

Abstract
Using the tris(3,5-diphenylpyrazol-1-yl)borate (Ph2Tp) supporting ligand, a series of mono- and dinuclear ferrous complexes containing hydroquinonate (HQate) ligands have been prepared and structurally characterized with X-ray crystallography. The monoiron(II) complexes serve as faithful mimics of the substrate-bound form of hydroquinone dioxygenases (HQDOs) – a family of nonheme Fe enzymes that catalyze the oxidative cleavage of 1,4-dihydroxybenzene units. Reflecting the variety of HQDO substrates, the synthetic complexes feature both mono- and bidentate HQate ligands. The bidentate HQates cleanly provide five-coordinate, high-spin Fe(II) complexes with the general formula [Fe(Ph2Tp)(HLX)] (1X), where HLX is a HQate(1-) ligand substituted at the 2-position with a benzimidazolyl (1A), acetyl (1B and 1C), or methoxy (1D) group. In contrast, the monodentate ligand 2,6-dimethylhydroquinone (H2LF) exhibited a greater tendency to bridge between two Fe(II) centers, resulting in formation of [Fe2(Ph2Tp)2(μ-LF)(MeCN)] [2F(MeCN)]. However, addition of one equivalent of “free” pyrazole (Ph2pz) ligand provided the mononuclear complex, [Fe(Ph2Tp)(HLF)(Ph2pz)] [1F(Ph2pz)], which is stabilized by an intramolecular hydrogen bond between the HLF and Ph2pz donors. Complex 1F(Ph2pz) represents the first crystallographically-characterized example of a monoiron complex bound to an untethered HQate ligand. The geometric and electronic structures of the Fe/HQate complexes were further probed with spectroscopic (UV-vis absorption, 1H NMR) and electrochemical methods. Cyclic voltammograms of complexes in the 1X series revealed an Fe-based oxidation between 0 and −300 mV (vs. Fc+/0), in addition to irreversible oxidation(s) of the HQate ligand at higher potentials. The one-electron oxidized species (1Xox) were examined with UV-vis absorption and electron paramagnetic resonance (EPR) spectroscopies.
Introduction
The degradation of single- and multi-ring aromatic hydrocarbons by bacteria is a key component of the global carbon cycle and the basis of bioremediation technologies. In aerobic environments, the catabolism of aromatic compounds is dependent on nonheme iron dioxygenases that cleave aromatic rings with incorporation of both atoms of O2 into the product.1 Such transformations are challenging due to the intrinsic stability of aromatic systems and the high activation barrier to reaction with triplet dioxygen. With the notable exception of the intradiol catechol dioxygenases, the active sites of ring-cleaving dioxygenases overcome these obstacles by coordinating both substrate and O2 to a single Fe(II) site.2 The iron center is typically attached to the protein by a facial array of one carboxylate (Asp or Glu) and two His residues (Scheme 1),3 although variants of this 2-His-1-carboxylate motif have recently been reported.4
Scheme 1.

While the well-studied extradiol catechol dioxygenases (ECDOs) are the prototypical ring-cleaving dioxygenases,2,5 members of this enzymatic family employ a remarkable variety of substrates, including protocatechuates,6 2-aminophenols,7 and salicylates.8 Of particular relevance to this manuscript are dioxygenases that cleave hydroquinones (HQs = 1,4-dihydroxybenzene and its derivatives). The HQ-cleaving dioxygenases (HQDOs) can be grouped into two categories. The first class oxidizes substrates with carboxylate groups at the 2-position of the aromatic ring, namely, gentisate9 and homogentisate10 (2,5-dihydroxybenzoate and 2,5-dihydroxyphenylacetate, respectively; Scheme 1). In these enzymes, the substrate likely binds to iron in a bidentate manner via the phenolate and carboxylate donors.11 In the second class, the substrate coordinates in a monodentate fashion, since the halogenated or unsubstituted HQs lack a metal-binding moiety at the ortho position. Examples include 2,6-dichlorohydroquinone 1,2-dioxygenase (PcpA),12 chlorohydroquinone dioxygenase (LinE),13 and hydroquinone 1,2-dioxygenase (MnpC).14
While there have been few mechanistic studies of the hydroquinone 1,2-dioxygenases, the proposed catalytic cycles largely follow the pattern derived from extensive studies of the ECDOs.12a Coordination of the deprotonated HQ substrate to the Fe(II) center displaces some or all of the H2O ligands found in the resting state (Scheme 1), thereby facilitating O2 binding to the iron center. Formation of a short-lived ferric-superoxo intermediate is thought to trigger the transfer of one electron from the substrate ligand to iron, resulting in a bound p-benzosemiquinone radical. The existence of this putative intermediate would likely require deprotonation of the distal –OH group by a second-sphere residue, although it is not clear whether these three events (O2 coordination, electron transfer, and proton transfer) occur in a stepwise or concerted manner. The degree of semiquinone character on the substrate ligand in the O2-bound form of the enzyme is also uncertain; for instance, a recent computational study by Ye and Neese15 has cast doubt on the existence of a superoxo-Fe(II)-semiquinone intermediate in the ECDO (and, by extension, the HQDO) mechanism. While the nature of this intermediate remains disputed, it is well-established that the next step of the catalytic cycle involves generation of an Fe(II)-alkylperoxo species, which undergoes a Criegee rearrangement and hydrolysis to eventually yield the ring-opened product.15–16
Unanswered questions regarding the HQDOs can be answered, in part, through the development of synthetic complexes that replicate the structure and/or function of the enzyme active site. Remarkably, a survey of the literature found only a single example of a crystallographically-characterized monoiron(II)-hydroquinonate complex: Fe(L)2, where is L is a deprotonated Schiff base of 2,5-dihydroxybenzaldehyde.17 The dearth of reported Fe/HQ complexes is partly due to the ability of hydroquinonate (HQate) ligands to adopt a bridging position between metal centers, as demonstrated by structures of diiron(III)-porphyrin and -salen complexes with bridging HQate dianions.18,19 Recently, Machonkin and Holland described the formation and 1H NMR characterization of a mononuclear iron(II)-2-methylhydroquinonate complex supported by the 1,3,5-tris(tolylideneimino)cyclohexane ligand;20 however, this species is unstable and it was not possible to obtain crystals suitable for crystallographic analysis.
In this manuscript, we report the synthesis and X-ray structural characterization of several monoiron(II) complexes containing HQate ligands. Each complex features the tris(3,5-diphenylpyrazol-1-yl)borate(1-) supporting ligand (Ph2Tp), as substituted Tp ligands are well-known to faithfully mimic the coordination environment of the 2-His-1-carboxylate facial triad.21 We found that inclusion of bulky phenyl groups at the 3-positions of the pyrazole rings generally discourages formation of the diiron(II)μ-hydroquinonate(2-) complexes, although dinuclear species were generated with certain HQs. As shown in Scheme 2, two types of HQ ligands were employed in this study: i) bidentate (or “tethered”) ligands that feature an ortho substituent capable of metal coordination (H2LA–E), and (ii) the monodentate (or “untethered”) ligand 2,6-dimethylhydroquinone (H2LF). These HQs were selected because they reflect the range of substrates oxidized by HQDOs, with the monodentate and bidentate ligands resembling (chloro)hydroquinones and (homo)gentisates, respectively.{NOTE: The series also includes 2-hydroxyacetophenone (H2LC) as a control to properly evaluate the role of the distal –OH group in tuning the structural and electronic properties of our HQDO models. Each of the resulting complexes was characterized with crystallographic, spectroscopic (UV-vis absorption, 1H NMR), and electrochemical techniques. Indeed, we report here the first X-ray structure of a mononuclear Fe complex featuring an untethered hydroquinonate ligand. We also employed spectroscopic methods, including electron paramagnetic resonance (EPR), to examine the ferric species generated upon one-electron oxidation of the monoiron(II) complexes. These results lay the foundation for future studies that will explore the O2 reactivity of complexes that mimic the enzyme-substrate intermediates of HQDOs.
Scheme 2.

Results and Discussion
1. Fe(II) Complexes with Tethered Hydroquinonate Ligands – Synthesis and Solid State Structures
The mononuclear iron(II) complexes 1A–D (Scheme 2) were prepared by mixing equimolar amounts of K(Ph2Tp) and FeX2 (X = Cl or OTf) with the singly-deprotonated ligands, –HLA–D, in MeCN (or MeCN/CH2Cl2 solvent mixture). The resulting air-sensitive complexes dissolve easily in CH2Cl2, but are largely insoluble in more polar solvents like MeCN and MeOH. With the exception of 1B, which contains a 2-acetylphenolate ligand, the FTIR spectrum of each complex exhibits a ν(O-H) feature arising from the distal hydroxyl group, indicating that the HQ ligands are monoanionic and coordinated to a single Fe center.
Crystals of 1A–D suitable for X-ray structure determination were obtained by layering concentrated CH2Cl2 solutions with either MeCN or pentane. Details concerning data collection and analysis are provided in Table 3, and selected bond distances and angles for 1A–D are shown in Table 1. As illustrated in Figure 1, each complex features a five-coordinate (5C) Fe(II) center bound to a facially coordinating Ph2Tp ligand and bidentate HLA–D group. The Fe-NTp bonds exhibit an average distance of 2.14 Å across the series, characteristic of high-spin (S = 2) ferrous complexes.21d, 22 The Fe1-O1 distances, which range between 1.927(1) and 1.961(1) Å, are also typical for iron(II)-phenolate units in 5C complexes.23
Table 3.
Summary of X-ray Crystallographic Data Collection and Structure Refinement
| 1A • CH2Cl2 | 1B • 2CH2Cl2 a | 1C | 1D • [HNEt3]OTf | 1F | 2E•2Et2O | [2F(MeCN)]•2DCE b | |
|---|---|---|---|---|---|---|---|
| empirical formula | C60H47BCl2FeN8O2 | C55H45BCl4FeN6O3 | C53H41BFeN6O2 | C59H57BF3FeN7O6S | C68H55BFeN8O2 | C106H96B2Fe2N12O6 | C104H87B2Cl4Fe2N13O2 |
| formula weight | 1049.62 | 1046.46 | 860.58 | 1115.84 | 1082.86 | 1767.27 | 1826.02 |
| crystal system | Monoclinic | Orthorhombic | Monoclinic | Triclinic | Monoclinic | Triclinic | Monoclinic |
| space group | P21/c | P212121 | P21/n | P -1 | P21/c | P -1 | Pn |
| a, Å | 14.5859(5) | 11.2362(4) | 10.4129(2) | 9.6832(4) | 17.8034(7) | 9.6359(10) | 13.4895(3) |
| b, Å | 13.6416(4) | 17.7643(6) | 30.8300(5) | 9.8868(5) | 22.3235(10) | 13.4413(14) | 10.0544(3) |
| c, Å | 25.2090(8) | 25.2782(8) | 13.1758(2) | 28.3671(15) | 13.6123(6) | 18.205(2) | 32.9607(9) |
| α, deg | 90 | 90 | 90 | 85.740(4) | 90 | 95.633(9) | 90 |
| β, deg | 93.779(3) | 90 | 90.634(2) | 86.952(4) | 99.746(4) | 105.298(10) | 96.806(3) |
| γ, deg | 90 | 90 | 90 | 81.003(4) | 90 | 99.002(9) | 90 |
| V, Å3 | 5005.0(3) | 5045.6(3) | 4229.6(1) | 2672.5(2) | 5331.9(4) | 2222.1(4) | 4438.9(2) |
| Z | 4 | 4 | 4 | 2 | 4 | 1 | 2 |
| Dcalc, g/cm3 | 1.393 | 1.351 | 1.351 | 1.387 | 1.349 | 1.321 | 1.342 |
| λ, Å | 1.5418 | 1.5418 | 0.7107 | 1.5418 | 0.7107 | 1.5418 | 1.5418 |
| μ, mm−1 | 3.831 | 4.518 | 0.408 | 3.209 | 0.340 | 3.131 | 4.018 |
| θ-range, deg | 7 to 148 | 7 to 148 | 7 to 59 | 9 to 148 | 7 to 59 | 7 to 148 | 7 to 148 |
| reflections collected | 35336 | 19096 | 58359 | 15029 | 62124 | 15573 | 23318 |
| independent reflections | 9913 [Rint = 0.0382] | 8984 [Rint = 0.0435] | 10931 [Rint = 0.0402] | 15029 | 13600 [Rint = 0.0409] | 8687 [Rint = 0.0622] | 12059 [Rint = 0.0406] |
| data / restraints / parameters | 9913 / 0 / 672 | 8984/0/634 | 10931 /0 / 569 | 15029 / 0 / 709 | 13600 / 0 / 731 | 8687 / 24 / 569 | 12059 / 31 / 1132 |
| GOF (on F2) | 1.041 | 1.103 | 1.027 | 1.074 | 1.058 | 1.026 | 1.024 |
| R1/wR2 (I > 2σ(I)) c | 0.0354 / 0.0891 | 0.0814 / 0.2410 | 0.0375 / 0.0891 | 0.0864 / 0.2278 | 0.0414 / 0.1005 | 0.0718 / 0.1807 | 0.0638 / 0.1662 |
| R1/wR2 (all data) c | 0.0449 / 0.0954 | 0.0838 / 0.2437 | 0.0462 / 0.0953 | 0.1008 / 0.2402 | 0.0552 / 0.1083 | 0.1152 / 0.2227 | 0.0821 / 0.1826 |
One of the solvate molecules in 1B • 2CH2Cl2 is only partially (77%) populated.
One of the solvate molecules in [2F(MeCN)]• 2DCE is only partially (68%) populated.
R1 = Σ ||Fo| − |Fc|| / Σ|Fo|; wR2 = [Σw(Fo2 − Fc2)2 / Σw(Fo2)2]1/2
Table 1.
Selected Bond Distances (Å) and Bond Angles (deg) from the X-ray Structures of Monoiron(II) Hydroquinonate Complexes 1A–D and 1F.
| 1A•CH2Cl2 | 1B•2CH2Cl2 | 1C | 1D•[HNEt3]OTf | 1F | |
|---|---|---|---|---|---|
| Fe-O1 | 1.961(1) | 1.937(4) | 1.927(1) | 1.931(3) | 1.893(1) |
| Fe-N1 | 2.122(1) | 2.131(5) | 2.108(1) | 2.130(4) | 2.085(1) |
| Fe-N3 | 2.148(2) | 2.089(5) | 2.093(1) | 2.129(4) | 2.085(1) |
| Fe-N5 | 2.147(2) | 2.185(5) | 2.228(1) | 2.186(4) | 2.285(1) |
| Fe-L a | 2.139(2) | 2.079(4) | 2.103(1) | 2.317(3) | 2.341(2) |
| O1-C46 | 1.341(2) | 1.303(7) | 1.304(2) | 1.337(6) | 1.338(2) |
| O2-C49 | 1.370(2) | 1.370(7) | 1.371(6) | 1.390(2) | |
| O1-Fe-N1 | 110.64(5) | 129.5(2) | 128.04(5) | 128.5(2) | 134.21(6) |
| O1-Fe-N3 | 153.34(5) | 139.9(2) | 140.01(5) | 130.0(2) | 131.98(6) |
| O1-Fe-N5 | 94.78(5) | 97.9(2) | 100.61(4) | 115.6(1) | 101.45(5) |
| O1-Fe-LX | 88.90(5) | 86.4(2) | 85.28(4) | 75.7(1) | 89.04(5) |
| N1-Fe-N3 | 95.65(6) | 90.6(2) | 91.50(4) | 94.2(1) | 93.61(5) |
| N1-Fe-N5 | 92.11(5) | 90.2(2) | 88.25(4) | 89.4(2) | 85.41(5) |
| N3-Fe-N5 | 79.38(6) | 81.2(2) | 84.39(4) | 85.6(2) | 83.65(5) |
| LX -Fe-N1 | 90.69(6) | 87.4(2) | 91.32(4) | 85.3(1) | 89.01(5) |
| LX -Fe-N3 | 95.37(6) | 95.3(2) | 88.47(4) | 84.5(1) | 87.53(5) |
| LX -Fe-N5 | 174.27(6) | 175.7(2) | 172.83(4) | 168.4(1) | 169.24(5) |
| τ-value b | 0.35 | 0.60 | 0.55 | 0.64 | 0.58 |
L is the N or O atom of the pendant donor of the HQ anion.
For a definition of the τ-value, see reference 24. A five-coordinate complex with ideal square-pyramidal geometry would have a τ-value of 0.0, while those with ideal trigonal bipyramidal geometry would have a value of 1.0.
Figure 1.

Thermal ellipsoid plots (50% probability) derived from the X-ray structures of 1A•CH2Cl2 (top), 1B•2CH2Cl2 (middle), and 1D•[HNEt3]OTf (bottom). Non-coordinating solvent molecules and most hydrogen atoms have been omitted for clarity, as well as the Ph-rings at the 5-positions of the Ph2Tp ligand. The HNEt3+ counter cation in the 1D•[HNEt3]OTf structure is not shown.
The coordination geometry of 1A is intermediate between square pyramidal and trigonal bipyramidal (τ = 0.3524), and the HLA ligand adopts a twisted conformation with a dihedral angle of 35° between the planes of the HQate and benzimidazolyl rings (Figure 1). This orientation is likely the result of π-stacking interactions between the benzimidazolyl moiety and a 3-phenyl substituent of the Ph2Tp ligand, in addition to steric repulsion between the HQate ring and a second phenyl group. Compared to 1A, the structures of 1B and 1C lie much further towards the trigonal-bipyramidal limit (τ = 0.60 and 0.55, respectively) with the acetyl group in an axial position trans to a pyrazole donor (N5). The metric parameters for 1B and 1C are nearly identical, suggesting that the structural effects of the para hydroxyl group are minimal. The O1-C46 distances in 1B and 1C are shorter than the corresponding distance in 1A (1.303 vs. 1.341 Å; Table 1) due to delocalization of the negative charge onto the 2-acetyl group. The O1-C46 bond of the acetophenone-derived ligands therefore acquires some double-bond character, whereas the twisted conformation of the HLA ligand indicates a lack of electronic conjugation between the π-systems.
In contrast to the HLA–C donors, the 2-methoxyhydroquinonate ligand (HLD) in 1D forms a five-membered ring chelate with the Fe(II) center. This fact, coupled with the intrinsically weak donating ability of methoxy substituents, results in a rather lengthy Fe1-O3 distance of 2.317(3) Å. Thus, in certain respects, 1D can be considered to possess an intermediate coordination number between 4 and 5. As evidence, the O1-Fe1-N5 angle increases from an average of 97.8° in 1A–C to 115.6° in 1D (with a corresponding decrease in the O3-Fe1-NTp angles), as the HQate donor shifts out of the equatorial plane (Table 1). Thus, if the weakly-bound –OCH3 group is ignored, 1D appears to adopt a distorted trigonal pyramidal geometry with the O1 donor in the axial position. Notably, complex 1D co-crystallizes with one equivalent of [HNEt3]OTf salt, and the triflate anion participates in a hydrogen-bonding interaction with the distal –OH group in the solid state (Figure 1; the O2···O4 distance is 2.782(6)). This feature is reminiscent of acid/base interactions between HQ substrates and conserved second-sphere residues that have been proposed to play an important role in HQDO catalysis.9b, 12a
The diiron(II) μ-LX complexes were never observed in preparations of 1A–D, and we initially attributed the lack of dinuclear side-products to the steric demands of the Ph2Tp ligand. To evaluate this hypothesis, we generated the compound 2,5-dimethoxyhydroquinone (H2LE), which is capable of coordinating two metal centers in a bidentate fashion. Interestingly, use of this ligand provides the diiron(II) complex 2E as the only isolated product even when the reactants are mixed in equimolar ratios, thereby proving that the Ph2Tp framework is capable of supporting dinuclear complexes. The X-ray structure of 2E is shown in Figure 2 and key metric parameters are listed in the caption. The complex is centrosymmetric with an Fe···Fe distance of 8.15 Å. The Fe-O/N distances of 2E are nearly identical to those of the analogous monoiron(II) complex 1D, although the position of the HQate ligand with respect to the NTp donors is somewhat different (e.g., ∠O1-Fe1-N5 = 98.1(2)° and 115.6(1)° in 2E and 1D, respectively). The fact that the 2,5-dimethoxylhydroquinonate ligand exclusively yields 2E, whereas ligands HLA–D favor monomeric species, suggests that the thermodynamic benefit of bidentate chelation at both Fe(II) centers is able to overcome the steric barrier to dimerization.
Figure 2.
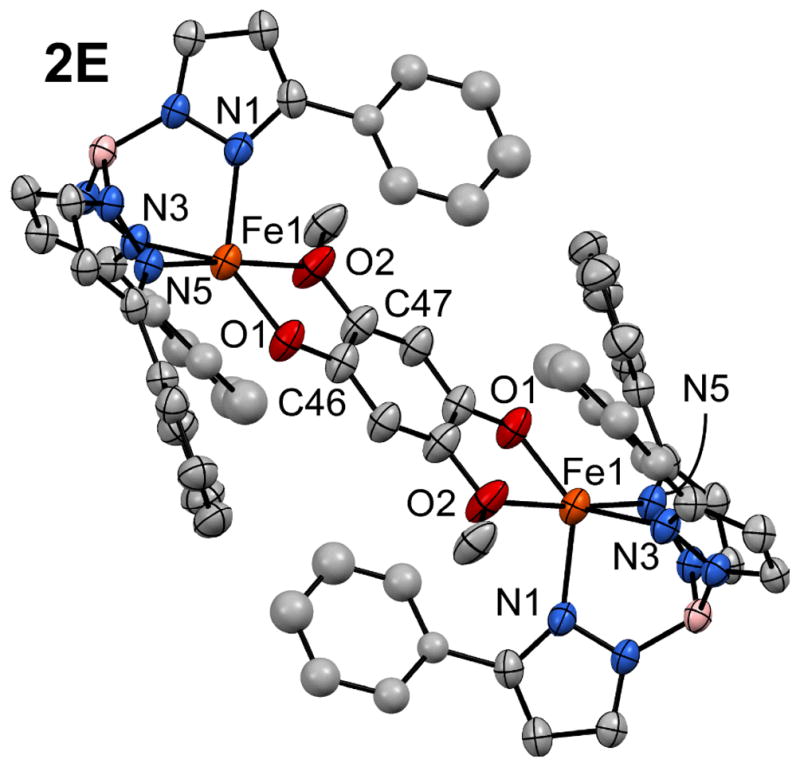
Thermal ellipsoid plot (50% probability) derived from the X-ray structure of 2E•CH2Cl2. Non-coordinating solvent molecules and hydrogen atoms have been omitted for clarity, in addition to Ph-rings at the 5-positions of the Ph2Tp ligand. Ellipsoids are not shown for four Ph rings due to disorder. Selected bond lengths (Å) and angles (°) [note: the complex is centrosymmetric]: Fe1-O1 1.904(3), Fe1-O2 2.328(3), Fe1-N1 2.107(4), Fe1-N3 2.108(3), Fe1-N5 2.213(4), O1-C46 1.329(5), O2-C47 1.388(6); O1-Fe1-O2 75.2(1), O1-Fe1-N1 131.9(2), O1-Fe1-N3 138.7(2), O1-Fe1-N5 98.9(1), O2-Fe1-N1 91.8(2), O2-Fe1-N3 97.9(1), O2-Fe1-N5 174.1(1), N1-Fe1-N3 88.3(1), N1-Fe1-N5 91.8(1), N3-Fe1-N5 86.9(1).
2. Fe(II) Complexes with an Untethered Hydroquinonate Ligand – Synthesis and Solid State Structures
As noted in the Introduction, several HQDOs oxidize “untethered” HQs that lack additional metal-coordinating groups. To replicate the monodentate binding mode of these HQ substrates, we employed the ligand 2,6-dimethylhydroquinone (H2LF). Reaction of H2LF with equimolar amounts of K(Ph2Tp), FeCl2, and NaOMe in MeCN generates a bright orange solid, which was recrystallized by slow diffusion of MeCN into a concentrated 1,2-dichloroethane (DCE) solution. X-ray analysis of the crystals revealed a diiron(II) structure with the formulation, [Fe2(Ph2Tp)2(μ-LF)(MeCN)] (2F(MeCN); Figure 3). Unlike 2E, the Fe(II) centers in 2F(MeCN) are not equivalent: Fe2 is 4C due to steric hindrance from the methyl substituents of the bridging LF dianion, and Fe1 is 5C with an additional solvent MeCN ligand. The Fe1 center exhibits a distorted trigonal bipyramidal coordination geometry (τ = 0.58), while the Fe2 geometry is best described as trigonal pyramidal (∠O2-Fe2-NTp = 125 ± 5°). The low Fe2 coordination number leads to relatively short metal-ligand bond lengths, especially the Fe2-O2 distance of 1.784(6) Å (see Figure 3 caption for additional metric parameters). The high-spin Fe ions are separated by 8.72 Å. While the initial synthesis of 2F employed equimolar amounts of reagents, the complex can also be prepared in greater yield by using only 0.5 equivalent of H2FF.
Figure 3.

Thermal ellipsoid plot (50% probability) derived from the X-ray structure of [2F(MeCN)]•2DCE. Non-coordinating solvent molecules, hydrogen atoms, and Ph-rings at the 5-positions of the Ph2Tp ligand have been omitted for clarity. Selected bond lengths (Å): Fe1-O1 1.852(6), Fe1-N1 2.111(5), Fe1-N3 2.136(6), Fe1-N5 2.187(5), Fe1-N13 2.289(8), O1-C93 1.348(10), Fe2-O2 1.784(6), Fe2-N7 2.105(5), Fe2-N9 2.119(5), Fe2-N11 2.136(5), O2-C96 1.352(10).
In an effort to prevent formation of 2F(MeCN), one equivalent of 3,5-diphenylpyrazole (Ph2pz) was included in the reaction mixture described above. Under these conditions, the reaction provided a yellow product that was recrystallized by DCE/pentane layering. X-ray diffraction analysis revealed that the crystals contain the 5C monoiron(II) complex, [Fe(Ph2Tp)(HLF)(Ph2pz)] [1F(Ph2pz)]. As shown in Figure 4, 1F(Ph2pz) features a trigonal bipyramidal coordination geometry (τ = 0.58) with the HQate and Ph2pz donors in equatorial and axial positions, respectively. These two ligands form an intramolecular hydrogen-bond that closes a five-membered ring, as evident in the O1···N8 distance of 2.840(2) Å and O1···H7 distance of 2.17(2) Å (the H2 and H7 atoms were found objectively and refined). The Fe1-O1 bond distance of 1.893(1) is shorter than the corresponding distances in the tethered complexes 1A–D, whereas the axial Ph2pz ligand is weakly bound with an Fe1-N7 distance of 2.341(2) Å (Table 1). As expected, HLF coordinates to the Fe(II) center via the more sterically-accessible O-atom at the 4-position of the HQ. Without the constraint of a pendant ligand, the HQate ring in 1F(Ph2pz) rotates away from the Fe center, as signified by the large Fe1-O1-C46 bond angle of 148.7(1)° (compared values of 125 ± 5° for 1A–1D).
Figure 4.

Thermal ellipsoid plot (50% probability) derived from the X-ray structure of 1F. Hydrogen atoms and Ph-rings at the 5-positions of the Ph2Tp ligand have been omitted for clarity. Selected bond lengths and angles are provided in Table 1.
3. Spectroscopic and Electrochemical Properties of Fe(II)-Hydroquinonate Complexes
Electronic absorption spectra of complexes 1A–D and 1F in CH2Cl2 are shown in Figure 5. Complexes 1B and 1C are both brightly colored due a weak absorption manifold (ε ~ 0.7 mM−1cm−1) in the visible region and an intense peak in the near-UV (ε ~5.5 mM−1cm−1). The application of time-dependent density functional theory (TD-DFT) to 2B revealed that the lower-energy band arises from an Fe(II)→HLB MLCT transition in which the acceptor molecular orbital (MO) has primarily acetyl(C=O*) character. The higher-energy feature is assigned to a HLB-based π→π* transition (see ESI† for details concerning the TD-DFT calculations). While 1A does not exhibit visible-region MLCT transitions like 1B and 1C, a very intense ligand-based π→π* band is observed with λmax = 369 nm (Figure 5). In contrast, complexes containing ligands derived from methoxy- and alkyl-substituted HQs (H2LD–F) have pale yellow colors due to broad UV absorption features that tail into the visible region (Figures 5 and S1, ESI†).
Figure 5.

Electronic absorption spectra of complexes 1A–D and 1F in CH2Cl2 at room temperature.
1H NMR spectra of complexes 1A–1D and 1F in CD2Cl2 display paramagnetically-shifted signals characteristic of high-spin monoiron(II) centers (Figure S2, ESI†). Peaks arising from the Ph2Tp supporting ligand are easily assigned by comparison to earlier literature reports (e.g., the signal from the 4-pyrzole protons consistently appears near 55 ppm).21d, 22a In each case, the resonance arising from the distal hydroxyl substituent was identified through H/D exchange with a small amount of added MeOH-d4. These peaks appear downfield with chemical shifts of 23 ± 3 ppm, although the hydroxyl proton is observed at 59 ppm in the 1D spectrum (Figure S2, ESI†). The observation of paramagnetically-shifted –OH resonances confirms that the HQate ligands do not adopt bridging positions in solution.
The electrochemical behavior of the monoiron(II) complexes 1A–D and 1F were studied by cyclic voltammetry in CH2Cl2 or THF solutions containing 100 mM [NBu4]PF6 as the supporting electrolyte. The cyclic voltammograms are displayed in Figure 6 and the results are summarized in Table 2. All redox potentials are referenced to the ferrocenium/ferrocene couple (Fc+/Fc). Complexes 1A–C display quasi-reversible one-electron oxidation waves between −290 and −30 mV that correspond to the Fe(II/III) couple. The Fe redox potential of 1A is significantly lower than those of 1B and 1C, reflecting the stronger donating ability of benzimidazolyl relative to acetyl groups. When the window is expanded to more positive potentials, both 1A and 1B exhibit a highly irreversible wave that likely corresponds to oxidation of the respective HQate ligands. The irreversible nature of the hydroquinonate-based oxidation is probably due to subsequent loss of the distal –OH proton to the surrounding medium. Notably, 1C is redox inactive are higher potentials, which is not surprising given that phenolates are intrinsically harder to oxidize than HQates.25
Figure 6.

Cyclic voltammograms of 1A–D and 1F. Data was collected in CH2Cl2 (1A–C) or THF (1D and 1F) with 100 mM (NBu4)PF6 as the supporting electrolyte and a scan rate of 100 mV/s. Each voltammogram was initiated by the anodic sweep.
Table 2.
Redox Potentials of Complexes 1A–D and 1F.a
| complex | solvent | Redox Potentials (mV vs Fc+/0) b |
|---|---|---|
|
| ||
| 1A | CH2Cl2 | E1/2 (ΔE) = −290 (110); Ep,a = +740 mV |
| 1B | CH2Cl2 | E1/2 (ΔE) = −110 (140); Ep,a = +1050 mV |
| 1C | CH2Cl2 | E1/2 (ΔE) = −30 (150) |
| 1D | THF | Ep,a = −50, +370, and +780 mV |
| 1F | THF | Ep,a = −100, +580, and +850mV |
Conditions: Solutions contained 100 mM (NBu4)PF6; scan rate of 100 mV s−1 at room temperature.
E1/2 and ΔE values are provided for (quasi)reversible processes; Ep,a values are given for irreversible oxidation events.
As shown in Figure 6, reversible electrochemical processes were not observed in the cyclic voltammograms of 1D and 1F; instead, each complex displays a weak anonic wave (Ep,a = −50 and −100 mV for 1D and 1F, respectively) that is assigned to one-electron oxidation of the Fe(II) center. The corresponding cathodic waves appear at much more negative potentials (Ep,c ~ −600 mV in both cases), suggesting an irreversible change following oxidation to Fe(III). Additional irreversible events arising from HQate-based oxidation are evident at higher potentials for 1D and 1F (Table 2; Figure 6). As expected, the potential of the first HQate-based oxidation shifts to more negative potentials as the HQate substituents become more electron-donating: E(HLD) < E(HLF) < E(HLA) < E(HLB). The ill-defined electrochemical behavior of 1D and 1F is likely a result of the greater conformational flexibility of their HQate ligands, which are not locked into a stable six-membered ring chelate like the HLA–C ligands.
Given that complexes 1A–1C display reversible Fe(II/III) redox couples, we sought to examine the corresponding ferric species, 1Xox, with spectroscopic methods. As shown in Figure 7, treatment of the Fe(II) complexes with one equivalent of a one-electron oxidant, such as acetylferrocenium or [N(C6H4Br-4)3]+, yields chromophores with broad, intense absorption features centered near 480 nm. Based on literature precedents,26 these bands are confidently assigned to HLA–C→Fe(III) LMCT transitions. The high intensities of the LMCT bands are indicative of strong Fe1-O1 covalency in the oxidized state, arising from overlap between the out-of-plane π-orbital of the phenolate ligand and the partially-occupied Fe(xy) orbital.27 EPR spectra of the oxidized species 1Aox-1Cox (Figure S3, ESI†) each reveal an intense derivative-shaped feature at g = 4.3 and a very weak peak near g = 9.4, characteristic of rhombic high-spin Fe(III) centers.
Figure 7.

Electronic absorption spectra of 1Aox-1Cox in CH2Cl2 at room temperature. The 1Xox species were obtained by treating the Fe(II) precursors with one equivalent of acetylferrocenuium (1Aox) or [N(C6H4Br-4)3]+ (1Box and 1Cox).
Conclusions
We have reported the synthesis and X-ray structure analysis of a series of monoiron(II) hydroquinonate complexes (1A–D and 1F) that represent the first crystallographically-characterized models of Fe/HQate interactions in HQDO active sites. The spectroscopic and electrochemical properties of the complexes were also described. The models employed bidentate (“tethered”) and monodentate (“untethered”) HQate ligands, since HQDOs oxidize both types of substrates. Although HQate ligands are known to bridge multiple metal centers, the tethered ligands (H2LA–D) cleanly provided 5C mononuclear complexes supported by the tridentate Ph2Tp framework. It was possible, though, to obtain the diiron(II) complex 2E by inclusion of an additional donor substituent at the 5-position of the HQate ring. Compared to the bidentate HQates, the untethered ligand, H2LF, readily adopted a bridging position between Fe(II) centers, as evident in the facile formation of 2F. Addition of one equivalent of free pyrazole (Ph2pz) to the reaction mixture, however, provided the complex 1F(Ph2pz) – the only structurally-characterized example of a monoiron(II) complex with an untethered HQate ligand reported to date. The stability of 1F(Ph2pz) is undoubtedly enhanced by an intramolecular hydrogen bond between the HQate and Ph2pz ligands (Figure 4). While crystallographic studies of substrate-bound HQDOs are not currently available, structures of ECDO:substrate complexes have revealed similar hydrogen-bonding interactions between the deprotonated O-atom of the catecholate ligand and second-sphere residues.2a, 28
Thus, 1F(Ph2pz) replicates important aspects of the enzymatic coordination environment. The results presented here provide a basis for future modeling studies of the HQDOs. As noted in the introduction, the non-innocent nature of HQate ligands is thought to play an important role in the HQDO mechanism. Indeed, the cyclic voltammograms of 1A and 1B reveal an irreversible wave that likely corresponds to HQate oxidation coupled to loss of the distal –OH proton. Detailed studies of the electron- and proton-transfer capabilities of our mono- and dinuclear HQate complexes are currently underway with the aim of generating novel Fe benzo(semi)quinone species. In addition, we will perform O2 reactivity studies to determine whether these excellent structural models also behave as functional models of the HQDOs.
Experimental Section
Materials and Methods
Unless otherwise noted, all reagents and solvents were purchased from commercial sources and used as received. Acetonitrile, dichloromethane, and tetrahydrofuran were purified and dried using a Vacuum Atmospheres solvent purification system. The synthesis and handling of air-sensitive materials were performed under inert atmosphere using a Vacuum Atmospheres Omni-Lab glovebox. The ligands K(Ph2Tp)29 and 2,5-dimethoxyhydroquinone (H2LE)30 were prepared according to literature procedures.
Elemental analyses were performed at Midwest Microlab, LLC in Indianapolis, IN. UV-vis absorption spectra were obtained with an Agilent 8453 diode array spectrometer equipped with a cryostat from Unisoku Scientific Instruments (Osaka, Japan) for temperature control. Fourier-transform infrared (FTIR) spectra of solid samples were measured with a Thermo Scientific Nicolet iS5 FTIR spectrometer equipped with the iD3 attenuated total reflectance accessory. 1H spectra were collected at room temperature with a Varian 400 MHz spectrometer. EPR experiments were performed using a Bruker ELEXSYS E600 equipped with an ER4415DM cavity resonating at 9.63 GHz, an Oxford Instruments ITC503 temperature controller, and an ESR-900 He flow cryostat. Electrochemical measurements were conducted in the glovebox with an epsilon EC potentiostat (iBAS) at a scan rate of 100 mV/s with 100 mM (NBu4)PF6. A three-electrode cell containing a Ag/AgCl reference electrode, a platinum auxiliary electrode, and a glassy carbon working electrode was employed for cyclic voltammetric (CV) measurements. Under these conditions, the ferrocene/ferrocenium (Fc+/0) couple has an E1/2 value of +0.52 V in CH2Cl2 and +0.61 V in THF.
2-(1-methyl-1H-benzimidazol-2-yl)hydroquinone (H2LA)
To 2,5-dihydroxybenzealdehyde (690 mg, 5.0 mmol) dissolved in 25 mL of ethanol, N-methyl-1,2-benzenediamine (0.56 mL, 5.0 mmol) in 15 mL of ethanol was added dropwise over the course of 30 minutes. The mixture was then stirred at 50 °C for two days. After cooling, 30 mL of H2O was added and the mixture was placed overnight in a freezer. The resulting brown precipitate was filtered and dried under vacuum to give the product (0.79 g, 66%). Anal. Calcd for C14H12N2O2 (MW = 240.26 g mol−1): C, 69.99; H, 5.03; N, 11.66. Found: C, 69.84; H, 5.15; N, 11.70. 1H NMR (δ, DMSO): 3.81 (s, 3H, NCH3), 6.83 (m, 2H), 7.02 (d, 1 H), 7.27 (m, 2H), 7.63 (m, 2H), 9.10 (br s, 1H, –OH), 10.43 (br s, 1H, –OH). 13C{1H} NMR (δ, DMSO): 31.6, 110.4, 116.0, 116.1 117.2, 118.5, 118.6, 121.9, 122.4, 135.8, 141.5, 149.1, 149.6, 151.9.
[Fe(Ph2Tp)(HLA)] (1A)
2-(1-methyl-1H-benzimidazol-2-yl)hydroquinone (H2LA) (120 mg, 0.50 mmol) was deprotonated by reaction with one equivalent of NaOMe in 10 mL of MeCN. To this solution was added FeCl2 (64.2 mg, 0.50 mmol) and K(Ph2Tp) (350 mg, 0.49 mmol). The mixture was stirred for overnight and the solvent removed under vacuum to give a yellow-brown solid. The crude product was dissolved in CH2Cl2 and filtered; the resulting solution yielded yellow crystals suitable for crystallographic analysis after standing for several days (0.37 g, 77%). Anal. Calcd for C59H45BFeN8O2 (MW = 964.70 g mol−1): C, 73.56; H, 4.70; N, 11.62. Found: C, 73.18; H, 4.87; N, 11.72. UV-vis [λmax, nm (ε, M−1cm−1) in CH2Cl2]: 369 (10,100). FTIR (cm−1, solution): 3592 (OH), 3047, 2988, 2901, 2611 (BH), 1543, 1484, 1415, 1332, 1243, 1171, 1070, 1007, 963, 914, 818, 760, 692.
[Fe(Ph2Tp)(HLB)] (1B)
Under an inert atmosphere, 182 mg (1.20 mmol) of 2′,5′-dihydroxyacetophenone (H2LB) was deprotonated by mixing with one equivalent of NaOMe in THF for 30 minutes, after which the solvent was removed to yield the Na(HLB) salt as a white solid. To this compound was added anhydrous FeCl2 (146 mg, 1.15 mmol) and K(Ph2Tp) (815 mg, 1.15 mmol) in 15 ml of MeCN. After stirring the reaction mixture overnight, the resulting solid was collected by vacuum filtration, dried, and redissolved in CH2Cl2. Layering with pentane provided reddish brown crystals suitable for X-ray diffraction (0.26 g, 26%). Anal. Calcd for C53H41BFeN6O3 (MW = 876.59 g mol−1): C, 72.62; H, 4.71; N, 9.59. Found: C, 72.49; H, 4.79; N, 9.73. UV-vis [λmax, nm (ε, M−1cm−1) in CH2Cl2]: 394 (5410), 485 (780), 527 (690). FTIR (cm−1, solid): 3559 (OH), 3058, 2608 (BH), 1604 (COacetyl), 1547, 1475, 1462, 1430, 1411, 1359, 1340, 1327, 1299, 1197, 1164, 1062, 1006, 965, 917, 810, 759, 693.
[Fe(Ph2Tp)(HLC)] (1C)
The method of preparation was similar to the one described for 1B, except that 2′-hydroxyacetophenone (H2LC) was substituted for H2LB. Orange crystals were obtained by layering a concentrated CH2Cl2 solution with MeCN. Yield = 24 %. Anal. Calcd for C53H41BFeN6O2 (MW = 860.59 g mol−1): C, 73.97; H, 4.80; N, 9.77. Found: C, 74.15; H, 4.92; N, 9.83. UV-Vis [λmax, nm (ε, M−1cm−1) in CH2Cl2]: 368 (5950), 441 (540), 485 (570). FTIR (cm−1, solid): 3060, 2618 (BH), 1613 (COacetyl), 1529, 1479, 1463, 1432, 1414, 1361, 1346, 1331, 1225, 1167, 1063, 1010, 966, 912, 863, 804, 753, 692.
[Fe(Ph2Tp)(HLD)] (1D)
This compound was prepared via two methods. Method A: 2-methoxyhydroquinone (H2LD, 151 mg, 1.1 mmol) and triethylamine (1.1 mmol) were stirred in MeCN, followed by addition of K(Ph2Tp) (710 mg, 1.0 mmol) and Fe(OTf)2 (372 mg, 1.05 mmol) dissolved in CH2Cl2 and MeCN, respectively. The mixture was stirred overnight, filtered, and the solvent removed under vacuum. The resulting solid was washed multiple times with MeCN to remove triflate salts and other impurities, then dried again. The solid was dissolved in CH2Cl2 and layered with hexane to yield a yellow crystalline powder (0.28 g, 33%). Anal. Calcd for C52H41BFeN6O3 (MW = 864.58 g mol−1): C, 72.24; H, 4.78; N 9.72. Found: C, 69.69; H, 5.65; N 10.63 (the discrepancies indicate the presence of small amounts of impurities). UV-Vis [λmax, nm (ε, M−1cm−1) in CH2Cl2]: 383 (1490). FTIR (cm−1, solid): 3563 (OH), 3056, 2931, 2615 (BH), 1543, 1495, 1477, 1461, 1410, 1357, 1305, 1260, 1226, 1164, 1060, 1008, 913, 818, 754, 690. Method B: Equimolar amounts of the four reagents – Fe(OTf)2, K(Ph2Tp), H2LD, and NEt3 – were mixed in CH2Cl2 and stirred overnight. The solution was filtered and the solvent removed under vacuum. The solid was taken up in CH2Cl2 and layered with pentane to yield yellow crystals suitable for X-ray diffraction.
[Fe(Ph2Tp)(HLF)(Ph2pz)] (1F)
3,5-diphenylpyrazole (236 mg, 1.04 mmol), K(Ph2Tp) (714 mg, 1.01 mmol), and 2,6-dimethylhydroquinone (H2LF, 164 mg, 1.19 mmol) were dissolved in a 3:1 mixture of DCE:MeCN. To this solution was added FeCl2 (129 mg, 1.02 mmol) in MeCN and NaOMe (0.23 mL of 4.37 M MeOH solution, 1.00 mmol). The reaction was stirred overnight. The solvent was evaporated under vacuum to give a pale orange solid. The crude solid was taken up into DCE and filtered, providing bright yellow solution. Yellow crystals were obtained by layering this DCE solution with pentane. Anal. Calcd for C68H55BFeN8O2 (MW = 1082.88 g mol−1): C, 75.42; H, 5.12; N 10.35. Found: C, 75.22; H, 5.00; N 10.21. UV-Vis [λmax, nm (ε, M−1cm−1) in CH2Cl2]: 374 (2530). FTIR (cm−1, solid): 3355 (OH), 3060, 3038, 2912, 2631 (BH), 1598, 1543, 1477, 1465, 1430, 1410, 1339, 1306, 1212, 1165, 1062, 1004, 967, 913, 851, 810, 754, 688.
[Fe2(Ph2Tp)2(μ-LE)] (2E)
2,5-dimethoxyhydroquinone (H2LE, 91 mg, 0.53 mmol) was first deprotonated by treatment with two equivalents of NaOMe in THF. After removal of the solvent, the resulting white solid Na2(LE) was mixed with FeCl2 (131 mg, 1.03 mmol) and K(Ph2Tp) (715 mg, 1.01 mmol) in MeCN, and the solution was stirred overnight. After removal of the solvent under vacuum, the yellow solid was taken up CH2Cl2 and the solution filtered to remove unwanted salts. Vapor diffusion of Et2O into this CH2Cl2 solution provided yellow-orange needles suitable for X-ray crystallography (0.11 g, 13%). Anal. Calcd for C98H76B2Fe2N12O4 (MW = 1619.07 g mol−1): C, 72.70; H, 4.73; N 10.38. Found: C, 72.45; H, 4.67; N, 10.36. UV-Vis [λmax, nm (ε, M−1cm−1) in CH2Cl2]: 317 (9300), 370 (sh), 444 (sh). FTIR (cm−1, solid): 3058, 2926, 2614 (BH), 1541, 1478, 1465, 1438, 1407, 1359, 1260, 1221, 1194, 1167, 1154, 1061, 1008, 888, 802, 756, 690.
[Fe2(Ph2Tp)2(μ-LF)(MeCN)] [2F(MeCN)]
Anhydrous FeCl2 (130 mg, 1.02 mmol) and K(Ph2Tp) (715 mg, 1.01 mmol) were combined with 0.5 equivalent of 2,6-dimethylhydroquinone (H2LF, 70.0 mg, 0.51 mmol) in 10 mL of MeCN. To this mixture was added 0.23 mL of 4.37 M solution of NaOMe (1.01 mmol). The reaction was stirred overnight, and the solvent removed under vacuum. The resulting solid was dissolved in DCE, filtered, and then layered with MeCN to provide reddish-brown needles (0.21 g, 26%) suitable for crystallographic analysis. The X-ray structure revealed two uncoordinated DCE molecules in the asymmetric unit, and elemental analysis suggest that a small amount of solvent (~0.8 equiv.) remains even after drying. Anal. Calcd for C100H79B2Fe2N13O2•0.8DCE (MW = 1707.27 g mol−1): C, 71.48; H, 4.85; N 10.67. Found: C, 71.47; H, 4.77; N, 10.37. UV-Vis [λmax, nm (ε, M−1cm−1) in CH2Cl2]: 288 (11,500), 377 (3600). FTIR (cm−1, solid): 3052, 2925, 2608 (BH), 1542, 1465, 1477, 1431, 1412, 1358, 1242, 1162, 1065, 1029, 1009, 969, 916, 847, 810.
Crystallographic Studies
Each complex was characterized with X-ray crystallography; details concerning the data collection and analysis are summarized in Table 3. The X-ray diffraction data were collected at 100 K with an Oxford Diffraction SuperNova kappa-diffractometer equipped with dual microfocus Cu/Mo X-ray sources, X-ray mirror optics, Atlas CCD detector and a low-temperature Cryojet device. The data were processed with CrysAlisPro program package (Oxford Diffraction Ltd., 2010) typically using a numerical Gaussian absorption correction (based on the real shape of the crystal) followed by an empirical multi-scan correction using SCALE3 ABSPACK routine. The structures were solved using the SHELXS program and refined with the SHELXL program31 within the Olex2 crystallographic package.32 All computations were performed on an Intel PC computer with Windows 7 OS. Some structures contain disorder that was detected in difference Fourier syntheses of electron density and accounted for using capabilities of the SHELX package. In most cases, hydrogen atoms were localized in difference syntheses of electron density but were refined using appropriate geometric restrictions on the corresponding bond lengths and bond angles within a riding/rotating model (torsion angles of methyl hydrogens were optimized to better fit the residual electron density).
Supplementary Material
Acknowledgments
We thank Dr. Brian Bennett for allowing us to perform EPR experiments at the National Biomedical EPR Center (supported by NIH P41 grant EB001980). A.T.F. also thanks Marquette University and the National Science Foundation (CAREER CHE-1056845) for generous financial support.
Notes and References
- 1.(a) Gibson DT, Parales RE. Curr Opin Biotechnol. 2000;11:236–243. doi: 10.1016/s0958-1669(00)00090-2. [DOI] [PubMed] [Google Scholar]; (b) Parales R, Resnick SM. In: Biodegradation and Bioremediation. Singh A, Ward OP, editors. Springer; Heidelberg: 2004. pp. 175–196. [Google Scholar]; (c) Furukawa K. Curr Opin Biotechnol. 2000;11:244–249. doi: 10.1016/s0958-1669(00)00091-4. [DOI] [PubMed] [Google Scholar]
- 2.(a) Vaillancourt FH, Bolin JT, Eltis LD. Crit Rev Biochem Mol Biol. 2006;41:241–267. doi: 10.1080/10409230600817422. [DOI] [PubMed] [Google Scholar]; (b) Costas M, Mehn MP, Jensen MP, Que L., Jr Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 3.Koehntop KD, Emerson JP, Que L., Jr J Biol Inorg Chem. 2005;10:87–93. doi: 10.1007/s00775-005-0624-x. [DOI] [PubMed] [Google Scholar]
- 4.(a) Straganz GD, Nidetzky B. Chembiochem. 2006;7:1536–1548. doi: 10.1002/cbic.200600152. [DOI] [PubMed] [Google Scholar]; (b) Diebold AR, Neidig ML, Moran GR, Straganz GD, Solomon EI. Biochemistry. 2010;49:6945–6952. doi: 10.1021/bi100892w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Lipscomb JD. Curr Opin Struct Biol. 2008;18:644–649. doi: 10.1016/j.sbi.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bugg TDH, Ramaswamy S. Curr Opin Chem Biol. 2008;12:134–140. doi: 10.1016/j.cbpa.2007.12.007. [DOI] [PubMed] [Google Scholar]; (c) Bruijnincx PCA, van Koten G, Gebbink RJMK. Chem Soc Rev. 2008;37:2716–2744. doi: 10.1039/b707179p. [DOI] [PubMed] [Google Scholar]; (d) Siegbahn PEM, Haeffner F. J Am Chem Soc. 2004;126:8919–8932. doi: 10.1021/ja0493805. [DOI] [PubMed] [Google Scholar]; (e) Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Chem Rev. 2000;100:235–349. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 6.Lipscomb JD, Orville AM. In: Met Ions Biol Syst. Sigel H, Sigel A, editors. Vol. 28. Marcel Dekker; New York: 1992. pp. 243–298. [Google Scholar]
- 7.(a) Li XW, Guo M, Fan J, Tang WY, Wang DQ, Ge HH, Rong H, Teng MK, Niu LW, Liu Q, Hao Q. Protein Sci. 2006;15:761–773. doi: 10.1110/ps.051967906. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang Y, Colabroy KL, Begley TP, Ealick SE. Biochemistry. 2005;44:7632–7643. doi: 10.1021/bi047353l. [DOI] [PubMed] [Google Scholar]; (c) Lendenmann U, Spain JC. J Bacteriol. 1996;178:6227–6232. doi: 10.1128/jb.178.21.6227-6232.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Matera I, Ferraroni M, Burger S, Scozzafava A, Stolz A, Briganti F. J Mol Biol. 2008;380:856–868. doi: 10.1016/j.jmb.2008.05.041. [DOI] [PubMed] [Google Scholar]; (b) Hintner JP, Lechner C, Riegert U, Kuhm AE, Storm T, Reemtsma T, Stolz A. J Bacteriol. 2001;183:6936–6942. doi: 10.1128/JB.183.23.6936-6942.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hintner JP, Remtsma T, Stolz A. J Biol Chem. 2004;279:37250–37260. doi: 10.1074/jbc.M313500200. [DOI] [PubMed] [Google Scholar]
- 9.(a) Harpel MR, Lipscomb JD. J Biol Chem. 1990;265:22187–22196. [PubMed] [Google Scholar]; (b) Chen J, Li W, Wang MZ, Zhu GY, Liu DQ, Sun F, Hao N, Li XM, Rao ZH, Zhang XC. Protein Sci. 2008;17:1362–1373. doi: 10.1110/ps.035881.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Veldhuizen EJA, Vaillancourt FH, Whiting CJ, Hsiao MMY, Gingras G, Xiao YF, Tanguay RM, Boukouvalas J, Eltis LD. Biochem J. 2005;386:305–314. doi: 10.1042/BJ20041370. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Titus GP, Mueller HA, Burgner J, Córdoba SRd, Penalva MA, Timm DE. Nat Struct Biol. 2000;7:542–546. doi: 10.1038/76756. [DOI] [PubMed] [Google Scholar]
- 11.Borowski T, Georgiev V, Siegbahn PEM. J Am Chem Soc. 2005;127:17303–17314. doi: 10.1021/ja054433j. [DOI] [PubMed] [Google Scholar]
- 12.(a) Machonkin TE, Doerner AE. Biochemistry. 2011;50:8899–8913. doi: 10.1021/bi200855m. [DOI] [PubMed] [Google Scholar]; (b) Machonkin TE, Holland PL, Smith KN, Liberman JS, Dinescu A, Cundari TR, Rocks SS. J Biol Inorg Chem. 2010;15:291–301. doi: 10.1007/s00775-009-0602-9. [DOI] [PubMed] [Google Scholar]; (c) Xu L, Resing K, Lawson SL, Babbitt PC, Copley SD. Biochemistry. 1999;38:7659–7669. doi: 10.1021/bi990103y. [DOI] [PubMed] [Google Scholar]; (d) Ohtsubo Y, Miyauchi K, Kanda K, Hatta T, Kiyohara H, Senda T, Nagata Y, Mitsui Y, Takagi M. FEBS Lett. 1999;459:395–398. doi: 10.1016/s0014-5793(99)01305-8. [DOI] [PubMed] [Google Scholar]
- 13.(a) Nagata Y, Endo R, Ito M, Ohtsubo Y, Tsuda M. Appl Microbiol Biotechnol. 2007;76:741–752. doi: 10.1007/s00253-007-1066-x. [DOI] [PubMed] [Google Scholar]; (b) Miyauchi K, Adachi Y, Nagata Y, Takagi M. J Bacteriol. 1999;181:6712–6719. doi: 10.1128/jb.181.21.6712-6719.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yin Y, Zhou NY. Curr Microbiol. 2010;61:471–476. doi: 10.1007/s00284-010-9640-3. [DOI] [PubMed] [Google Scholar]
- 15.Christian GJ, Ye SF, Neese F. Chem Sci. 2012;3:1600–1611. [Google Scholar]
- 16.(a) Kovaleva EG, Lipscomb JD. Science. 2007;316:453–457. doi: 10.1126/science.1134697. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bugg TDH, Lin G. Chem Commun. 2001;11:941–953. [Google Scholar]
- 17.Becker JM, Barker J, Clarkson GJ, van Gorkum R, Johal GK, Walton RI, Scott P. Dalton Trans. 2010;39:2309–2326. doi: 10.1039/b905706d. [DOI] [PubMed] [Google Scholar]
- 18.(a) Heistand RH, II, Roe AL, Que L., Jr Inorg Chem. 1982;21:676–681. [Google Scholar]; (b) Maroney MJ, Day RO, Psyris T, Fleury LM, Whitehead JP. Inorg Chem. 1989;28:173–175. [Google Scholar]
- 19.Rheingold AL, Miller J. Private communication to Cambridge Structural Database. 2003. [Google Scholar]
- 20.Rocks SS, Brennessel WW, Machonkin TE, Holland PL. Inorg Chem. 2010;49:10914–10929. doi: 10.1021/ic101377u. [DOI] [PubMed] [Google Scholar]
- 21.(a) Paria S, Que L, Paine TK. Angew Chem Intl Ed. 2011;50:11129–11132. doi: 10.1002/anie.201103971. [DOI] [PubMed] [Google Scholar]; (b) Mukherjee A, Cranswick MA, Chakrabarti M, Paine TK, Fujisawa K, Munck E, Que L. Inorg Chem. 2010;49:3618–3628. doi: 10.1021/ic901891n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bruijnincx PCA, Lutz M, Spek AL, Hagen WR, Weckhuysen BM, vanKoten G, Gebbink RJMK. J Am Chem Soc. 2007;129:2275–2286. doi: 10.1021/ja064816x. [DOI] [PubMed] [Google Scholar]; (d) Mehn MP, Fujisawa K, Hegg EL, Que L., Jr J Am Chem Soc. 2003;125:7828–7842. doi: 10.1021/ja028867f. [DOI] [PubMed] [Google Scholar]; (e) Ogihara T, Hikichi S, Akita M, Morooka Y. Inorg Chem. 1998;37:2614–2615. doi: 10.1021/ic9716215. [DOI] [PubMed] [Google Scholar]
- 22.(a) Park H, Baus JS, Lindeman SV, Fiedler AT. Inorg Chem. 2011;50:11978–11989. doi: 10.1021/ic201115s. [DOI] [PubMed] [Google Scholar]; (b) Siewert I, Limberg C. Angew Chem Intl Ed. 2008;47:7953–7956. doi: 10.1002/anie.200802955. [DOI] [PubMed] [Google Scholar]
- 23.(a) Paria S, Halder P, Chakraborty B, Paine TK. Indian J Chem, Sect A. 2011;50:420–426. [Google Scholar]; (b) Fujisawa K, Tada N, Nishida Y, Miyashita Y, Okamoto K. Inorg Chem Commun. 2008;11:381–384. [Google Scholar]
- 24.Addison AW, Rao TN, Reedijk J, Vanrijn J, Verschoor GC. J Chem Soc, Dalton Trans. 1984:1349–1356. [Google Scholar]
- 25.Warren JJ, Tronic TA, Mayer JM. Chem Rev. 2010;110:6961–7001. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.(a) Ito M, Amagai H, Fukui H, Kitajima N, MoroOka Y. Bull Chem Soc Jpn. 1996;69:1937–1945. [Google Scholar]; (b) Pyrz JW, Roe AL, Stern LJ, Que L., Jr J Am Chem Soc. 1985;107:614–620. [Google Scholar]
- 27.Davis MI, Orville AM, Neese F, Zaleski JM, Lipscomb JD, Solomon EI. J Am Chem Soc. 2002;124:602–614. doi: 10.1021/ja011945z. [DOI] [PubMed] [Google Scholar]
- 28.(a) Sato N, Uragami Y, Nishizaki T, Takahashi Y, Sazaki G, Sugimoto K, Nonaka T, Masai E, Fukuda M, Senda T. J Mol Biol. 2002;321:621–636. doi: 10.1016/s0022-2836(02)00673-3. [DOI] [PubMed] [Google Scholar]; (b) Vaillancourt FH, Barbosa CJ, Spiro TG, Bolin JT, Blades MW, Turner RFB, Eltis LD. J Am Chem Soc. 2002;124:2485–2496. doi: 10.1021/ja0174682. [DOI] [PubMed] [Google Scholar]
- 29.Kitajima N, Fujisawa L, Fujimoto C, Moro-oka Y, Hashimoto S, Kitagawa T, Toriumi K, Tatsumi K, Nakamura A. J Am Chem Soc. 1992;114:1277–1291. [Google Scholar]
- 30.Hanss D, Walther ME, Wenger OS. Chem Commun. 2010;46:7034–7036. doi: 10.1039/c0cc01591a. [DOI] [PubMed] [Google Scholar]
- 31.Sheldrick GM. Acta Crystallogr Sect A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 32.Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H. J Appl Crystallogr. 2009;42:339–341. doi: 10.1107/S0021889811041161. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.