

Abstract
MicroRNAs (miRs) are small noncoding RNAs that typically act as regulators of gene expression by base pairing with the 3′ UTR of messenger RNAs (mRNAs) and either repressing their translation or initiating degradation. As of this writing over 24,500 distinct miRs have been identified, but the functions of the vast majority of these remain undescribed. This paper represents a summary of our in depth analysis of the genomic origins of miR loci, detailing the formation of 1,213 of the 7,321 recently identified miRs and thereby bringing the total number of miR loci with defined molecular origin to 3,605. Interestingly, our analyses also identify evidence for a second, novel mechanism of miR locus generation through describing the formation of 273 miR loci from mutations to other forms of noncoding RNAs. Importantly, several independent investigations of the genomic origins of miR loci have now supported the hypothesis that miR hairpins are formed by the adjacent genomic insertion of two complementary transposable elements (TEs) into opposing strands. While our results agree that subsequent transcription over such TE interfaces leads to the formation of the majority of functional miR loci, we now also find evidence suggesting that a subset of miR loci were actually formed by an alternative mechanism—point mutations in other structurally complex, noncoding RNAs (e.g., tRNAs and snoRNAs).
Keywords: LINE, microRNA, miR, miRNA, noncoding RNA, repetitive, retrotransposon, SINE, transposable, transposon
Introduction
MicroRNAs (miRs) are short (approximately 20 nucleotide) noncoding RNAs (ncRNAs) involved in the regulation of gene expression.1 Functioning much like small interfering RNAs (siRNAs), miRs bind to complimentary messenger RNA (mRNA) resulting in the repression of translation.2,3 MiRs are initially transcribed in the nucleus as portions of larger precursor molecules called pri-miRs. These initial transcripts are processed by Drosha to generate ~70 nucleotide stem loops (pre-miRs) that are exported into the cytosol where Dicer ultimately trims these dsRNA pre-miRs into functional single stranded miRs.2,4 Following this, complementary sequence association between a miR and mRNA target leads to inhibited translation of the mRNA molecule.2
To date, our principal obstacle to deciphering miR function has proven to be our inability to accurately predict miR association with target mRNAs. This is principally the result of the capacity of miRs to associate with mRNAs bearing imperfect sequence complementarities.5 Although accurately determining which mRNAs a miR targets has been an extremely active area of research, no universal model of target prediction has been widely adopted. Unlike siRNAs which associate with targets through perfect complementarity, miR association requires as little as seven complementary nucleotides.6 Usually located at the 5′ end of a miR, these seven complementary nucleotides are known as “seeds” and their complement in target mRNAs are known as “seed matches.”7 This complementarity between seeds and seed matches is the core requirement for the majority of currently utilized miR target prediction algorithms after which programs largely differ in how much they weight target sequence conservation across species and how they score additional complementarity between the mRNA and the remainder of the miR.8-15
While the genomic origins for the majority of miRs and their mRNA target sites remain undescribed, a relationship between miRs and TEs has long been suggested and the random nature of TE colonization has caused several groups to adopt a similar mechanism for the establishment of functional miRs.16-20 As approximately half of the human genome is composed of TEs21 and as much as 80% of plant genomes18 are comprised of TE sequences, widespread TE colonization is a hallmark of the genomes of higher, more complex organisms. Although long thought to be detrimental to genomic integrity (as TE insertions into coding sequences are usually detrimental), strikingly over 50% of metazoan transcripts bear at least some amount of TE sequence (typically in their 3′ or 5′UTRs).22,23 Smalheiser and Torvik,20 however, were the first to describe a genomic origin for miR loci describing a novel advantage associated with TE genomic colonization: the formation of miRs from the adjacent insertions of two related TEs (Fig. 1). Now confirmed in several subsequent analyses,16-20,25-27 their model indicates that transcription across neighboring TE insertions on opposite strands, followed by DICER processing and RISC incorporation, was initially responsible for the formation of the majority of functional miR loci. In addition, a transposable element-based miR origin suggests an additionally advantageous role for continued genome colonization—the miRs formed by this mechanism would likely be able to repress any mRNAs bearing sequences obtained from that miR’s progenitor TE.2 This suggests that a network of target mRNAs bearing a common TE is likely formed prior to the creation of the miR locus that ultimately regulates it (Fig. 2). While there are several additional implications of miRs originating from TE sequences (e.g., determining transcriptional regulation16), our recently developed miR target prediction algorithm (OrbId: Origin-based identification of microRNA targets19) suggests that a particularly advantageous utility for this information is the refinement of target prediction through restricting putative targets to mRNAs which contain the TE giving rise to a particular miR. Therefore, in light of our having previously defined the genomic events behind the formation of over 2,300 distinct miRs24 and having successfully developed a novel miR target prediction algorithm utilizing this information to accurately predict mRNAs targeted by miRs clearly formed from TE sequences,19 this report now details our continuing analysis of this topic - a comprehensive update examining the over 7,000 novel miRs described since our initial study.28 To our surprise, in addition to characterizing over 1,000 new miR molecular origins from TEs, we also find evidence suggesting that a subset of miRs actually arose from a distinct, previously undescribed mechanism - RNA structural alteration resulting from point mutations to noncoding RNAs such as tRNAs and snoRNAs. In contrast to miRs formed from TEs (which likely target mRNAs bearing their progenitor TE sequences), it is tempting to speculate that instead of regulating mRNA expression like the majority of characterized miRs, this novel subset of noncoding RNA-derived miRs may actually be charged with regulating the activity of their progenitor noncoding RNAs.
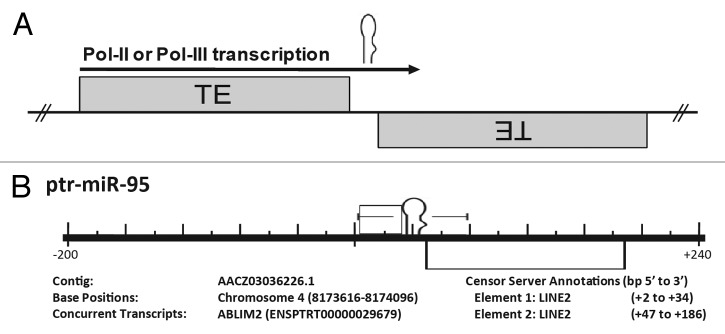
Figure 1. Formation of miRs typically occurs when two complementary TEs inserted into opposing strands are then subsequently transcribed. (A) Cartoon representation illustrating the proposed origin of many miRs. A miR hairpin is depicted just above an arrow indicating read through transcription from a positive strand transposable element (TE) into an adjacent negative strand TE. Transcriptional read through would result in an imperfect RNA hairpin being produced which could potentially be recognized and processed by the RNAi machinery with each stem corresponding to the terminal nucleotides of the contributing TEs. (B) Pan troglodytes miR-95 alignment to the RepBase data set. All repetitive elements taken from RepBase (indicated by open rectangles) occurring within 200 bp (5′ and 3′) have been included in the scale diagram. The repetitive element annotations23 are described immediately beneath the diagram as “Element 1, Element 2, etc…” as they occur 5′ to 3′. “Base Positions” refers to base pair location in the genome occupied by the miR hairpin (according to the current Ensembl assembly; www.ensembl.org). All loci have been shown with respect to the positive strand and the orientation of internal repetitive elements illustrated by their relative position above (5′ to 3′) or below (3′ to 5′) the center line. Repetitive element base pair positions are relative to the distance (±) from the first nucleotide of the pre-miR (as occurring on the positive strand). Figure directly adapted from reference 24.

Figure 2. Cartoon illustration of the genomic events believed to be responsible for the formation of many miRs. Origin of numerous miRs occurs when random TE insertions gives rise to a beneficial regulatory adaptation within the organism. Subsequent transcription of this TE interface by RNA polymerase followed by RISC processing can lead to miR establishment if the resulting small RNA confers some advantage in gene expression.
Results
To determine if any of the 7,321 miRs identified since the completion of our initial analysis24 were formed from TE sequences, we screened these miR loci against all known repetitive elements22,29 and noncoding RNAs30 using an established BLAST-based strategy.31 Through these analyses, we were able to define the molecular events responsible for 1,213 miR origins. In agreement with previous studies,17,18,20,24-27 our results indicate that the predominant mechanism for miR formation from TE sequences occurred via the model illustrated in Figure 1. Encouragingly, we find the results of our current analysis of 7,321 miRs to be strikingly similar to our previous analysis of over 15,000 distinct miR loci. In our initial analysis of 15,176 miRs, we were able to successfully define the molecular origins of 2,392 (or 15.8%) of miR genomic loci with defining alignments averaging 82.9% identity over 85.7 bps.24 Similarly, in this analysis we were able to characterize the molecular origins of 1,213 of 7,321 (or 16.6%) miR genomic loci with defining alignments averaging 82.4% identity over 57.0 bps (Table S1).
Transposable element origins
There are three distinct categories of TEs: DNA transposons, LTR (long-terminal repeat) retrotransposons, and non-LTR retrotransposons.32-34 DNA transposons are generally flanked by simple inverted repeats and consist of at least two genes encoding the proteins necessary for making and inserting DNA copies of itself elsewhere in the genome.34 In agreement with our earlier findings, DNA transposons were responsible for the formation of the largest number of definable miR loci in our analysis (517 origins) with related satellite DNA repeats being responsible for the formation of nine additional miR loci. The next largest number of definable miR loci in our analysis corresponded to miRs formed from non-LTR retrotransposons which contain genes encoding gag and pol-like ORFs.33 In all, we were able to identify 307 distinct miR loci arising from non-LTR retrotransposon sequences with 187 of these corresponding to long interspersed repeats (LINEs) and 120 to short interspersed repeated elements (SINEs) (Table 1). Following this, we find a significant portion of miR loci defined in our analysis corresponded to related LTR retrotransposons which also encode gag and pol-like ORFs but are additionally characterized by being flanked by ~400 bp highly structured long-terminal repeats (or LTRs).32 In all, our current analyses were able to identify 236 distinct miR loci formed from LTR retrotransposon sequences.
Table 1. Summary of miR loci transposable element origins.
| Repeat type | Sequence based alignment | Familial inclusion | Total MiRs 2013 | Total MiRs 2011 | Total # of MiRs |
|---|---|---|---|---|---|
| DNA transposon | 467 | 50 | 517 | 891 | 1408 |
| LTR retrotransposon | 235 | 1 | 236 | 414 | 650 |
| Non-LTR retrotransposon | 305 | 2 | 307 | 814 | 1121 |
| -LINE | 186 | 1 | 187 | 461 | 648 |
| -SINE | 119 | 1 | 120 | 353 | 473 |
| Noncoding RNAs | 128 | 0 | 128 | 61 | 189 |
| tRNAs | 33 | 0 | 33 | 51 | 84 |
| Satellites | 6 | 3 | 9 | 137 | 146 |
| Other | 39 | 1 | 40 | 24 | 64 |
| Total | 1156 | 57 | 1213 | 2392 | 3605 |
Repeat Type, RepBase classification.22 Sequence Based Alignment, total number of unique miR origins identified through alignment to a consensus transposable element. Familial Inclusion, total number of unique miR origins determined through familial inclusion. Total MiRs 2013, total origins determined through both sequence based alignment and familial inclusion in the current study. Total MiRs 2011, total origins determined through both sequence based alignment and familial inclusion in our initial analysis.24 Total # of MiRs, total origins determined through both sequence based alignment and familial inclusion in both the current analysis and 2011 study. Others, miRs significantly aligning to characterized RNA structural elements (e.g., IRES elements) contained within the RFAM data set.35,36
Familial inclusions
In addition to sequence-based alignment, ~4.7% of the origins described in this study were defined by familial inclusion and placed into 13 distinct miR families as previously described.24 In brief, miRs were grouped into common families using accepted miRBase28 nomenclature following sequence based annotation. Following this, a common molecular origin was assigned to all of the miRs within a given family if both of the following criteria were met: 1) at least two of the miRs within a family were described as arising from the same TE and 2) at least 75% of the members with origins defined through sequence-based alignment were identified as being formed from the same TE. Utilizing this strategy we were able to additionally define 4.7% (57) of the 1,213 miR origins described in this report (Table S2). Figure 3 illustrates the utility of familial inclusion by demonstrating how the origins of six unique miR-444 loci were obtained utilizing this strategy. As our initial criteria for sequence based alignment are particularly stringent, only two miR-444 hairpins in our analysis were initially defined as arising from a common Gypsy repeat (each bearing over 80% identity to 40 base pairs of the hairpin). Figure 3 however illustrates the striking degree of sequence conservation between the 8 members of the miR-444 family strongly suggesting they share a common molecular origin—the initial formation of miR-444 in an ancestral species.

Figure 3. Alignment of microRNA-444 family. Alignment of the eight miR-444 hairpins is illustrated. Each individual hairpin sequence is shown with the associated species on the right and the miRBase28 identifier on the left. In all, six unique miR-444 hairpin origins were characterized by familial inclusion. *, indicates 100% conservation of the nucleotide. Grey shading indicates specific miR hairpins initially identified as bearing significant sequence complementarity to Gypsy repeats through sequence based alignment.
Taxon-specific miR expansions
We identified a total of 344 miR loci that likely arose from transposable elements in taxon-specific expansions.
Primates
In all we found several taxa-specific but no species-specific expansions during this analysis with all taxonomic expansions involving at least three different primate species. Analysis of taxonomic expansions in primate groups revealed expansions in the miR-1260, miR-1273, miR-151, miR-3154, miR-378, miR-4536, miR-548, miR-6127, and miR-6130 families. These include a total of 45 human (Homo sapiens), 10 Western gorilla (Gorilla gorilla), 10 Bornean orangutan (Pongo pygmaeus), 8 common chimpanzee (Pan troglodytes), and 6 crab-eating macaque (Macaca fascicularis) loci defined (Table S1). The expansion of Mariner/Tc1 derived miR-548 in primates was the largest expansion identified in all of the animal kingdom with a total of 47 loci.
Fish
We identified two species-specific taxonomic expansions, one in Danio rerio (zebrafish) and one in Oryzias latipes (Japanese killifish). Our analyses identify 12 distinct miR-812 loci in the Japanese killifish genome arising from a variety of sources. The three distinct miR-2190 in the zebrafish, however, were found to have arisen from Atlantys-2-I-OS Gypsy Oryza elements (Table S1).
Mice/Rat
A total of five taxonomic expansions were found in the mouse/rat grouping, with a total of 42 distinct hits. Our sequenced base alignments for the mouse (Mus musculus) show that the nine miR-467 loci were formed from TSEEEEBEII Mariner/Tc 1 elements, and the ten miR-669 loci was formed from CR1-18-HM CR1 Hydra elements. Four possible origins for MIR-466 were also uncovered for Mus musculus and two for the rat Ratticus norvicus (Table S1).
Plants
Our analysis was able to define 201 loci across 31 distinct miR families in plant genomes. The largest taxonomic expansion identified in plants was for miR-156. There were 19 definitions across 13 species. The miR-5565 and miR-5568 families were populated solely by Sorghum bicolor. Thirteen definitions were found across these two families, with nine definitions corresponding to Helitron-N11-SBi Helitron Sorghum elements and four corresponding to STOWAWAY22-SB DNA elements. Twenty-nine definitions for Medicago truncatula were found across six miR families (miR-5298, miR-5291, miR-5272, miR-5741, miR-2592, and miR-2590). ShaMUDRAV2-MT MuDR Medicago, RF00028; Intron-gpI; AY098639.1/6-303, and MtPH-M-I-Ia Harbinger Medicago elements were common origins across these families (Table S1).
Other
Four loci from the miR-2284 family in the bull (Bos taurus) genome were identified and three loci from the miR-3015 family in the pea aphid (Acrthosiphon pisum) genome apparently arising from DNA3-4-AP DNA elements (Table S1).
Non-transposable element origins
Although ~87% of our annotations identified through sequence-alignment based strategies clearly indicated that TE genomic insertions led to the formation of these miRs, we also identified 161 miRs of the 7,321 miRs in this analysis with striking sequence similarity to known noncoding RNA sequences (Table S1; Table 1). In light of this we, we also elected to revisit our initial analysis of miR genomic origins24 resulting in the identification of an additional 112 miR::noncoding RNA relationships which had been previously overlooked. In all, we have now identified 273 potential, non-transposable element, sequence-based origins for miRs from various forms of noncoding RNAs with 60 miRs likely arising from snoRNAs, 16 from scaRNAs, 41 from rRNAs, 84 from tRNAs, 11 from snRNAs, 38 from various ribozymes and 23 from other forms of noncoding RNAs including antisense RNAs, long noncoding RNAs, tmRNAs and sRNAs (Fig. 4; Table 1). RNA structural analyses37 suggest these miR loci may have been formed through mutations that created substrates which could be processed by DICER (Fig. 5; Fig. S1). Whether the function of these miRs is 3′UTR mRNA binding to regulate protein translation or instead the modulation of complex secondary structures remains to be determined. In either event these findings suggest an entirely novel mechanism for microRNA locus generation.
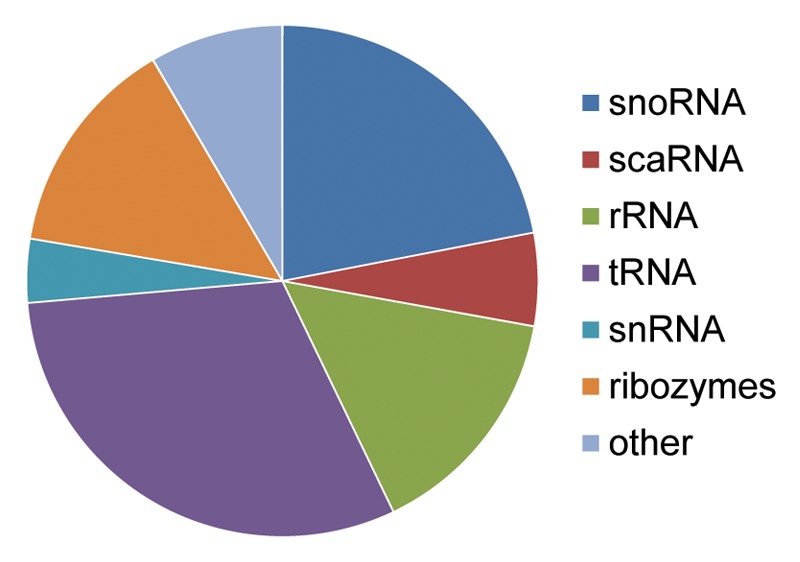
Figure 4. Depiction of various noncoding RNAs where distinct mutations resulted in microRNA formation. The distribution of 273 noncoding RNA miR alignments indicating non-transposable element origins identified in our analyses are depicted. In all, 60 corresponded to snoRNAs, 16 to scaRNAs, 41 to rRNAs, 84 to tRNAs, 11 to snRNAs, 38 to various ribozymes and 23 to other forms of noncoding RNAs such as antisense RNAs, long noncoding RNAs, tmRNAs, and sRNAs.

Figure 5. Formation of a miR through tRNA mutation. (A) Structural diagram illustrating the potential effects of a single point mutation to tRNA secondary structure. The most thermodynamically stable conformations of two tRNA sequences are shown. The two sequences resulting in the distinct structural conformations are identical except that the uracil at position 50 in the endogenous tRNA shown on the left has been replaced by a cytosine at position 50 on the right. Secondary structures and thermodynamic stabilities were computed using Mfold.37 (B) Caenorhabditis elegans miR-4937 alignment to the RFAM data set. All repetitive elements and noncoding RNAs (open rectangles) occurring within 500 bp (5′ and 3′) have been included in the scale diagram as depicted in Figure 1B. While we find numerous loci arising by the mechanism depicted in Figure 1A, we find others (like miR-4937) do not. Significantly aligning to a C. elegans tRNA (tRNAArgTCG), we propose an additional mechanism (point mutation(s) resulting in an alteration of normal tRNA secondary structure gave rise to pre-miR-4937.
MicroRNA target prediction
As we have now described the TEs responsible for forming over 3,300 distinct microRNA genomic loci24 and have previously demonstrated the utility of this information in predicting human miR targets,19 we next examined origin-based target prediction in additional species. To achieve this, we selected Gallus gallus (chicken) miR-6672 (which we found was originally formed from CR1 sequences) and Sus scrufa (pig) miR-4331(which we found was originally formed from PRE1 sequences) to predict mRNA targets utilizing our OrBId methodology which requires a shared origin common to both a miR and its target.19 Importantly, this strategy requires the sites targeted by a particular miR to contain a complete miR seed match and at least 50% of sequence identity between sequences immediately flanking a mature miR (in the pre-miR) and the sequences immediately flanking a putative mRNA target site. Examples of alignments between each miR, progenitor TEs and putative targets are illustrated in Figure 6. While these results will ultimately require experimental validation, as the miR locus and proposed target sites were apparently formed from a common ancestral genomic element, we suggest the targets illustrated in Figure 6 likely depict functional interactions.
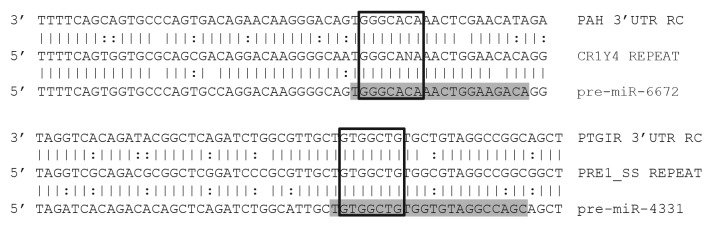
Figure 6. Alignments of miRs with predicted targets. Illustration shows a predicted miR target 3′UTRs on top, a consensus transposable element in the middle, and the corresponding miR sequence on the bottom. Mature miRs are highlighted in gray. Open boxes indicate perfect seed matches. To qualify as a 3′UTR match alignments were required to 1) contain a perfect seed match, 2) match ≥ 50% of the flanking sequence used in the target query, and 3) occur within a 3′UTR sequence aligning to a miR’s progenitor TE sequence. Vertical lines indicate base identity with the TE consensus sequence. Dotted lines indicate purine/pyrimidine conservation. PAH, Gallus gallus phenylalanine-4-hydroxylase ENSGALG00000012754. PTGIR, Sus scrufa prostaglandin I2 (prostacyclin) receptor ENSSSCG00000026602. RC, reverse complemented
Discussion
The primary objective of this work was to define the molecular origins of the 7,321 additional miR genomic loci identified since completing our initial analysis.24,28 In all, we were able to successfully define the molecular origins of 1,213 newly characterized miR genomic loci (bringing the total number of defined miR genomic origins to 3,605) and also demonstrate the utility of this information in predicting miR targets across species (Fig. 6). Importantly, we find our results largely in agreement with the growing body of evidence indicating that the majority of functional miRs were created by mobile element transposition events17,18,20,24-27 and suggest that future informatic analyses will continue to characterize additional miR-TE relationships as miR discovery remains ongoing (as evidenced by the 7,321 miR characterized in the two years since our initial analysis24,28). In addition, using our current computational methodology, we consistently find we are able to characterize the genomic origins of ~15% of miR loci. That said, we suggest this represents a marked underestimate of the percentage of miR loci formed from TEs as: 1) the high degree of stringency we required for our alignments resulted in our discarding thousands of likely origins likely representing miRs whose nonessential sequences have simply degenerated more than younger miR loci having been formed earlier in evolutionary time, and 2) our ability to identify transposable elements will also improve as the RepBase22,23,29 data set is updated with novel TEs.
Excitingly, in addition to characterizing the origins of over 1,000 new miR loci from transposable element sequences, we have now also identified 273 miRs likely formed from noncoding RNA mutations. Interestingly, despite apparently having arisen through entirely distinct mechanisms, a preliminary analysis of the genomic distributions of, and available chromatin interactome data on,38 these miRs find no significant differences between noncoding RNA-derived miRs, TE-derived miRs or miR distributions as a whole. We speculate that point mutations to noncoding RNA secondary structures resulted in the formation of stable hairpins suitable for processing by the RNAi machinery. Indeed, mfold37 RNA structural analyses support this hypothesis as we find single point mutations to noncoding RNAs (e.g., tRNAs) can result in conformational changes that create substrates potentially processed by DICER (Fig. 5; Fig. S1). Unlike miRs formed from TE sequences, however, we see no clear rationale for assuming miRs formed from noncoding RNAs would target mRNAs for translational repression. Whereas miRs formed from TEs would clearly be expected to target any mRNAs bearing their progenitor TE in their UTRs (Fig. 2), the distinct molecular origin for miRs formed from noncoding RNAs does not provide a mechanism for the establishment of a mRNA target network concurrent with miR creation. Strikingly, however, we find miRs identified as likely arising from noncoding RNA mutation often differ from their suggested progenitor noncoding RNAs by only a few nucleotides (Fig. 7) suggesting their sequences are under considerable evolutionary constraint. Due to this and the lack of a rationale for mRNA targeting, we find it tempting to speculate that these miRs may be charged with regulating the RNAs involved with maintaining basic cellular metabolism that these miRs do share sequence complementarity with their progenitor noncoding RNAs. Importantly, several groups have recently, independently reported finding short RNAs excised from various noncoding RNAs associated with Ago proteins in assembled RISC complexes (e.g., snoRNAs,39,40 snRNAs,40 vault RNAs41 and tRNAs40,42). Whether these short RNAs correspond to miR precursors formed through mutation as we have suggested in this work, or if instead some percentage of noncoding RNAs are at times processed by the RNAi machinery to participate in normal cellular regulation will ultimately require further examination. That said, RISC complexes carrying these noncoding RNA pieces likely target sequences through the same means as those bearing their traditional miR counterparts—through sequence complementarity. If so, we suggest simple association of these ~20 nt RNAs with their ~100 nt corresponding progenitor full-length noncoding RNAs would likely function by rendering the targeted ncRNAs inactive through disruption of their essential structural motifs similar to several characterized riboswitches (reviewed in refs. 43-45). However, whether the function of these miRs is to regulate protein translation through mRNA 3′UTR binding or instead noncoding regulation through conformational modulation due to miR association will for now remain undetermined. In either event, the relationships between these miRs and related noncoding RNAs identified in this work suggest a second, previously undescribed mechanism potentially responsible for miR locus formation.

Figure 7. Noncoding RNA miR origins. Alignments showing the high degree of sequence conservation between select miRs (top) and progenitor noncoding RNA (bottom) sequences. miR-1839, cgr-mir-1839 mi0020426; scaRNA, RF00426 SCARNA15 ABDC01642996.1/97-188; miR-6173, hbr-MIR6173 mi0021483; rRNA, RF01959 SSU rRNA AF166114.1/117516-116014; miR-1903, cgr-mir-1903 mi0020435; tRNA, RF00005 tRNA AAHX01000286.1/15293-15364; miR-1291, ggo-mir-1291 mi0020755; snoRNA, RF00410 SNORA2 CEC01039678.1/1523-1387; miR-3473b, mmu-mir-3473b mi0016997; U4, RF00015 U4 AAHX01028334.1/65497-65654.
Materials and Methods
MiR and 3′UTR sequence retrieval
FASTA files containing complete sets of mature and stem loop miR sequences were obtained from miRBase28 (http://www.mirbase.org/). Full sets of ENSEMBL 3′UTR sequences and miR loci flanking genomic sequences were obtained using the Biomart utility46 (www.ensembl.org/biomart/martview).
Screening miR loci for repetitive origins
As a control, all annotations and alignment analyses were run identically in parallel by two distinct individuals and then compared for verification. FASTA files containing individual miR stem loops as well as miRs with 500 nucleotides of flanking sequence both upstream and downstream (when available) were aligned against the full RepBase22 annotated repetitive elements data set, the RFAM noncoding RNA collection35 (www.sanger.ac.uk/Software/Rfam), and tRNAscan-SE47 (lowelab.cse.ucsc.edu/GtRNAdb) using stand-alone BLAST31 (BLASTN 2.2.15 with -FF, -W7, -e.1 flags). Alignments recognized as positive relationships were strictly defined as ≥ 80% identity to at least 40 nt or ≥ 70% identity to at least 50 nt of a miR hairpin. The highest scoring alignment for each pre-miR (averaging 82.4% identity over 57.0 nts) was taken to correspond to initial miR origins. Upon completion of sequence based origin characterization, miRs were sorted into familial clusters using miRBase nomenclature.28 Common familial origins were defined if: 1) the same TE scored the best alignment to multiple miR family members and 2) at least 75% of all family members produced significant alignments to the best scoring TE.
MiR target prediction
MiR hairpin sequences were screened against the corresponding set of 3′ UTRs currently available in Ensembl Biomart46 for that species using BLASTN31 2.2.15 with -FF,, -S2, -W7 flags. Putative targets were required to 1) contain perfect miR seed matches, 2) contain ≥ 50% identity to at least 50 base pairs of flanking sequences, and 3) be contained within 3′UTR sequences annotated as being the same TE as the miR’s progenitor TE by RepBase.29
RNA structural conformation modeling
Secondary structures and thermodynamic stabilities were computed using Mfold.37 The most thermodynamically stable conformation of individual miRNA sequences were determined when unaltered as well as with single nucleotide changes at positions differing from identified progenitors.
Supplementary Material
Disclosure of Potential Conflicts of Interest
We, the authors, declare no financial or nonfinancial conflicts of interest.
Acknowledgments
This work was funded by the Department of Biology, the College of Arts and Sciences at the University of South Alabama.
Glossary
Abbreviations:
- Ago
argonaute
- bp
base pair
- LINE
long interspersed repeated element
- LTR
long terminal repeat
- miR
microRNA
- mRNA
messenger RNAs
- ncRNA
noncoding RNA
- nt
nucleotide
- OrBId
origin based identification of microRNA targets algorithm
- ORF
open reading frame
- RISC
RNA-Induced Silencing Complex
- RNAi
RNA interference
- rRNA
ribosomal RNA
- scaRNA
small Cajal body-specific RNA
- SINE
short interspersed repeated elements
- siRNA
small interfering RNA
- snoRNA
small nucleolar RNA
- snRNA
spliceosomal RNA
- sRNA
short regulatory RNA
- TE
transposable element
- tmRNA
transfer messenger RNA
- tRNA
transfer RNA
- UTR
untranslated region
Citation: Roberts JT, Cooper EA, Favreau CJ, Howell JS, Lane LG, Mills JE, Newman DC, Perry TJ, Russell ME, Wallace BM, et al. Continuing analysis of microRNA origins: Formation from transposable element insertions and noncoding RNA mutations. Mobile Genetic Elements 2014; 3:e27755; 10.4161/mge.27755
Footnotes
Previously published online: www.landesbioscience.com/journals/mge/article/27755
References
- 1.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–54. doi: 10.1016/0092-8674(93)90529-Y. [DOI] [PubMed] [Google Scholar]
- 2.Hutvágner G, Zamore PD. A microRNA in a multiple-turnover RNAi enzyme complex. Science. 2002;297:2056–60. doi: 10.1126/science.1073827. [DOI] [PubMed] [Google Scholar]
- 3.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–11. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 4.Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA. 2004;10:1957–66. doi: 10.1261/rna.7135204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smalheiser NR, Torvik VI. Complications in mammalian microRNA target prediction. Methods Mol Biol. 2006;342:115–27. doi: 10.1385/1-59745-123-1:115. [DOI] [PubMed] [Google Scholar]
- 6.Zeng Y, Wagner EJ, Cullen BR. Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol Cell. 2002;9:1327–33. doi: 10.1016/S1097-2765(02)00541-5. [DOI] [PubMed] [Google Scholar]
- 7.Lai EC. Micro RNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat Genet. 2002;30:363–4. doi: 10.1038/ng865. [DOI] [PubMed] [Google Scholar]
- 8.Burgler C, Macdonald PM. Prediction and verification of microRNA targets by MovingTargets, a highly adaptable prediction method. BMC Genomics. 2005;6:88. doi: 10.1186/1471-2164-6-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krüger J, Rehmsmeier M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006;34:W451-4. doi: 10.1093/nar/gkl243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–98. doi: 10.1016/S0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 11.Nam JW, Shin KR, Han J, Lee Y, Kim VN, Zhang BT. Human microRNA prediction through a probabilistic co-learning model of sequence and structure. Nucleic Acids Res. 2005;33:3570–81. doi: 10.1093/nar/gki668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rehmsmeier M, Steffen P, Hochsmann M, Giegerich R. Fast and effective prediction of microRNA/target duplexes. RNA. 2004;10:1507–17. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, Bartel DP. Prediction of plant microRNA targets. Cell. 2002;110:513–20. doi: 10.1016/S0092-8674(02)00863-2. [DOI] [PubMed] [Google Scholar]
- 14.Saetrom O, Snøve O, Jr., Saetrom P. Weighted sequence motifs as an improved seeding step in microRNA target prediction algorithms. RNA. 2005;11:995–1003. doi: 10.1261/rna.7290705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X, Wang X. Systematic identification of microRNA functions by combining target prediction and expression profiling. Nucleic Acids Res. 2006;34:1646–52. doi: 10.1093/nar/gkl068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borchert GM, Lanier W, Davidson BL. RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol. 2006;13:1097–101. doi: 10.1038/nsmb1167. [DOI] [PubMed] [Google Scholar]
- 17.Devor EJ, Peek AS, Lanier W, Samollow PB. Marsupial-specific microRNAs evolved from marsupial-specific transposable elements. Gene. 2009;448:187–91. doi: 10.1016/j.gene.2009.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diao XM, Lisch D. Mutator transposon in maize and MULEs in the plant genome. Yi Chuan Xue Bao. 2006;33:477–87. doi: 10.1016/S0379-4172(06)60075-9. [DOI] [PubMed] [Google Scholar]
- 19.Filshtein TJ, Mackenzie CO, Dale MD, Dela-Cruz PS, Ernst DM, Frankenberger EA, He C, Heath KL, Jones AS, Jones DK, et al. OrbId: Origin-based identification of microRNA targets. Mob Genet Elements. 2012;2:184–92. doi: 10.4161/mge.21617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smalheiser NR, Torvik VI. Mammalian microRNAs derived from genomic repeats. Trends Genet. 2005;21:322–6. doi: 10.1016/j.tig.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 21.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. The sequence of the human genome. Science. 2001;291:1304–51. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 22.Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–7. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 23.Tempel S, Jurka M, Jurka J. VisualRepbase: an interface for the study of occurrences of transposable element families. BMC Bioinformatics. 2008;9:345. doi: 10.1186/1471-2105-9-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Borchert GM, Holton NW, Williams JD, Hernan WL, Bishop IP, Dembosky JA, Elste JE, Gregoire NS, Kim JA, Koehler WW, et al. Comprehensive analysis of microRNA genomic loci identifies pervasive repetitive-element origins. Mob Genet Elements. 2011;1:8–17. doi: 10.4161/mge.1.1.15766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Piriyapongsa J, Jordan IK. A family of human microRNA genes from miniature inverted-repeat transposable elements. PLoS One. 2007;2:e203. doi: 10.1371/journal.pone.0000203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan Y, Zhang Y, Yang K, Sun Z, Fu Y, Chen X, Fang R. Small RNAs from MITE-derived stem-loop precursors regulate abscisic acid signaling and abiotic stress responses in rice. Plant J. 2011;65:820–8. doi: 10.1111/j.1365-313X.2010.04467.x. [DOI] [PubMed] [Google Scholar]
- 27.Yao C, Zhao B, Li W, Li Y, Qin W, Huang B, Jin Y. Cloning of novel repeat-associated small RNAs derived from hairpin precursors in Oryza sativa. Acta Biochim Biophys Sin (Shanghai) 2007;39:829–34. doi: 10.1111/j.1745-7270.2007.00346.x. [DOI] [PubMed] [Google Scholar]
- 28.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–7. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kohany O, Gentles AJ, Hankus L, Jurka J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics. 2006;7:474. doi: 10.1186/1471-2105-7-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, Eddy SR, Gardner PP, Bateman A. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2013;41:D226–32. doi: 10.1093/nar/gks1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bushman FD. Targeting survival: integration site selection by retroviruses and LTR-retrotransposons. Cell. 2003;115:135–8. doi: 10.1016/S0092-8674(03)00760-8. [DOI] [PubMed] [Google Scholar]
- 33.Konkel MK, Batzer MA. A mobile threat to genome stability: The impact of non-LTR retrotransposons upon the human genome. Semin Cancer Biol. 2010;20:211–21. doi: 10.1016/j.semcancer.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ni J, Clark KJ, Fahrenkrug SC, Ekker SC. Transposon tools hopping in vertebrates. Brief Funct Genomic Proteomic. 2008;7:444–53. doi: 10.1093/bfgp/eln049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, Bateman A. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005;33:D121–4. doi: 10.1093/nar/gki081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucleic Acids Res. 2003;31:439–41. doi: 10.1093/nar/gkg006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–15. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen D, Fu LY, Zhang Z, Li G, Zhang H, Jiang L, Harrison AP, Shanahan HP, Klukas C, Zhang HY, et al. Dissecting the chromatin interactome of microRNA genes. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt1294. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ender C, Krek A, Friedländer MR, Beitzinger M, Weinmann L, Chen W, Pfeffer S, Rajewsky N, Meister G. A human snoRNA with microRNA-like functions. Mol Cell. 2008;32:519–28. doi: 10.1016/j.molcel.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 40.Burroughs AM, Ando Y, de Hoon MJ, Tomaru Y, Suzuki H, Hayashizaki Y, Daub CO. Deep-sequencing of human Argonaute-associated small RNAs provides insight into miRNA sorting and reveals Argonaute association with RNA fragments of diverse origin. RNA Biol. 2011;8:158–77. doi: 10.4161/rna.8.1.14300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Persson H, Kvist A, Vallon-Christersson J, Medstrand P, Borg A, Rovira C. The non-coding RNA of the multidrug resistance-linked vault particle encodes multiple regulatory small RNAs. Nat Cell Biol. 2009;11:1268–71. doi: 10.1038/ncb1972. [DOI] [PubMed] [Google Scholar]
- 42.Haussecker D, Huang Y, Lau A, Parameswaran P, Fire AZ, Kay MA. Human tRNA-derived small RNAs in the global regulation of RNA silencing. RNA. 2010;16:673–95. doi: 10.1261/rna.2000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Henkin TM. Riboswitch RNAs: using RNA to sense cellular metabolism. Genes Dev. 2008;22:3383–90. doi: 10.1101/gad.1747308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Breaker RR. Riboswitches and the RNA world. Cold Spring Harb Perspect Biol. 2012;4:4. doi: 10.1101/cshperspect.a003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Montange RK, Batey RT. Riboswitches: emerging themes in RNA structure and function. Annu Rev Biophys. 2008;37:117–33. doi: 10.1146/annurev.biophys.37.032807.130000. [DOI] [PubMed] [Google Scholar]
- 46.Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, Huber W. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005;21:3439–40. doi: 10.1093/bioinformatics/bti525. [DOI] [PubMed] [Google Scholar]
- 47.Chan PP, Lowe TM. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 2009;37:D93–7. doi: 10.1093/nar/gkn787. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.