

Abstract
MicroRNAs (miRNAs) are endogenous small non-coding ribonucleotides that regulate expression of target genes governing diverse biological functions. Mechanistically, miRNA binding to the target complimentary sequences on the mRNA results in degradation or inhibition of protein translation. The short guiding and binding sequence of miRNA allows them to target a large repertoire of transcripts altering expression of many proteins. These miRNA targets are not restricted to specific signaling pathways but to a diverse group of transcripts, which harbor the target complimentary sequence. miRNA targeting of these diverse transcripts result in regulation of multiple signaling pathways establishing miRNAs as regulators of systems biomolecular networks. Accumulating evidence shows that miRNAs play an important role in cardiac development, hypertrophy, and failure, thereby are integral to regulating adaptive and maladaptive remodeling. Since cardiac remodeling and failure is a complex phenotype, it is apparent that global biomolecular networks and miRNAs profiles would be altered. Indeed, the miRNA profiles are varied with different etiologies of heart failure indicating that miRNAs could be the global regulators. Although the idea of miRNA being global regulators is not new, we believe that the time is ripe to discuss the role of miRNAs in regulating biomolecular networks. We discuss in the review, the use of Ingenuity Pathways Analysis algorithms with predicted targets of altered miRNA in dilated cardiomyopathy to computationally determine the alterations in canonical functional pathways and to generate biomolecular networks.
Keywords: MicroRNAs, Cardiac Hypertrophy, Dilated Cardiomyopathy, Heart Failure, Signaling Networks, Cardiovascular Phenotype
Introduction
The pathophysiology of cardiac dysfunction is a complex phenotype that ensues with alterations in the cellular signaling with progression of the pathology [1-3]. The phenotype is complex in part, due to the dynamic response of the cardiovascular system to diverse workloads including hemodynamic or neuroendocrine stresses emanating from hypertension, aortic stenosis, valvular dysfunction, or myocardial infarction. In response to multiple stresses, the heart undergoes adaptive compensatory remodeling [3-5] that augments the immediate needs of enhanced workload. Despite being beneficial in the short run, prolonged adaptive remodeling is deeply deleterious to the myocyte [3]. A growing body of evidence indicates that multiple signaling pathways are altered upon cardiac insult suggesting global changes accounting for both the complexity of stresses and the resultant phenotype [6-8]. Therefore, it is imperative to start understanding global alterations in cardiac pathology to a myriad of stresses rather than understanding the regulation of individual genes/proteins in a signaling pathway. It has long been appreciated that global analysis is key to better understanding the complex pathology of heart failure [9, 10]. In this regard, only transcription factors have been studied in that realm [11-14] due to its ability to regulate generation of multiple proteins [12-14]. In-depth studies on transcription factors regulating cardiac signaling pathways still cannot completely elucidate pathological manifestations suggesting the presence of other global regulators. miRNA with their ability to regulate expression of multiple molecules in non-canonical manner could be one of the global regulators.
MicroRNAs
MicroRNAs (miRNAs) are endogenously expressed 18–25 nucleotide-long non-coding single-stranded RNA that regulates target gene expression in a sequence-specific manner [15]. miRNAs are transcribed from DNA as long pre-miRNA transcripts that are processed into pre-miRNA hairpin by RNase-III enzyme Drosha [16] and transported out of the nucleus [15]. The hairpin loops of pre-miRNA are cleaved by ribonuclease Dicer generating mature double-stranded RNA [16]. Single strand from this mature miRNA is incorporated into RNA-induced silencing complex (RISC) that enables for interaction with target mRNA transcripts [15]. miRNAs encoded from the introns of the genes (also referred as mirtrons) [17, 18] escape the Drosha-mediated cleavage and are thought to be recognized by Dicer machinery (due to looped spliceosome intermediates) to be processed for loading into the RISC complex [17].
miRNA-loaded RISC complex is an active site of translational repression. The miRNA–RISC complex presented correctly leads to partial complementary binding with the mRNAs 3′ untranslated region (UTR) that results in translational repression of the target mRNA [15]. Significantly, the 3′ UTR can be bound by multiple RISC complexes that may in turn bring about cooperative synergistic repression of the target mRNA. Although the mechanisms of repression are only partially understood, it is believed that RISC complexes on the 3′ UTR may alter the structural organization of the mRNA sterically hindering the ribosomal machinery movement [19, 20]. In addition to translational repression, the RISC-targeted mRNAs are also directed towards degradation. More recently, studies have shown that miRNA may also stimulate transcription [21] thereby, suggesting that more work needs to be done to dissect the mechanisms by which miRNAs regulate gene expression. Therefore, alteration in miRNAs leads to changes in gene expression, and the sequence-based mechanism provides miRNAs with a large arsenal of targets qualifying them as a new generation of global regulators. Indeed, studies in mouse models from Olson’s group have shown that alteration in expression of single miRNA notably mimics the complex phenotype of cardiac hypertrophy and failure [22] indicating that miRNAs may critically regulate cardiac pathology.
miRNAs—Cardiac Function/Dysfunction and Heart Failure
From the time miRNAs were first identified to be altered in cardiac disease [22] 5 years ago, there has been a dramatic increase in the number of studies linking miRNAs to cardiac development, dysfunction, and heart failure [23-26]. Genome-wide miRNA profiles have identified specific miRNA signatures in mouse models and human heart failure [27-30] in which specific miRNAs are either up- or downregulated with etiologies of heart failure (Fig. 1) [29]. We and others have reported in patients with heart failure upregulation of miRNAs −21, −23a, −125b, −195, −199A, −214, and −342 and downregulation of miRNAs −1, −7, −29b, −30, −133, −150, and −378 [27-31]. Studies have been carried to specifically define the function for some of the altered miRNAs seen in human heart failure using neonatal cardiomyocytes, transgenic and knockout mice (detailed in Table 1), and some of the critical mice studies are discussed below. Cardiac overexpression of miRNA-195 in mice leads to hypertrophic growth and myocyte disarray resulting in dilated cardiomyopathy (DCM) and heart failure [22]. Knockout mice for miRNA-133 (double knockout of miRNA-133a 1 and 2) showed that embryos had septal defects, and mice surviving till adulthood had severe dilated cardiomyopathy [32]. Intriguingly, miRNA-133 is downregulated in mouse hearts following induction of hypertrophy, and this hypertrophy can be rescued by adenoviral overexpression of miRNA-133 [33]. Conversely, just the infusion of anti-sense oligo-nucleotide for miRNA-133 was sufficient to induce hypertrophic growth, activation of fetal gene program, and electrophysiological abnormalities [33]. Subjecting miRNA-133a transgenic mice to banding did not inhibit cardiac hypertrophy but was associated with less myocardial fibrosis and apoptosis [34]. Furthermore, overexpression of miRNA-133 increased the QT intervals in electrocardiographic recordings in isolated ventricular myocytes suggesting a broader role for miRNA-133a in the modulating cardiac function [34]. Similarly, electrophysiological abnormalities are also observed in the mice with deletion of miRNA-1, which is attributed to upregulation of Irx5, a transcription factor that negatively regulates Kv4.2 potassium channel [35]. Surprisingly, cardiac overexpression of miRNA-214 had no cardiac hypertrophic phenotype [22] despite its consistent dysregulation in multiple human heart failure studies [27-30]. It is also interesting to note that miRNA-214 is one of the first miRNAs to be altered following transverse aortic banding [29], and yet, overexpression of miRNA-214 does not have a hypertrophic phenotype [22]. This suggests that either the right phenotypic read out for miRNA-214 is yet to be determined, or it could potentially be a miRNA that would be an “effect” and not the “cause” of cardiac dysfunction following initial insult on the heart. miRNA-21 and miRNA-29 are preferentially expressed in cardiac fibroblasts and are altered with cardiac stress leading to profound effects on the cardiac function. miRNA-29 is downregulated in cardiac hypertrophy leading to upregulation of its target extracellular matrix proteins causing fibrosis [36]. While miRNA-21 is progressively upregulated with congestive heart failure leading to inhibition of its target SPRY1 resulting in enhanced extracellular regulated signaling (ERK) providing increased fibroblast survival [37].
Fig. 1.

Heat map of significantly altered miRNAs (miRNome) of individual patient samples from non-failing and failing human hearts diagnosed with dilated cardiomyopathy (DCM) [29]
Table 1.
miRNAs experimentally determined to play a role in cardiac hypertrophy/ cardiomyopathy
| microRNA | Experiment | Phenotype |
|---|---|---|
| miRNA-1-2 | Mouse knockout | Cardiac septal defects, hyperplasia and delay between atrial and ventricular repolarizations (PR interval) was shortened [35] |
| miRNA-1 | Neonatal cardiomyocyte | Inhibits FBS/endothelin/isoproterenol overexpression-mediated hypertrophy [42] |
| miRNA-21 | TAC and isoproterenol induced cardiac hypertrophy | Upregulated in compensatory hypertrophy and downregulated in decompensation [54] |
| Neonatal cardiomyocyte overexpression | Outgrowths in the cardiomyocytes accompanied by connections via gap junctions [54] | |
| Transgenic cardiomyocyte-specific expression | No specific phenotype indicating minimal role for miRNA21 in cardiomyocytes [37] | |
| Cardiac fibroblast overexpression | Anti-apoptotic [37] | |
| miRNA-23a | Antagomir infusion using minipumps | Isoproterenol-induced cardiac hypertrophy is attenuated with miRNA23a antagomirs [55] |
| Neonatal cardiomyocyte overexpression | Induces hypertrophy [22, 55] | |
| miRNA-23b | Neonatal cardiomyocyte overexpression | Induces hypertrophy [22] |
| miRNA-24 | Antagomir infusion using minipumps | Isoproterenol-induced cardiac hypertrophy is not altered [55] |
| Neonatal cardiomyocyte overexpression | Induces hypertrophy [22] | |
| miRNA-27 | Antagomir infusion using minipumps | Isoproterenol-induced cardiac hypertrophy is not attenuated [55] |
| miRNA-92 | miRNA inhibitor treatment of neonatal cardiomyocytes | Minimal effect on fetal gene expression [31] |
| miRNA-100 | miRNA mimic treatment of neonatal cardiomyocytes | Results in re-expression of fetal genes [31] |
| miRNA-129 | Neonatal cardiomyocyte transfection | Induces hypertrophy [30] |
| miRNA-133 | Neonatal cardiomyocyte transfection | Inhibited hypertrophy [33] |
| Antagomir infusion using minipumps | Induces cardiac hypertrophy [33] | |
| Double knockout of miRNA-133-a/b | Embryonic myocyte proliferation, septal defects, and surviving adults have severe dilated cardiomyopathy [32] | |
| Transgenic cardiomyocyte specific expression | Inhibitor of cardiomyocyte proliferation [32] | |
| Transgenic cardiomyocyte-specific expression subjected to TAC | Cardiac hypertrophy is not inhibited, but decreases myocardial fibrosis and cardiomyocyte apoptosis [34] | |
| miRNA-195 | Neonatal cardiomyocyte overexpression | Induces hypertrophy [22] |
| Transgenic cardiomyocyte specific expression | Induces cardiac hypertrophy and dilated cardiomyopathy [22] | |
| miRNA-199a | Neonatal cardiomyocyte overexpression | Cardiomyocyte enlargement [22] |
| miRNA-208 | Knockout mice subjected to TAC or bred to mouse model of hypertrophy | No hypertrophic response in both the cases [38] |
| miRNA-214 | Neonatal cardiomyocyte overexpression | Cardiomyocyte hypertrophy [22] |
| Transgenic cardiomyocyte specific expression | No phenotype [22] |
MiRNA-208 is unique with regards to cardiac function as it specifically expressed only in the cardiac myocytes as a part of the α-MHC gene. Importantly, miRNA-208 knockout mice neither showed any hypertrophic response with transverse aortic constriction nor did it not undergo pathological remodeling upon being bred with mouse model of pathological hypertrophy [38]. Understanding the role of individual miRNAs is critical as it is understood that miRNAs may be quick responders to stress [28] thereby altering the expression of target proteins leading to changes in homeostasis. Therefore, changes in miRNAs could thereby bring about translational alteration of multiple genes modulating their expression in a non-canonical manner. Indeed, majority of the in vivo studies discussed above have assessed the complex cardiac hypertrophic response as a measure of function upon alteration of a miRNA and yet have evaluated only few molecules at molecular level as targets. The ability of miRNAs to alter the complex hypertrophic response suggests that miRNAs may have a much broader role to play in cardiac pathology and could regulate various processes rather than targeting single gene products. Our study has used this concept to assess the global signaling pathways modulated by predicated targets of altered miRNAs from end-stage human heart failure [29] using the tools described below.
Global Regulation of Signaling by miRNAs
As the role of miRNAs in regulating single gene to multiple genes is emerging, it is becoming clear that miRNAs could be regulators of global processes resulting in a specific phenotypic outcome [39]. Consistent with their global role, studies have shown that miRNAs alter the protein output of multiple gene products [29] thereby regulating signaling networks in a non-classical manner. Indeed, miRNA signature profile varies with etiology as observed in aortic stenosis, dilated cardiomyopathy, and ischemic cardiomyopathy [27]. Therefore, the process leading to the phenotype appears to be regulated not by a single miRNA but the net effect on gene expression by subset of altered miRNAs in each of the etiologies. Translational repression brought about by miRNAs may affect both the beneficial as well as deleterious pathways [40, 41]. The net effect on a phenotype is thought to be a balance between the deleterious and compensatory signaling pathways regulated by miRNA targeting. Thus a concept for simultaneous alterations in miRNAs upsetting the homeostasis orienting the global signaling networks towards progression of pathology places a systems perspective on the disease regulated by miRNA. Further supporting the idea that miRNAs potentially play a role in global pathways comes from recent comprehensive studies on miRNA and mRNA profiles carried out on human heart failure samples pre- and post left ventricular assist device (LVAD) placement [28]. Interestingly, the authors observed a significant reversal in the miRNA profile following LVAD compared to mRNA profile suggesting that miRNAs are more sensitive responders to stress [28]. Therefore, miRNAs may be the critical quick responders regulating biomolecular networks in response to changes in the homeostasis.
miRNA-based regulation adds a new layer of control (as schematized in Fig. 2), altering the influence of transcriptome on a phenotype. All these components of the multi-layered regulation (Fig. 2) play a critical role in maintenance of homeostasis. Acute alteration in any of these drives the system towards dysfunction as inputs from all these modulators (Fig. 2) are integrated into the manifestation of a phenotype. Although it is known that each of the layers cross-talk with each other, it is believed that one of the most powerful regulators for sustained changes in homeostasis leading to pathology are the transcription factors [13]. The effects of transcription factors are global as their activity manifests in upregulation or downregulation of transcript, and in turn, proteins affecting the signaling pathways and physiology [12, 14]. It is important to note in this context that miRNA generation itself is regulated by transcriptional factors, and the generated miRNAs themselves can regulate production of proteins including transcriptional factors [42]. Although input from global regulators is not independent in the systems biomolecular network, it is however important to determine the contributions of each of regulators in a given phenotype.
Fig. 2.
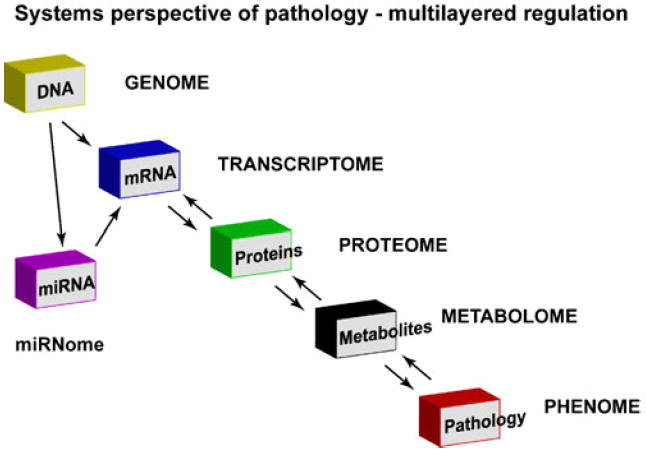
Systems perspective of complex disease pathology encompassing integrated analysis at each level. The scheme represents a multilayered regulation that has been effectively studied to provide a global analysis at each layer. Contribution by miRNA “miRNome” has been added to the multilayered regulation to give another layer of regulation in complex global signaling network
Understanding global alterations of proteins by miRNAs is still at its infancy; our recent studies in human DCM has shown that a specific set of signaling networks that contain predicted targets of altered miRNAs in DCM are altered [29]. All the predicted targets of altered miRNA in human dilated cardiomyopathy were used in the analysis as an unbiased approach toward understanding global regulation of signaling networks. Computational analysis of these predicted targets using Ingenuity Pathway knowledge base determined that the most over-represented functional network with highest significance was the cardiovascular system development and function [29] (Table 2). The confirmation that ~1,800 predicted targets of altered miRNAs regulate cardiovascular function using the unbiased computational approach suggests that this bioinformatic approach is a great tool for global analysis of signaling networks. Although the unbiased computational approach identified cardiovascular function to be altered, it is to be noted that predicted targets of altered miRNAs were used in the analysis. Recent studies [43] have identified that there is a high rate of false positives in the predicted targets database and therefore is a limitation in these studies. Despite this limitation on the use of predicted targets, identification that these predicted targets regulated cardiovascular function suggest that in our case, a significant portion of the predicted targets may be actual targets. Similar over-representative canonical pathways analysis was carried out using predicted targets of dysregulated miRNAs in primary muscle disorders [44]. In this study, 39 miRNAs were found to be altered, and use of all the predicted targets for the 39 mRNAs (~4,400 genes) leads to over-representation of pathways that regulated various aspects of muscular disorders [44]. The most well-represented pathways targeted by miRNA being cellular pathways regulating cell motility, communication, and degradation [44]. These studies reinforce the idea that inclusion of all the predicted targets in the canonical pathway function analysis could result in identification of functionally relevant pathways that are regulated by miRNA targeting. Identification of relevant pathways to the disease condition is of great significance as these pathways can be resurrected by therapeutically targeting the miRNAs.
Table 2.
Expression of potential targets and validated miRNA in human cardiomyopathy

|
Reference: [29]
A network view of pathological process reveals that identification of specific canonical pathways is just a cog in the big wheel of signaling networks where in cross-talk between pathways and molecules are integral to defining the phenotype. It is important to appreciate that miRNAs altered in cardiovascular pathology would have simultaneous targeting effects globally. With growing body of evidence that miRNAs could regulate cardiovascular phenotype [25, 36, 40], it is time to integrate “miRNome” (miRNA profile) as another regulatory layer to already multilayered system controlling the phenotypic outcome (Fig. 2). Consistently, miRNome in different cardiac etiologies like aortic stenosis, dilated cardiomyopathy, or ischemic cardiomyopathy is different [27] suggesting that these subset of altered miRNAs may target different signaling pathways thereby contributing to phenotypic differences. Studies by Thum et al., [30] show that a combination of miRNAs or a selective miRNome is sufficient to initiate fetal gene expression and morphological changes similar to those observed in heart failure. Indeed, overexpression of single miRNA [22] alters cellular hypertrophy, strengthening the idea that the miRNome alterations will have implications in global biomolecular signaling networks.
Lack of integrated miRNome and proteomic analysis in different cardiac etiologies at present is a limitation that we anticipate will be overcome. Currently, the networks that are built have used an unbiased approach of using all the predicted targets as comprehensive identification and validation of targets for each of the miRNAs is not available. We believe that this limitation should not markedly alter the targeted biomolecular networks by miRNome in dilated cardiomyopathy because inclusion of all predicted targets for canonical function analysis showed that the predicted targets regulate cardiac function (as described above). Importantly, intense efforts are currently required to develop high-throughput methods to identify and validate the targets for each of the miRNA, which will allow for determination of biomolecular networks targeted by altered miRNome with etiology. As more efforts are put into identification of targets for various miRNAs, miRNome will be another regulatory layer deciding the phenotypic outcomes like in embryonic development or stages in pathology as these stages have specific miRNA profiles [22, 23, 27-29]. Since miRNAs in general reduce the expression of the proteins by targeting the transcripts, these target proteins can therefore be used in analytical algorithmic tools to assess for potential causality of the pathology. Such an analysis can be referred to gene expression-trait causality correlationship that in our case is primarily driven by miRNome. Many databases and analytical tools have been developed to understand the function contributions of genes/proteins in a given process. Gene set enrichment analysis [45], Database for Annotation Visualization and Integrated Discovery [46, 47], and Ingenuity Pathway Analysis (IPA) [48] are some of the tools developed to test for enrichment of specific biological processes and molecular function of the genes involved in those processes. These analytical capabilities provide us with powerful tools to globally understand dysregulation of pathways in a given pathology.
Multiple tools/algorithms are available to globally determine the biomolecular networks associated with complex phenotypes, and all of them are based on two modeling approaches, forward or reverse modeling. The forward network modeling approach applies a set of equations generated a priori from previously defined biological relationships that are then tested and revised as needed. The reverse approach does not apply predefined set of relationship, but rather utilizes mathematical tools for network construction and lets the data define the relationship among elements studied [49]. IPA analytical tool used in our studies to an extent is based on the forward networking approach. The Bayesian networking model uses the probabilistic approach to a certain extent based on reverse networking model. We have used the IPA tool to generate the networks as they have the sophistication/flexibility to handle large datasets to generate networks based on the comprehensive IPA knowledge base [48, 50]. The IPA tool generates systems-based signaling networks by modeling relationship based on the various elements in the system (in our case the predicted targets for the miRNome in DCM). Each predicted target is assessed for network eligibility using the Ingenuity knowledge base that utilizes the ability of the identified molecule to interact and cross-talk with molecules in the database. The network eligible molecules serve as seeds for generating networks. The network eligible molecules are combined to maximize their specific connectivity that generates a network. An example of the network is shown in Fig. 3 from our studies on human DCMs and represents the basic unit for systems-wide approach. According to the algorithm used by IPA, a maximum of 35 molecules can be a part of the network; and therefore, individual networks may have up to 35 molecules depicting their connectivity or cross-talk. Although the IPA networking tool has great capabilities, there are limitations associated with it as it uses a curated database to generate network; and therefore, the networks may be biased towards well-studied molecules. Another limitation of using IPA is the use of the fixed number of molecules (35) to determine individual networks as it is potentially possible that the actual networks in the cell may be larger than this imposed restriction. Despite these limitations, studies on the effects of butyrates in kidney epithelial cells have shown that IPA analysis/tool can lead to linking of metabolic networks previously thought to be independent [51].
Fig. 3.

A representative network showing NF-κB as the hub or high connectivity node. The hub is the center of the web of signaling connections and NF-κB is connected to nearly all the molecules in the network. Altered miRNAs in end-stage heart failure are overlaid with their respective predicted targets. miRNA represented in green are downregulated and in red are upregulated in end-stage human dilated cardiomyopathy. Importantly, NF-κB is not a predicted target to any of the altered miRNAs “miRNome” in DCM, yet it could be regulated by alterations in miRNA targets [29]
Networks are composed of elements that give its topology. Elements in the network include specific transcripts/proteins (also known as nodes) and the connections (relationship) among them (edges) [50, 52]. The edges indicate a relationship between two elements in the network that could involve regulation at transcript level, protein interaction pattern, or any other measurement that describes a meaningful association [52]. Since IPA uses biological data, a network can be composed of a small number of highly connected molecules and many more with far few connections. Critically, the molecules that are high connected tend to behave more similarly to one another compared to the less-connected ones (in the network) with regards to a phenotype [53]. It is known that high-degree connectivity nodes are more essential than nodes that have fewer interaction partners as they maintain the overall connectivity of the network. These high connectivity nodes are called hubs, which play a central role in the network as targeting the hub genes disrupts the network and likely impacts the biological process [50, 52, 53]. The hub molecule NF-κB had a high degree of connectivity with many molecules in our representative network (Fig. 3) [29]. It is important to note that the less-connected molecule like HDGF in the network (Fig. 3) may also be critical in the biomolecular network as they provide for connectivity to a node on the neighboring network that could be described as a meaningful association between two elements in a system.
Since a single network cannot regulate the physiological process in isolation, the global regulation involves integrative cross-talk between the networks that manifests in a phenotype. The IPA algorithm can be used to assess the connectivity between the networks, and the interacting networks can be merged to give a global biomolecular network. Each network like the one represented in Fig. 3 was then merged with other networks by sub-network connectivity to generate a DCM miRNome-regulated biomolecular network [29]. The predicted targets for the DCM miRNome were represented in 43 networks from the total of 75 networks encompassing the complete global biomolecular network. Important limitations of these studies are a) the use of predicted targets to generate the network and b) the use of curated IPA knowledge database as already mentioned in the previous section. Despite these limitations, the association of DCM miRNome predicted targets with 43 of the 75 networks [29] suggests that not random but a specific set of pathways may be operationally altered manifesting in the DCM phenotype.
As all the targets for miRNAs are not identified and validated, an alternative strategy to obtain information on network is to assess the expression of all the predicted targets from the publicly available database for the pathology and assess the expression pattern relationship to its cognate miRNA. This is based on the assumption that upregulation of miRNA results in downregulation of its targets and vice versa. Therefore, proteins identified in expression studies to have inverse correlationship with the miRNAs expression profile may potentially be actual targets accounting for identification that these predicted proteins alter cardiac signaling [29]. An overlay of these molecules onto the network generates potentially targeted pathways by the miRNome in that given pathology and is a method to understand the underpinnings of miRNome-regulated pathways. Such strategies have been used by us and others to generate information on potential pathways that miRNAs could specifically regulate determining the progression of the disease [29, 44]. In this context of the overlay of the miRNA targets, it has to be explained that regulation by miRNAs is not included into the IPA algorithm as regulatory mechanism connecting two nodes in the network. Therefore, integration of the miRNA targets into the network will provide for new connectivity between the nodes and can be included into the algorithm once they are validated as targets. It is important to note that miRNA-mediated regulation of the various targets is different than the classical regulatory mechanisms like phosphorylation–dephosphorylation mediated by kinases–phosphatases or feed-back mechanisms in the metabolic cascades. The regulation by transcriptional regulators, kinases, phosphatases, signaling cascades, and ubiquitination is already integrated into the algorithm to generate the network topology, while miRNA-mediated regulation is not integrated into the algorithm and will therefore be another regulation on the nodes of the network that may potentially alter the topology of the network.
We have used the representative NF-κB network to discuss how miRNA-mediated regulation may alter the network, and it provides some interesting insights into the way miRNAs could regulate molecules that may not be obvious using the classical analytical approaches. For instance, DCM miRNome did not directly target NF-κB, but members of the NF-κB network were targets of multiple miRNAs. Mutual interactions within the network are bound to change as a result of altered miRNAs providing the explanation for NF-κB deregulation in DCM [29]. Therefore, depending on the individually altered miRNAs in the miRNome, the gene transcript will be regulated and have consequences on the protein function. An overlay shows that lot of molecules in the NF-κB network are predicted targets for the altered miRNAs in human DCM (Fig. 3). While expression analysis in human DCM showed that four of the predicted targets in the network were altered, of which, only three of the predicted targets could be validated. Critically, alteration in these three seems to be sufficient to alter expression of the hub NF-κB molecule [29]. These studies now provide unique miRNA-based regulation of molecules in the network, which needs to be integrated into the network building algorithm. Many of the altered proteins assessed from cardio genomics expression database are hub molecules [29] and are predicted targets of altered miRNAs in DCM. A significant number of these predicted targets showed inverse correlationship of expression with their respective miRNAs. Although the analysis is circumstantial, it definitely provides new information on the regulation of miRNAs globally. The miRNA-mediated regulation is going to be a critical determinant in signaling mechanism/physiological responses as miRNAs are known to be more sensitive than mRNAs to hemodynamic changes [28]. The sensitivity of miRNAs to detect changes portrays them to be a critical player in physiological responses. Therefore, based on important role, miRNAs are going in play in physiology; it is critical that efforts are invested in identifying and validating miRNA targets so that they can be used for network analysis instead of using predicted targets.
Conclusion and Future Direction
The role and regulation of individual genes/proteins in cardiovascular diseases have progressed at a great pace, but understanding and integrating global changes into the complex cardiovascular phenotype have significantly lagged behind. Although we are a long way from understanding the cardiovascular phenotype from systems perspective, generation of new analytical tools, high-throughput expression arrays (to define proteome, metabolme), and importantly, miRNomes, the missing link in the puzzle of global interacting networks will accelerate this process. We believe that with development of new technology and tools, complex cardiovascular phenotype modulated by multiple elements can be better understood, and higher order interactions will routinely include all players/elements involved in this process. A key research goal of network analysis beyond various “omics” is formulating universal characteristics of nonrandom networks associated with systems pathology. We need to explain the formation of sub-network ties and hub centrality to explain how network functions merge in vivo and how randomness is suppressed to transgress towards pathology. Breaking such nonrandom networks into a more diffuse network structure should be more conducive to collectively reversing the system to a healthy phenotype.
Acknowledgments
This work was supported in part by NIH RO1 HL089473 (S.V.NP) and RO1 HL083243 (S.S.K).
Footnotes
Conflict of interest The authors declare not conflict of interest.
Contributor Information
Sathyamangla V. Naga Prasad, Department of Molecular Cardiology, Lerner Research Institute, Cleveland Clinic Foundation, 9500 Euclid Avenue, Cleveland, OH 44195, USA Molecular Cardiology, Lerner Research Institute, Cleveland Clinic Foundation, Cleveland, OH 44195, USA, prasads2@ccf.org.
Sadashiva S. Karnik, Department of Molecular Cardiology, Lerner Research Institute, Cleveland Clinic Foundation, 9500 Euclid Avenue, Cleveland, OH 44195, USA
References
- 1.Bristow MR. Beta-adrenergic receptor blockade in chronic heart failure. Circulation. 2000;101:558–569. doi: 10.1161/01.cir.101.5.558. [DOI] [PubMed] [Google Scholar]
- 2.Mann DL, Deswal A, Bozkurt B, Torre-Amione G. New therapeutics for chronic heart failure. Annual Review of Medicine. 2002;53:59–74. doi: 10.1146/annurev.med.53.082901.104004. [DOI] [PubMed] [Google Scholar]
- 3.Opie LH, Commerford PJ, Gersh BJ, Pfeffer MA. Controversies in ventricular remodelling. Lancet. 2006;367:356–367. doi: 10.1016/S0140-6736(06)68074-4. [DOI] [PubMed] [Google Scholar]
- 4.Del Monte F, Hajjar RJ. Intracellular devastation in heart failure. Heart Failure Reviews. 2008;13:151–162. doi: 10.1007/s10741-007-9071-9. [DOI] [PubMed] [Google Scholar]
- 5.Latronico MV, Elia L, Condorelli G, Catalucci D. Heart failure: Targeting transcriptional and post-transcriptional control mechanisms of hypertrophy for treatment. International Journal of Biochemistry and Cell Biology. 2008;40:1643–1648. doi: 10.1016/j.biocel.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 6.Arad M, Seidman JG, Seidman CE. Phenotypic diversity in hypertrophic cardiomyopathy. Human Molecular Genetics. 2002;11:2499–2506. doi: 10.1093/hmg/11.20.2499. [DOI] [PubMed] [Google Scholar]
- 7.Chien KR. Stress pathways and heart failure. Cell. 1999;98:555–558. doi: 10.1016/s0092-8674(00)80043-4. [DOI] [PubMed] [Google Scholar]
- 8.Dorn GW, 2nd, Robbins J, Sugden PH. Phenotyping hypertrophy: Eschew obfuscation. Circulation Research. 2003;92:1171–1175. doi: 10.1161/01.RES.0000077012.11088.BC. [DOI] [PubMed] [Google Scholar]
- 9.Brown JH, Del Re DP, Sussman MA. The Rac and Rho hall of fame: A decade of hypertrophic signaling hits. Circulation Research. 2006;98:730–742. doi: 10.1161/01.RES.0000216039.75913.9e. [DOI] [PubMed] [Google Scholar]
- 10.Frey N, Olson EN. Cardiac hypertrophy: The good, the bad, and the ugly. Annual Review of Physiology. 2003;65:45–79. doi: 10.1146/annurev.physiol.65.092101.142243. [DOI] [PubMed] [Google Scholar]
- 11.Dorn GW, 2nd, Matkovich SJ. Put your chips on transcriptomics. Circulation. 2008;118:216–218. doi: 10.1161/CIRCULATIONAHA.108.789933. [DOI] [PubMed] [Google Scholar]
- 12.Hobert O. Common logic of transcription factor and microRNA action. Trends in Biochemical Sciences. 2004;29:462–468. doi: 10.1016/j.tibs.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 13.McKinsey TA, Olson EN. Toward transcriptional therapies for the failing heart: Chemical screens to modulate genes. Journal of Clinical Investigation. 2005;115:538–546. doi: 10.1172/JCI24144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Potthoff MJ, Olson EN. MEF2: A central regulator of diverse developmental programs. Development. 2007;134:4131–4140. doi: 10.1242/dev.008367. [DOI] [PubMed] [Google Scholar]
- 15.Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 16.Hammond SM. Dicing and slicing: The core machinery of the RNA interference pathway. FEBS Letters. 2005;579:5822–5829. doi: 10.1016/j.febslet.2005.08.079. [DOI] [PubMed] [Google Scholar]
- 17.Okamura K, Hagen JW, Duan H, Tyler DM, Lai EC. The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila. Cell. 2007;130:89–100. doi: 10.1016/j.cell.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruby JG, Jan CH, Bartel DP. Intronic microRNA precursors that bypass Drosha processing. Nature. 2007;448:83–86. doi: 10.1038/nature05983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brennecke J, Stark A, Russell RB, Cohen SM. Principles of microRNA-target recognition. PLoS Biology. 2005;3:e85. doi: 10.1371/journal.pbio.0030085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nature Review Genetics. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 21.Vasudevan S, Tong Y, Steitz JA. Switching from repression to activation: microRNAs can up-regulate translation. Science. 2007;318:1931–1934. doi: 10.1126/science.1149460. [DOI] [PubMed] [Google Scholar]
- 22.van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, et al. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:18255–18260. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cordes KR, Srivastava D. MicroRNA regulation of cardiovascular development. Circulation Research. 2009;104:724–732. doi: 10.1161/CIRCRESAHA.108.192872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Divakaran V, Adrogue J, Ishiyama M, Entman ML, Haudek S, Sivasubramanian N, et al. Adaptive and maladptive effects of SMAD3 signaling in the adult heart after hemodynamic pressure overloading. Circ Heart Fail. 2009;2:633–642. doi: 10.1161/CIRCHEARTFAILURE.108.823070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Latronico MV, Condorelli G. MicroRNAs and cardiac pathology. Nature Reviews in Cardiology. 2009;6:419–429. doi: 10.1038/nrcardio.2009.56. [DOI] [PubMed] [Google Scholar]
- 26.van Rooij E, Olson EN. Searching for miR-acles in cardiac fibrosis. Circulation Research. 2009;104:138–140. doi: 10.1161/CIRCRESAHA.108.192492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ikeda S, Kong SW, Lu J, Bisping E, Zhang H, Allen PD. Altered microRNA expression in human heart disease. Physiol Genomics. 2007;31:367–373. doi: 10.1152/physiolgenomics.00144.2007. [DOI] [PubMed] [Google Scholar]
- 28.Matkovich SJ, Van Booven DJ, Youker KA, Torre-Amione G, Diwan A, Eschenbacher WH, et al. Reciprocal regulation of myocardial microRNAs and messenger RNA in human cardiomyopathy and reversal of the microRNA signature by biomechanical support. Circulation. 2009;119:1263–1271. doi: 10.1161/CIRCULATIONAHA.108.813576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Naga Prasad SV, Duan ZH, Gupta MK, Surampudi VS, Volinia S, Calin GA, et al. Unique microRNA profile in end-stage heart failure indicates alterations in specific cardiovascular signaling networks. Journal of Biological Chemistry. 2009;284:27487–27499. doi: 10.1074/jbc.M109.036541. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, et al. MicroRNAs in the human heart: A clue to fetal gene reprogramming in heart failure. Circulation. 2007;116:258–267. doi: 10.1161/CIRCULATIONAHA.107.687947. [DOI] [PubMed] [Google Scholar]
- 31.Sucharov C, Bristow MR, Port JD. miRNA expression in the failing human heart: Functional correlates. Journal of Molecular and Cellular Cardiology. 2008;45:185–192. doi: 10.1016/j.yjmcc.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu N, Bezprozvannaya S, Williams AH, Qi X, Richardson JA, Bassel-Duby R, et al. MicroRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes and Development. 2008;22:3242–3254. doi: 10.1101/gad.1738708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, et al. MicroRNA-133 controls cardiac hypertrophy. Nature Medicine. 2007;13:613–618. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 34.Matkovich SJ, Wang W, Tu Y, Eschenbacher WH, Dorn LE, Condorelli G, et al. MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure-overloaded adult hearts. Circulation Research. 2010;106:166–175. doi: 10.1161/CIRCRESAHA.109.202176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, et al. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129:303–317. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 36.van Rooij E, Marshall WS, Olson EN. Toward microRNA-based therapeutics for heart disease: The sense in antisense. Circulation Research. 2008;103:919–928. doi: 10.1161/CIRCRESAHA.108.183426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–984. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- 38.van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- 39.Bartel DP, Chen CZ. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nature Review Genetics. 2004;5:396–400. doi: 10.1038/nrg1328. [DOI] [PubMed] [Google Scholar]
- 40.Thum T, Catalucci D, Bauersachs J. MicroRNAs: Novel regulators in cardiac development and disease. Cardiovascular Research. 2008;79:562–570. doi: 10.1093/cvr/cvn137. [DOI] [PubMed] [Google Scholar]
- 41.Wang N, Zhou Z, Liao X, Zhang T. Role of microRNAs in cardiac hypertrophy and heart failure. IUBMB Life. 2009;61:566–571. doi: 10.1002/iub.204. [DOI] [PubMed] [Google Scholar]
- 42.Ikeda S, He A, Kong SW, Lu J, Bejar R, Bodyak N, et al. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Molecular and Cellular Biology. 2009;29:2193–2204. doi: 10.1128/MCB.01222-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yousef M, Showe L, Showe M. A study of microRNAs in silico and in vivo: Bioinformatics approaches to microRNA discovery and target identification. FEBS Journal. 2009;276:2150–2156. doi: 10.1111/j.1742-4658.2009.06933.x. [DOI] [PubMed] [Google Scholar]
- 44.Eisenberg I, Eran A, Nishino I, Moggio M, Lamperti C, Amato AA, et al. Distinctive patterns of microRNA expression in primary muscular disorders. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:17016–17021. doi: 10.1073/pnas.0708115104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang da W, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, et al. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biology. 2007;8:R183. doi: 10.1186/gb-2007-8-9-r183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang da W, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Research. 2007;35:W169–175. doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, et al. A network-based analysis of systemic inflammation in humans. Nature. 2005;437:1032–1037. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- 49.Schadt EE, Lum PY. Thematic review series: Systems biology approaches to metabolic and cardiovascular disorders. Reverse engineering gene networks to identify key drivers of complex disease phenotypes. Journal of Lipid Research. 2006;47:2601–2613. doi: 10.1194/jlr.R600026-JLR200. [DOI] [PubMed] [Google Scholar]
- 50.Ma’ayan A. Insights into the organization of biochemical regulatory networks using graph theory analyses. Journal of Biological Chemistry. 2009;284:5451–5455. doi: 10.1074/jbc.R800056200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li CJ, Li RW, Wang YH, Elsasser TH. Pathway analysis identifies perturbation of genetic networks induced by butyrate in a bovine kidney epithelial cell line. Function and Integrative Genomics. 2007;7:193–205. doi: 10.1007/s10142-006-0043-2. [DOI] [PubMed] [Google Scholar]
- 52.Wu S, Lusis AJ, Drake TA. A systems-based framework for understanding complex metabolic and cardiovascular disorders. Journal of Lipid Research. 2009;50(Suppl):S358–S363. doi: 10.1194/jlr.R800067-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zotenko E, Mestre J, O’Leary DP, Przytycka TM. Why do hubs in the yeast protein interaction network tend to be essential: Reexamining the connection between the network topology and essentiality. PLoS Computational Biology. 2008;4:e1000140. doi: 10.1371/journal.pcbi.1000140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sayed D, Rane S, Lypowy J, He M, Chen IY, Vashistha H, et al. MicroRNA-21 targets Sprouty2 and promotes cellular outgrowths. Molecular Biology of the Cell. 2008;19:3272–3282. doi: 10.1091/mbc.E08-02-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin Z, Murtaza I, Wang K, Jiao J, Gao J, Li PF. miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:12103–12108. doi: 10.1073/pnas.0811371106. [DOI] [PMC free article] [PubMed] [Google Scholar]