50.Perros P, McCrimmon RJ, Shaw G, Frier BM. Diabetic Medicine. 1995;12:622. doi: 10.1111/j.1464-5491.1995.tb00553.x. [DOI] [PubMed] [Google Scholar]
- 51.Kadiyala R, Peter R, Okosieme OE. International Journal of Clinical Practice. 2010;64:1130. doi: 10.1111/j.1742-1241.2010.02376.x. [DOI] [PubMed] [Google Scholar]
- 52.Dimitriadis GD, Raptis SA. Exp Clin Endocrinol Diabetes. 2001;109:S225. doi: 10.1055/s-2001-18584. [DOI] [PubMed] [Google Scholar]
- 53.Dimitriadis G, Baker B, Marsh H, Mandarino L, Rizza R, Bergman R, Haymond M, Gerich J. Am J Physiol Endocrinol Metab. 1985;248:E593. doi: 10.1152/ajpendo.1985.248.5.E593. [DOI] [PubMed] [Google Scholar]
- 54.Flynn RWV, MacDonald TM, Jung RT, Morris AD, Leese GP. Journal of Clinical Endocrinology & Metabolism. 2006;91:2159. doi: 10.1210/jc.2005-1833. [DOI] [PubMed] [Google Scholar]
- 55.Jornayvaz FR, Lee HY, Jurczak MJ, Alves TC, Guebre-Egziabher F, Guigni BA, Zhang D, Samuel VT, Silva JE, Shulman GI. Endocrinology. 2012;153:583. doi: 10.1210/en.2011-1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Senese R, Valli V, Moreno M, Lombardi A, Busiello RA, Cioffi F, Silvestri E, Goglia F, Lanni A, de Lange P. Pflugers Arch. 2011;461:153. doi: 10.1007/s00424-010-0892-3. [DOI] [PubMed] [Google Scholar]
- 57.Silvestri E, Moreno M, Lombardi A, Ragni M, de Lange P, Alexson SEH, Lanni A, Goglia F. FEBS Letters. 2005;579:1639. doi: 10.1016/j.febslet.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 58.Gong DW, He Y, Karas M, Reitman M. Journal of Biological Chemistry. 1997;272:24129. doi: 10.1074/jbc.272.39.24129. [DOI] [PubMed] [Google Scholar]
- 59.Lanni A, Beneduce L, Lombardi A, Moreno M, Boss O, Muzzin P, Giacobino JP, Goglia F. FEBS Letters. 1999;444:250. doi: 10.1016/s0014-5793(99)00061-7. [DOI] [PubMed] [Google Scholar]
- 60.Bézaire V, Seifert EL, Harper ME. The FASEB Journal. 2007;21:312. doi: 10.1096/fj.06-6966rev. [DOI] [PubMed] [Google Scholar]
- 61.Estey C, Seifert EL, Aguer C, Moffat C, Harper ME. Experimental Gerontology. 2012;47:361. doi: 10.1016/j.exger.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Angulo P. Alimentary Pharmacology & Therapeutics. 2007;25:883. doi: 10.1111/j.1365-2036.2007.03246.x. [DOI] [PubMed] [Google Scholar]
- 63.Farrell GC, van Rooyen D, Gan L, Chitturi S. Gut Liver. 2012;6:149. doi: 10.5009/gnl.2012.6.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lanni A, Moreno M, Lombardi A, de Lange P, Silvestri E, Ragni M, Farina P, Chieffi Baccari G, Fallahi P, Antonelli A, Goglia F. The FASEB Journal. 2005;19:1552. doi: 10.1096/fj.05-3977fje. [DOI] [PubMed] [Google Scholar]
- 65.Cable EE, Finn PD, Stebbins JW, Hou J, Ito BR, van Poelje PD, Linemeyer DL, Erion MD. Hepatology. 2009;49:407. doi: 10.1002/hep.22572. [DOI] [PubMed] [Google Scholar]
- 66.Heimberg M, Olubadewo JO, Wilcox HG. Endocrine Reviews. 1985;6:590. doi: 10.1210/edrv-6-4-590. [DOI] [PubMed] [Google Scholar]
- 67.Cachefo A, Boucher P, Vidon C, Dusserre E, Diraison F, Beylot M. Journal of Clinical Endocrinology & Metabolism. 2001;86:5353. doi: 10.1210/jcem.86.11.7981. [DOI] [PubMed] [Google Scholar]
- 68.Klieverik LP, Coomans CP, Endert E, Sauerwein HP, Havekes LM, Voshol PJ, Rensen PC, Romijn JA, Kalsbeek A, Fliers E. Endocrinology. 2009;150:5639. doi: 10.1210/en.2009-0297. [DOI] [PubMed] [Google Scholar]
- 69.Biondi B, Kahaly GJ. Nat Rev Endocrinol. 2010;6:431. doi: 10.1038/nrendo.2010.105. [DOI] [PubMed] [Google Scholar]
- 70.Franklyn JA, Boelaert K. Lancet. 2012;379:1155. doi: 10.1016/S0140-6736(11)60782-4. [DOI] [PubMed] [Google Scholar]
- 71.Olshausen KV, Bischoff S, Kahaly G, Mohr-Kahaly S, Erbel R, Beyer J, Meyer J. The American Journal of Cardiology. 1989;63:930. doi: 10.1016/0002-9149(89)90142-2. [DOI] [PubMed] [Google Scholar]
- 72.Biondi B, Palmieri EA, Fazio S, Cosco C, Nocera M, Saccà L, Filetti S, Lombardi G, Perticone F. Journal of Clinical Endocrinology & Metabolism. 2000;85:4701. doi: 10.1210/jcem.85.12.7085. [DOI] [PubMed] [Google Scholar]
- 73.Forfar JC, Miller HC, Toft AD. The American Journal of Cardiology. 1979;44:9. doi: 10.1016/0002-9149(79)90243-1. [DOI] [PubMed] [Google Scholar]
- 74.Frost L, Vestergaard P, Mosekilde L. Archives of Internal Medicine. 2004;164:1675. doi: 10.1001/archinte.164.15.1675. [DOI] [PubMed] [Google Scholar]
- 75.Kahaly GJ, Nieswandt J, Mohr-Kahaly S. Thyroid. 1998;8:1165. doi: 10.1089/thy.1998.8.1165. [DOI] [PubMed] [Google Scholar]
- 76.Baxter JD, Dillmann WH, West BL, Huber R, Furlow JD, Fletterick RJ, Webb P, Apriletti JW, Scanlan TS. The Journal of Steroid Biochemistry and Molecular Biology. 2001;76:31. doi: 10.1016/s0960-0760(01)00052-8. [DOI] [PubMed] [Google Scholar]
- 77.Forrest D, Vennstrom B. Thyroid. 2000;10:41. doi: 10.1089/thy.2000.10.41. [DOI] [PubMed] [Google Scholar]
- 78.Perra A, Simbula G, Simbula M, Pibiri M, Kowalik MA, Sulas P, Cocco MT, Ledda-Columbano GM, Columbano A. The FASEB Journal. 2008;22:2981. doi: 10.1096/fj.08-108464. [DOI] [PubMed] [Google Scholar]
- 79.Ocasio CA, Scanlan TS. ACS Chem Biol. 2006;1:585. doi: 10.1021/cb600311v. [DOI] [PubMed] [Google Scholar]
- 80.Grijota-Martinez C, Samarut E, Scanlan TS, Morte B, Bernal J. Endocrinology. 2011;152:1136. doi: 10.1210/en.2010-0813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zaraibn-Herzberg A, Marques J, Sukovich D, Periasamy M. J Biol Chem. 1994;269:1460. [PubMed] [Google Scholar]
- 82.Tsika RW, Bahl JJ, Leinwand LA, Morkin E. Proc Natl Acad Sci USA. 1990;87:379. doi: 10.1073/pnas.87.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dillmann WH. The American journal of medicine. 1990;88:626. doi: 10.1016/0002-9343(90)90530-q. [DOI] [PubMed] [Google Scholar]
- 84.Johansson C, Vennstrom B, Thoren P. Am J Physiol. 1998;275:R640. doi: 10.1152/ajpregu.1998.275.2.R640. [DOI] [PubMed] [Google Scholar]
- 85.Wikstrom L, Johansson C, Salto C, Barlow C, Campos Barros A, Baas F, Forrest D, Thoren P, Vennstrom B. EMBO J. 1998;17:455. doi: 10.1093/emboj/17.2.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Grasselli E, Voci A, Demori I, Canesi L, De Matteis R, Goglia F, Lanni A, Gallo G, Vergani L. J Endocrinol. 2012;212:149. doi: 10.1530/JOE-11-0288. [DOI] [PubMed] [Google Scholar]
- 87.Grasselli E, Voci A, Canesi L, Goglia F, Ravera S, Panfoli I, Gallo G, Vergani L. J Endocrinol. 2011;210:59. doi: 10.1530/JOE-11-0074. [DOI] [PubMed] [Google Scholar]
- 88.Moreno M, Silvestri E, De Matteis R, de Lange P, Lombardi A, Glinni D, Senese R, Cioffi F, Salzano AM, Scaloni A, Lanni A, Goglia F. The FASEB Journal. 2011;25:3312. doi: 10.1096/fj.11-181982. [DOI] [PubMed] [Google Scholar]