

Abstract
Mitochondrial dysfunction leading to deficits in energy production, Ca2+ uptake capacity, and free radical generation has been implicated in the pathogenesis of familial amyotrophic lateral sclerosis (ALS) caused by mutations in Cu, Zn superoxide dismutase (SOD1). Numerous studies link UCP2, a member of the uncoupling protein family, to protection of neurons from mitochondrial dysfunction and oxidative damage in various mouse models of acute stress and neurodegeneration, including Parkinson’s disease. Here, we tested the potential neuroprotective effects of UCP2 and its ability to modulate mitochondrial function, in the G93A mutant SOD1 mouse model of familial ALS. Disease phenotype, mitochondrial bioenergetics, and Ca2+ uptake capacity were investigated in the central nervous system of double transgenic mice, expressing both human mutant G93A SOD1 and human UCP2 (hUCP2). Unexpectedly, hUCP2 expression accelerated the disease course of SOD1 mutant mice. In addition, we did not observe a classical uncoupling effect of hUCP2 in G93A brain mitochondria, although we did detect a decrease in reactive oxygen species (ROS) production from mitochondria challenged with the respiratory chain inhibitors rotenone and antimycin A. We also found that mitochondrial Ca2+ uptake capacity was decreased in the double transgenic mice, as compared to G93A mice. Taken together our results indicate that the neuroprotective role of UCP2 in neurodegeneration is disease-specific and that, while a mild uncoupling by UCP2 in brain mitochondria may protect against neurodegeneration in some injury paradigms, the mitochondrial damage and the disease caused by mutant SOD1 cannot be ameliorated by UCP2 overexpression.
Keywords: ALS, mitochondria, UCP2, SOD1
Introduction
Mitochondrial uncoupling protein 2 (UCP2) is involved in protection against oxidative stress associated with several types of neuronal injury and with neurodegenerative diseases (Andrews et al., 2009; Andrews et al., 2005; Andrews et al., 2008; Conti et al., 2005; Deierborg Olsson et al., 2008; Della-Morte et al., 2009; Haines and Li, 2012; Haines et al., 2010; Islam et al., 2012; M et al., 2012; Nakase et al., 2007). UCP2 localizes across the inner mitochondrial membrane of several tissues, including the CNS, where it has been shown to inhibit reactive oxygen species (ROS) generation and promote survival of dopaminergic neurons in a model of Parkinson’s disease (Andrews et al., 2005). Although the precise biochemical function of UCP2 is still a matter of debate (Brand and Esteves, 2005; Divakaruni and Brand, 2011; Starkov, 2006), accumulating literature shows that mitochondrial UCP2 levels inversely correlate with ROS production (Andrews and Horvath, 2009; Arsenijevic et al., 2000; Brand et al., 2002; Casteilla et al., 2001; Echtay et al., 2002; Kowaltowski et al., 1998; Nègre-Salvayre et al., 1997; Nicholls and Budd, 2000), suggesting a regulatory role in mitochondrial bioenergetics. In addition, studies that used overexpression, knock down, and mutagenesis approaches showed that UCP2 and UCP3 were necessary for ruthenium red–sensitive mitochondrial uptake of endoplasmic reticulum Ca2+ released in response to histamine stimulation (Trenker et al., 2007). Other possible functions are critically reviewed in (Divakaruni and Brand, 2011; Starkov, 2006), but the general opinion is that up-regulation of UCP2 could be neuroprotective.
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease, which starts generally in the 4th and 5th decades, when loss of spinal cord and cortical motor neurons leads to progressive paralysis and premature death (Cozzolino and Carrì, 2012). Increased oxidative radical damage is thought to be causally involved in motor neuron death in ALS (Barber et al., 2006). Furthermore, mitochondrial oxidative damage has been demonstrated in patients affected by sporadic ALS (Shaw et al., 1995; Shibata et al., 2002) and in transgenic mice expressing a familial ALS-linked mutant Cu, Zn superoxide dismutase (SOD1) (Shibata, 2001). In transgenic mouse models of SOD1 familial ALS, oxidative stress precedes motor neuron loss (Kong and Xu, 1998; Panov et al., 2011) and it is associated with mitochondrial bioenergetics deficits in the spinal cord (Jung et al., 2002; Kirkinezos et al., 2005; Mattiazzi et al., 2002), primary astrocytes (Cassina et al., 2008), and the motor cortex (Loizzo et al., 2010; Mattiazzi et al., 2002). In addition, mitochondrial Ca2+ uptake capacity is affected in ALS mice prior to motor neuron dysfunction (Damiano et al., 2006). However, it remains unclear whether mitochondrial dysfunction is a cause or a consequence of oxidative damage.
Because of the proposed metabolic and oxidative damage components of the disease, therapeutic strategies tested in the ALS mouse models have often broadly focused on bioenergetics and antioxidant agents, such as vitamin E (Gurney et al., 1996), creatine (Klivenyi et al., 1999), and catalase (Reinholz et al., 1999), with mixed outcomes (for a review see (Turner and Talbot, 2008)). In the present study, we crossed a human UCP2 (hUCP2) transgenic mouse with the G93A mutant SOD1 mouse, to test whether UCP2 overexpression could specifically decrease mitochondrial ROS production, modulate bioenergetics and calcium uptake, and afford neuroprotection in a familial ALS model. In addition, we expected that metabolic investigations in the double transgenic mice would shed new light on the functions of UCP2 in the healthy and diseased CNS.
Materials and Methods
Genetically modified mice
G93A mutant human SOD1 mice in a C57BL/6J genetic background were obtained from Jackson Laboratories (strain B6.Cg-Tg(SOD1-G93A)1Gur/J). C57BL/6J mice overexpressing human UCP2 under the control of its endogenous promoter were generous gifts from Dr. Tamas L. Horvath (Yale University). Overexpression of human UCP2 in the brain was assessed by real time PCR as previously described (Horvath et al., 2003). Double transgenic mice expressing SOD1 G93A and hUCP2 (hUCP2 G93A) were generated by crossing female hUCP2+/+ with male SOD1 G93A+/− mice. Resulting Females hUCP2+/− SOD1 G93A−/− were crossed with male SOD1 G93A+/− mice to yield hUCP2+/− SOD1 G93A+/−, SOD1 G93A+/−, hUCP2+/−, and non-transgenic control mice (ntg). Mice were genotyped by PCR of tail DNA at 21 days of age as previously described, (Horvath et al., 2003; Kim et al., 2012). Central nervous system UCP2 and SOD1 mRNA overexpression was confirmed by quantitative real time PCR. All animal experiments were carried out in sibling- and gender-matched pairs after approval by the Institutional Animal Care and Use Committee (IACUC).
Mouse phenotypes
Survival, body weight, and motor performance on an accelerating rod were determined as previously described (Kim et al., 2012). When mice became unable to right themselves within 20 s of being placed on their side they were euthanized and age at time of death was recorded. Body weight and physical performance on an accelerating rod (Rotarod, Columbus Instruments) were assessed every 2 weeks starting at 80 days of age.
Oxygen consumption and carbon dioxide production rates (VO2 and VCO2, respectively) were determined at resting conditions (absence of exercise, no dietary restrictions) for 5 minutes by placing animals in a 2 L sealed chamber with dual gas sensors (Vernier Soft. Tech. LLC). The rates were plotted as mL gas/min/kg at 120, 130, and 140 days of age.
Isolation of brain mitochondria and measurement of mitochondrial ATP synthesis, ROS emission, Ca2+ uptake, and membrane potential
Isolation and purification of mouse brain mitochondria was performed by differential centrifugation of homogenates on a discontinuous percoll gradient as previously described (Damiano et al., 2006). Pure mitochondria were extracted from the non-synaptosomal percoll gradient layer and washed three times in buffer containing 75 mM sucrose, 225 mM mannitol, 10 mM HEPES; 2 mM EDTA pH 7.4. All reagents were from Sigma (Sigma-Aldrich, Co, LLC), unless otherwise stated.
ATP synthesis was measured in purified brain mitochondria using a luciferase/luciferin-based approach, as previously described (Manfredi et al., 2002). The following measurements were carried out in a water bath-equipped (37°C) F-7000 spectrofluorometer (Hitachi). ROS emission was measured as Amplex Red (Invitrogen) fluorescence (555 nm excitation and 581 nm emission wavelengths) in presence of exogenous horseradish peroxidase and mitochondrial H2O2 as described (Starkov, 2010). Briefly, 100 μg mitochondria were added to 1mL incubation buffer (125 mM KCl, 20 mM Hepes, 0.2 mM EGTA, 2 mM KH2PO4, 200 μg/mL BSA, 1 μM Amplex Red, 4 U horseradish peroxidase, pH 7.2). Standard curves were used to calculate H2O2 emission rates after sequential addition of substrate (5mM glutamate, 2mM malate), 1 μM rotenone, and 1.8 μM antimycin A. Mitochondrial Ca2+ uptake was estimated fluorimetrically with Fura-6F (340/380 nm excitation and 510 nm emission wavelengths) (Molecular Probes) upon repetitive additions of 10 nmol of Ca2+ to the incubation medium (125 mM KCl, 20 mM Hepes, 1 mM MgCl2, 2 mM KH2PO4, 0.2 mM ATP, 1 μM rotenone, 5 mM succinate, 0.3 μM Fura-6, pH 7.2). Mitochondrial membrane potential was estimated using safranin O. Both procedures were performed as described (Damiano et al., 2006). Mitochondrial membrane potential (Δψm) was estimated using the fluorescence of safranin O with excitation and emission wavelengths of 495 nm and 586 nm, respectively, as described (Figueira et al., 2012). Incubation buffer was 125 mM KCl, 20 mM Hepes, 1 mM MgCl2, 2 mM KH2PO4, 0.2 mM ATP, 200 μg/mL BSA, 5 mM glutamate, 2mM malate, 2 μM Safranin O, pH 7.2). Δψm inhibition curves were obtained by repetitive additions of 25 nmol Ca2+ or 2 – 16 nM respiratory chain uncoupler SF6847.
Results
hUCP2 expression effect on disease progression and survival of SOD1 G93A mice
We investigated the effects of hUCP2 overexpression on disease progression by comparing lifespan, motor performance, and body weight of age and gender matched non-transgenic (ntg) and transgenic mice (hUCP2, G93A, and hUCP2 G93A). Equal numbers of male and female mice were used for each group. The lifespan of hUCP2 mice was unchanged compared to ntg (not shown), while the survival of hUCP2 G93A mice was reduced compared to G93A mice (average survival 166 ± 2.7 days and 172 ± 1.8 days, respectively; p = 0.047; n = 24; figure 1A, B).
Figure 1. hUCP2 worsens survival and exercise performance of G93A mice.

(A) Kaplan–Meier survival curve and (B) mean survival histograms of G93A and hUCP2–G93A mice (n = 24). (C, D) Time spent on an accelerating rod (2 rpm/sec) at indicated ages (n = 13). Data are presented as mean ± SEM.
Motor impairment assessment in a subset of the mice in each group showed a trend for decreased rotarod performance in hUCP2, as compared to ntg mice, but this difference did not reach statistical significance at any of the time points analyzed in the study (Figure 1C). In both G93A and hUCP2 G93A mice, a decline in rotarod performance was observed starting at 136 days of age. This decline was significantly accelerated in hUCP2 G93A, as compared to G93A mice (p = 0.002, and 0.006 at 136 and 150 days, respectively; n = 13; figure 1D).
The body weight of hUCP2 mice was lower than ntg mice, in accordance with previous studies (Horvath et al., 2003), but it remained stable over time (figure 2A). Conversely, the body weight of both G93A and hUCP2 G93A mice declined starting at 130 days of age, and there was no significant difference between these two groups. To assess whether UCP2 expression resulted in abnormal metabolic rates at the level of the whole organism, we measured respiratory quotients (VCO2/VO2) at different time points (figure 2B). We did not observe significantly differences amongst ntg, hUCP2, G93A, and hUCP2 G93A mice, which suggest that the changes in body weight in the ALS mice relative to ntg mice were not attributable to a change in substrates utilization (e.g. from high carbohydrate to high protein catabolism) and that the overexpression of UCP2 did not affect substrate utilization.
Figure 2. hUCP2 induces weight loss but does not affect the basic metabolic rates of pre- and symptomatic mice.
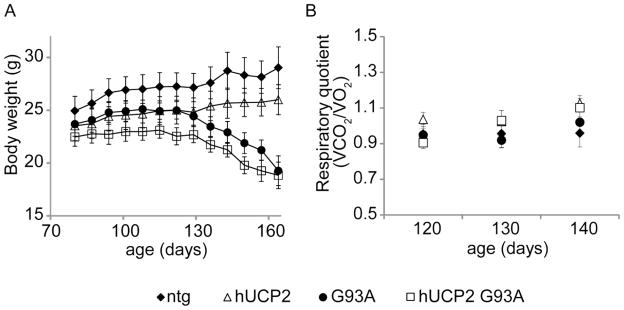
(A) Body weight determinations every two weeks from 80 to 164 days of age (n = 13). (B–D) Oxygen consumption, carbon dioxide production, and respiratory quotients, respectively, at the indicated ages (n = 13). Data are presented as mean ± SEM.
Taken together these results indicated that UCP2 overexpression worsens the disease phenotype in the G93A mutant SOD1 mouse, by accelerating onset and decreasing survival.
hUCP2 effects on brain mitochondrial function, ROS production, and calcium uptake
It has been previously shown by our group and others that a cohort of mitochondrial functions including ATP synthesis (Mattiazzi et al., 2002), ROS emission (Panov et al., 2011), and Ca2+ handling (Damiano et al., 2006; Kim et al., 2012) are altered in spinal cord and brain mitochondria from mice and rats harboring the G93A SOD1 mutation. These functional alterations are thought to be determining factors in the onset and progression of ALS (Cozzolino and Carrì, 2012; Martin, 2011). Therefore, we examined mitochondrial bioenergetics in purified brain mitochondria of 100 days old mice. We used brain as a source of mitochondria for two reasons. First, brain mitochondria undergo the same functional deficits found in the spinal cord of ALS mice and rats (Cassina et al., 2008; Cozzolino and Carrì, 2012; Damiano et al., 2006; Kim et al., 2012; Martin, 2011). Second, brain preparations yield much larger amounts of mitochondria, which minimize animal utilization. Additionally, brain preparations yield more reproducible biochemical results and contain mitochondria from neurons and glia, such as astrocytes, which are relevant to ALS pathogenesis. The age of 100 days was chosen because it reflects a pre-symptomatic disease stage, at which mitochondrial functional abnormalities are already detectable (Damiano et al., 2006).
ATP synthesis rates of ntg and hUCP2 brain mitochondria were similar (90.5 ± 2.9 vs. 93.8 ± 2.5 nmol/min/mg mitochondrial protein, respectively), but were significantly decreased in G93A and hUCP2 G93A, as compared to the rates of ntg mitochondria (68.1 ± 10.5 nmol/min/mg and 68.3 ± 7.7 nmol/min/mg, respectively, p = 0.04, Figure 3). There was no significant difference between the ATP synthesis rates of G93A and hUCP2 G93A mitochondria.
Figure 3. ATP synthesis rates of brain mitochondria from SOD1 G93A mice are decreased regardless of hUCP2 expression.

Histogram bars represent rates of ATP synthesis from purified brain mitochondria (160 μg/mL) of 100-day-old mice (n = 4). Rates were obtained according to the luciferin-luciferase assay described in materials and methods. Data are presented as mean ± SEM of brain mitochondria preparations per group.
We then measured emission of H2O2 from pure brain mitochondria to determine the effects of hUCP2 on ROS production. H2O2 emission rates were estimated before and after sequential addition of complexes I and III inhibitors (rotenone and antimycin A, respectively), in the presence of different substrates. Representative graphs show that Amplex Red fluorescence (an H2O2 indicator) increased over time upon sequential addition of mitochondria, substrate, rotenone, and antimycin A in the presence of glutamate and malate (figure 4A and 4B) or succinate (figure 5A and 5B). Hydrogen peroxide emission in hUCP2 was decreased as compared to emission from ntg mitochondria (32.5 ± 1.35 vs. 36 ± 0.9 pmol/min/mg protein; p = 0.006; figure 4C). Interestingly, H2O2 emission was lowered in hUCP2 G93A as compared to ntg mitochondria (31.6 ± 2.1; p=0.03), but was similar to G93A (30.3 ± 2.4). After addition of rotenone (figure 4D), H2O2 emission of ntg mitochondria increased as expected (137 ± 3.8), but less so in hUCP2 (120 ± 5.2, p = 0.014), G93A (113.5 ± 4.5, p = 0.002), and hUCP2 G93A mitochondria (101 ± 2.6, p < 0.001). With rotenone inhibition, hUCP2 G93A mitochondria emitted less H2O2 as compared G93A ones (p = 0.017). Similar results were obtained after addition of antimycin A - H2O2 emission of ntg mitochondria reached maximum levels (162 ± 2.5) but was lower in hUCP2 (141 ± 10.7, p = 0.05), G93A (139.1 ± 2.7, p = 0.01), and hUCP2 G93A (130 ± 3.3, p = 0.002) mitochondria (figure 4E). Like rotenone, antimycin A also elicited lower H2O2 emission in hUCP2 G93A relative to G93A mitochondria (p = 0.05). Analyses of mitochondria respiring with succinate as a substrate produced similar results, where hUCP2 G93A showed decreased ROS compared to G93A mitochondria, under inhibited (i.e., rotenone and antimycin A) conditions (figure 5A–E).
Figure 4. hUCP2 decreases ROS emission driven by glutamate and malate utilization.

(A, B) Sample curves of hydrogen peroxide emission from purified brain mitochondria. In the presence of horse radish peroxidase, Amplex Red fluorescence is directly proportional to H2O2. Substrate (5 mM glutamate, 2 mM malate), 1 μM rotenone (complex I inhibitor), and 1 μM antimycin A (complex III inhibitor) additions are indicated by dashed arrows. (C–E) Histogram bars represent rates of H2O2 emission in presence of substrate (C) and after addition of rotenone (D) and antimycin A (E); n = 4 for ntg and hUCP2; n = 5 for G93A and hUCP2 G93A. Data are presented as mean ± SEM.
Figure 5. hUCP2 decreases ROS emission driven by succinate utilization.

(A, B) Sample curves of hydrogen peroxide emission from purified brain mitochondria. Substrate (5mM succinate), 1 μM rotenone (complex I inhibitor), and 1 μM antimycin A (complex III inhibitor) additions are indicated by dashed arrows. (C–E) Histogram bars represent rates of H2O2 emission in presence of substrate (C) and after addition of rotenone (D) and antimycin A (E). Data are presented as mean ± SEM of n = 8 for ntg and hUCP2 and n = 4 for G93A and hUCP2 G93A.
Taken together, these results confirmed that UCP2 has a protective effect on ROS production, but they also showed that, surprisingly, G93A SOD1 causes a decrease, rather than an increase, in ROS production from brain mitochondria. Furthermore, they indicated that UCP2 has an additive effect in decreasing ROS production in mitochondria treated with respiratory chain inhibitors.
We examined the effects of hUCP2 overexpression on mitochondrial Ca2+ uptake capacity by measuring Fura-6F fluorescence after bolus Ca2+ additions to purified brain mitochondria at 100 days of age. Maximal Ca2+ uptake capacity was expressed as the total amount of Ca2+ (nmol Ca2+/mg protein) at which uptake ceased (i.e., the rate of uptake was zero). As expected, Ca2+ uptake capacity in G93A mitochondria was lower relative to that of ntg and hUCP2 (figure 6A, B, (Kim et al., 2012)). However, contrary to hUCP2, which had a higher uptake capacity than ntg mitochondria (898 ± 48 nmol Ca2+/mg protein vs 809 ± 44, respectively, p = 0.03, n = 5), hUCP2 G93A had lower Ca2+ uptake capacity than G93A mitochondria (721 ± 31 vs. 593 ± 50, p = 0.018; n = 5). This result suggested the intriguing possibility that in ntg and bio-energetically defective G93A mitochondria, UCP2 has opposite regulatory effects on Ca2+ uptake capacity.
Figure 6. hUCP2 effects on mitochondrial Ca2+ uptake capacity and membrane potential.

(A) Kinetics of Ca2+ uptake in brain mitochondria measured by monitoring the change of Fura-6F fluorescence ratio (340/380 nm excitation, 510 nm emission) on Ca2+ loading (250 nmol of Ca2+/mg protein in each addition, indicated by arrows). The fluorescence peaks corresponds to increase in extra-mitochondrial Ca2+, whereas the decreases in fluorescence reflects mitochondrial Ca2+ uptake. The sustained increase at the end of trace shows that mitochondria are unable to further accumulate Ca2+. In the example, ntg and hUCP2 mitochondria took up six Ca2+ additions, whereas G93A and G93A hUCP2 mice mitochondria only took five Ca2+ additions. (B) Average brain mitochondrial Ca2+ uptake capacity in nmol Ca2+/mg of mitochondrial protein. Data are mean ± SEM of n = 5 brain mitochondria preparations per group. (C) Δψm response to 25 nmol Ca2+ bolus (250 nmol of Ca2+/mg protein) calculated as [(1/FLN nmol Ca2+)/(1/FL0 nmol Ca2+) × 100], where FLN is fluorescence of safranin-O at N nmol Ca2+. Safranin-O fluorescence inversely correlates with Δψm. Data are mean of n = 4 brain mitochondria preparations per group. (D) Δψm response to respiratory chain uncoupler SF6847 calculated as in C. Data are presented as mean of n =3 for hUCP2 and n = 4 for hUCP2 G93A. IC50values in C and D were calculated from the fitted curves of the Hill equation y=xn/(kn+xn), where x is Ca2+ or SF6847 concentration, n is the Hill coefficient, and k is the IC50 value
Saturation of Ca2+ uptake is accompanied by a loss of membrane potential (Δψm) in brain mitochondria (Chalmers and Nicholls, 2003). To assess whether hUCP2 expression affects depolarization induced by Ca2+ uptake, we used safranin-O fluorescence as a means to estimate changes in Δψm at increasing concentrations of Ca2+. hUCP2 and ntg mitochondria had similar sensitivities to Ca2+ induced depolarization (IC50, i.e. the Ca2+ concentration at which 0.1 mg of mitochondria lost 50% of the initial Δψm, was 889 ± 43 vs. 849 ± 45 nmol Ca2+/mg protein, respectively, n = 4, figure 6C). Furthermore, Ca2+-induced depolarization in G93A mitochondria did not differ from that of ntg controls (IC50 752 ± 45). However, hUCP2 G93A mitochondria were significantly more sensitive to Ca2+-induced depolarization than controls were (IC50 661 ± 37, p = 0.007). To assess whether the cause for enhanced sensitivity in hUCP2 G93A, but not in G93A mitochondria, was due to an uncoupling effect of UCP2, we measured Δψm changes at increasing concentrations of the respiratory chain uncoupler SF6847 (figure 6D). The response to the uncoupler was similar in G93A and hUCP2 G93A mitochondria (IC50 4.3 ± 0.2 vs. 4.4 ± 0.2 nmol SF6847/mg protein; n = 4).
Taken together, these results suggested that UCP2 does not cause uncoupling of brain mitochondria and that the differences in Ca2+ uptake capacity associated with its expression are likely related to a direct effect of UCP2 on the regulation of mitochondrial Ca2+ uptake.
Discussion
Numerous reports suggested that UCP2 is involved in neuroprotection against oxidative stress in ischemia-reperfusion injury as well as in animal models of neurodegenerative diseases (Andrews et al., 2009; Andrews et al., 2008; Conti et al., 2005; Deierborg Olsson et al., 2008; Della-Morte et al., 2009; Haines and Li, 2012; Haines et al., 2010; Islam et al., 2012; M et al., 2012; Nakase et al., 2007). For example, overexpression of hUCP2 in adult fly neurons increased uncoupled respiration, decreased oxidative damage, and extended lifespan (Fridell et al., 2005). Another study showed that transgenic overexpression of hUCP2 prolonged the life span of Mn, SOD knockout mice, presumably by slowing down the oxidative damage to mitochondria (Andrews and Horvath, 2009; Cozzolino and Carrì, 2012).
Here, we tested whether hUCP2 expression was able to protect mitochondrial function and slow down disease progression in a mouse model of familial ALS associated with mutant SOD1. Our results indicate that overexpression of hUCP2 in SOD1 G93A mice did not improve disease symptoms and survival rates, but rather it caused an acceleration of disease progression. These results highlighted the still undetermined function of UCP2 in the CNS, and prompted us to investigate how hUCP2 affects metabolism and CNS mitochondrial function in control and SOD1 mutant mice. hUCP2 mice have been shown to have lower amounts of body fat than non-transgenic (ntg) littermates, despite having a slightly higher food intake rate (Horvath et al., 2003). Accordingly, we found that hUCP2 had lower body weight than ntg, which matched the weight of G93A mice, prior to the terminal stages of disease (figure 2B). Interestingly, hUCP2 G93A double transgenic mice had lower body weight than the other groups, even at pre-symptomatic stages. We examined the basal metabolic rates and found no significant changes in RQs, indicating that hUCP2-expressing animals did not display significant changes in substrate utilization (i.e., carbohydrate vs. proteins).
In this work, we chose to investigate the bioenergetics and mitochondrial functions in brain mitochondria, because they undergo the same functional deficits found in the spinal cord of ALS mice (Cassina et al., 2008; Cozzolino and Carrì, 2012; Damiano et al., 2006; Kim et al., 2012; Martin, 2011), but provide a much more abundant, reproducible, and consistent source of material for biochemical studies. Brain mitochondria ATP synthesis was decreased in G93A mice, but not further decreased by hUCP2 co-expression with mutant SOD1, contrary to what might have been expected from the overexpression of an uncoupling protein.
A previous study found that G93A rat brain mitochondria had increased rates of ROS emission, although the age of the rats was not mentioned (Panov et al., 2011). We examined ROS emission from 100 days old mouse respiring brain mitochondria, before and after the sequential addition of rotenone and antimycin A. Contrary to expectations, we found decreased ROS emission in G93A mitochondria. While we cannot account for the discrepancy between G93A rat (Panov et al., 2011) and mouse brain mitochondria, the lower emission we observed may be due to a faster secondary conversion of H2O2 into •OH− radicals previously reported for G93A SOD1 (Bogdanov et al., 1998; Yim et al., 1996). An ever stronger •OH− radical generation activity was determined for A4V SOD1, one of the most common and severe mutations associated with familial ALS (Yim et al., 1997). Interestingly, in hUCP2 G93A double transgenic, but not in hUCP2 single transgenic mitochondria, there was a further decrease in ROS after the addition of rotenone or antimycin A. This suggests that mutant SOD1 could act in concert with hUCP2, in an additive or cooperative manner, to decrease ROS production under inhibited respiratory chain conditions.
Our results showing that hUCP2 expression increased Ca2+ uptake capacity in control brain mitochondria (figure 6A and 6B) was in agreement with an earlier study demonstrating that UCP2 expression increased Ca2+ uptake capacity and that its ablation had the opposite effect (Trenker et al., 2007). However, hUCP2 expression in G93A mice, not only failed to reverse the defect in Ca2+ uptake capacity caused by mutant SOD1, but it paradoxically increased it. To gain further insight into the mechanisms of this phenomenon we measured Δψm in response to Ca2+ loading. While ntg and hUCP2 mitochondria had similar Ca2+ IC50 values, hUCP2 G93A mitochondria were substantially more sensitive to Ca2+-induced depolarization (figure 6C). In contrast, when a different, non-Ca2+ dependent, depolarizing agent (SF6847) was tested, G93A, and hUCP2 G93A mitochondria had the same sensitivity to uncoupling (figure 6D). These results suggested that the role of UCP2 in SOD1 mutant brain mitochondria is not simply related to a classical uncoupling effect, but is possibly associated with regulation of Ca2+ handling. Based on these results, it could be speculated that mutant SOD1 in mitochondria alters the aforementioned functional interaction between UCP2 and the mitochondrial calcium uniporter (Trenker et al., 2007), resulting in further diminished rather than enhanced Ca2+ uptake capacity. Future studies focused on the interactions of SOD1 with the mitochondrial calcium uniporter and its regulatory components will be necessary to further demonstrate this hypothesis.
Mild mitochondrial uncoupling has been proposed as a mechanism to decrease Ca2+ overload and ROS emission, especially under conditions of excitotoxic injury. The rationale behind these effects is based on the “uncoupling-to-survive” hypothesis (Brand, 2000), which states that increased uncoupling leads to higher oxygen consumption and reduced proton motive force, which then reduces ROS generation. UCP2-induced mild uncoupling has been extensively documented and is generally thought to underlie the mechanisms of neuroprotection against oxidative injury (Andrews et al., 2009; Andrews et al., 2008; Conti et al., 2005; Deierborg Olsson et al., 2008; Della-Morte et al., 2009; Haines and Li, 2012; Haines et al., 2010; Islam et al., 2012; M et al., 2012; Nakase et al., 2007). Despite the fact that we did not find a classical uncoupling effect of hUCP2 in the mouse brain, we did observe a decrease in ROS production and a regulation of mitochondrial Ca2+ handling in concert with mutant SOD1.
Taken together, this work highlights the importance of using a combination of genetic and biochemical approaches to test broadly proposed, but seldom mechanistically investigated, pathogenesis hypotheses, Based on the results obtained in this study of hUCP2 G93A SOD1 double transgenic mice, we propose that the neuroprotection afforded by UCP2 might be specific for certain types of injury. Further, in the case of familial ALS, UCP2 overexpression may worsen the pathogenic effects of mutant SOD1 on mitochondria. Lowering mitochondrial ROS output by UCP2 overexpression did not protect against mitochondria functional damage and disease progression, suggesting the dissociation between mitochondrial ROS production and the biochemical and clinical phenotypes caused by mutant SOD1 in vivo.
Acknowledgments
This work was supported by grants: NS051419 and NS062055, The Packard Center for ALS Research, The Muscular Dystrophy Association.
Abbreviations list
- ALS
amyotrophic lateral sclerosis
- hUCP2
human uncoupling protein 2
- SOD1
superoxide dismutase 1
- ROS
reactive oxygen species
- CNS
central nervous system
- ntg
non-transgenic
- RQ
respiratory quotient
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andrews ZB, Erion D, Beiler R, Liu ZW, Abizaid A, Zigman J, Elsworth JD, Savitt JM, DiMarchi R, Tschöp M, Roth RH, Gao XB, Horvath TL. Ghrelin Promotes and Protects Nigrostriatal Dopamine Function via a UCP2-Dependent Mitochondrial Mechanism. The Journal of Neuroscience. 2009;29:14057–14065. doi: 10.1523/JNEUROSCI.3890-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews ZB, Horvath B, Barnstable CJ, Elsworth J, Yang L, Beal MF, Roth RH, Matthews RT, Horvath TL. Uncoupling protein-2 is critical for nigral dopamine cell survival in a mouse model of Parkinson’s disease. J Neurosci. 2005;25:184–191. doi: 10.1523/JNEUROSCI.4269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews ZB, Horvath TL. Uncoupling protein-2 regulates lifespan in mice. Am J Physiol Endocrinol Metab. 2009;296:E621–627. doi: 10.1152/ajpendo.90903.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, Tschop MH, Shanabrough M, Cline G, Shulman GI, Coppola A, Gao XB, Horvath TL, Diano S. UCP2 mediates ghrelin/’s action on NPY/AgRP neurons by lowering free radicals. Nature. 2008;454:846–851. doi: 10.1038/nature07181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenijevic D, Onuma H, Pecqueur C, Raimbault S, Manning BS, Miroux B, Couplan E, Alves-Guerra MC, Goubern M, Surwit R, Bouillaud F, Richard D, Collins S, Ricquier D. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nature Genetics. 2000;26:435–439. doi: 10.1038/82565. [DOI] [PubMed] [Google Scholar]
- Barber SC, Mead RJ, Shaw PJ. Oxidative stress in ALS: A mechanism of neurodegeneration and a therapeutic target. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2006;1762:1051–1067. doi: 10.1016/j.bbadis.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Bogdanov MB, Ramos LE, Xu Z, Beal MF. Elevated “Hydroxyl Radical” Generation In Vivo in an Animal Model of Amyotrophic Lateral Sclerosis. Journal of Neurochemistry. 1998;71:1321–1324. doi: 10.1046/j.1471-4159.1998.71031321.x. [DOI] [PubMed] [Google Scholar]
- Brand MD. Uncoupling to survive? The role of mitochondrial inefficiency in ageing. Exp Gerontol. 2000;35:811–820. doi: 10.1016/s0531-5565(00)00135-2. [DOI] [PubMed] [Google Scholar]
- Brand MD, Esteves TC. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell metabolism. 2005;2:85–93. doi: 10.1016/j.cmet.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Brand MD, Pamplona R, Portero-Otín M, Requena JR, Roebuck SJ, Buckingham JA, Clapham JC, Cadenas S. Oxidative damage and phospholipid fatty acyl composition in skeletal muscle mitochondria from mice underexpressing or overexpressing uncoupling protein 3. Biochemical Journal. 2002;368:597–603. doi: 10.1042/BJ20021077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassina P, Cassina A, Pehar M, Castellanos R, Gandelman M, de León A, Robinson KM, Mason RP, Beckman JS, Barbeito L, Radi R. Mitochondrial Dysfunction in SOD1G93A-Bearing Astrocytes Promotes Motor Neuron Degeneration: Prevention by Mitochondrial-Targeted Antioxidants. The Journal of Neuroscience. 2008;28:4115–4122. doi: 10.1523/JNEUROSCI.5308-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casteilla L, Rigoulet M, Pénicaud L. Mitochondrial ROS Metabolism: Modulation by Uncoupling Proteins. IUBMB Life. 2001;52:181–188. doi: 10.1080/15216540152845984. [DOI] [PubMed] [Google Scholar]
- Chalmers S, Nicholls DG. The Relationship between Free and Total Calcium Concentrations in the Matrix of Liver and Brain Mitochondria. Journal of Biological Chemistry. 2003;278:19062–19070. doi: 10.1074/jbc.M212661200. [DOI] [PubMed] [Google Scholar]
- Conti B, Sugama S, Lucero J, Winsky-Sommerer R, Wirz SA, Maher P, Andrews Z, Barr AM, Morale MC, Paneda C, Pemberton J, Gaidarova S, Behrens MM, Beal F, Sanna PP, Horvath T, Bartfai T. Uncoupling protein 2 protects dopaminergic neurons from acute 1,2,3,6-methyl-phenyl-tetrahydropyridine toxicity. Journal of Neurochemistry. 2005;93:493–501. doi: 10.1111/j.1471-4159.2005.03052.x. [DOI] [PubMed] [Google Scholar]
- Cozzolino M, Carrì MT. Mitochondrial dysfunction in ALS. Progress in Neurobiology. 2012;97:54–66. doi: 10.1016/j.pneurobio.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Damiano M, Starkov AA, Petri S, Kipiani K, Kiaei M, Mattiazzi M, Flint Beal M, Manfredi G. Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. Journal of Neurochemistry. 2006;96:1349–1361. doi: 10.1111/j.1471-4159.2006.03619.x. [DOI] [PubMed] [Google Scholar]
- Deierborg Olsson T, Wieloch T, Diano S, Warden CH, Horvath TL, Mattiasson G. Overexpression of UCP2 protects thalamic neurons following global ischemia in the mouse. J Cereb Blood Flow Metab. 2008;28:1186–1195. doi: 10.1038/jcbfm.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della-Morte D, Dave KR, DeFazio RA, Bao YC, Raval AP, Perez-Pinzon MA. Resveratrol pretreatment protects rat brain from cerebral ischemic damage via a sirtuin 1–uncoupling protein 2 pathway. Neuroscience. 2009;159:993–1002. doi: 10.1016/j.neuroscience.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divakaruni AS, Brand MD. The regulation and physiology of mitochondrial proton leak. Physiology. 2011;26:192–205. doi: 10.1152/physiol.00046.2010. [DOI] [PubMed] [Google Scholar]
- Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S, Stuart JA, Harper JA, Roebuck SJ, Morrison A, Pickering S, Clapham JC, Brand MD. Superoxide activates mitochondrial uncoupling proteins. Nature. 2002;415:96–99. doi: 10.1038/415096a. [DOI] [PubMed] [Google Scholar]
- Figueira TR, Melo DR, Vercesi AE, Castilho RF. Safranine as a Fluorescent Probe for the Evaluation of Mitochondrial Membrane Potential in Isolated Organelles and Permeabilized Cells. T Mitochondrial Bioenergetics. 2012:103–117. doi: 10.1007/978-1-61779-382-0_7. [DOI] [PubMed] [Google Scholar]
- Fridell YW, Sanchez-Blanco A, Silvia BA, Helfand SL. Targeted expression of the human uncoupling protein 2 (hUCP2) to adult neurons extends life span in the fly. Cell metabolism. 2005;1:145–152. doi: 10.1016/j.cmet.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Cutting FB, Zhai P, Doble A, Taylor CP, Andrus PK, Hall ED. Benefit of vitamin E, riluzole, and gababapentin in a transgenic model of familial amyotrophic lateral sclerosis. Annals of Neurology. 1996;39:147–157. doi: 10.1002/ana.410390203. [DOI] [PubMed] [Google Scholar]
- Haines B, Li PA. Overexpression of Mitochondrial Uncoupling Protein 2 Inhibits Inflammatory Cytokines and Activates Cell Survival Factors after Cerebral Ischemia. PLoS ONE. 2012;7:e31739. doi: 10.1371/journal.pone.0031739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines BA, Mehta SL, Pratt SM, Warden CH, Li PA. Deletion of mitochondrial uncoupling protein-2 increases ischemic brain damage after transient focal ischemia by altering gene expression patterns and enhancing inflammatory cytokines. J Cereb Blood Flow Metab. 2010;30:8. doi: 10.1038/jcbfm.2010.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath TL, Diano S, Miyamoto S, Barry S, Gatti S, Alberati D, Livak F, Lombardi A, Moreno M, Goglia F, Mor G, Hamilton J, Kachinskas D, Horwitz B, Warden CH. Uncoupling proteins-2 and 3 influence obesity and inflammation in transgenic mice. Int J Obes Relat Metab Disord. 2003;27:433–442. doi: 10.1038/sj.ijo.0802257. [DOI] [PubMed] [Google Scholar]
- Islam R, Yang L, Sah M, Kannan K, Anamani D, Vijayan C, Kwok J, Cantino ME, Beal MF, Fridell YWC. A neuroprotective role of the human uncoupling protein 2 (hUCP2) in a Drosophila Parkinson’s Disease model. Neurobiology of Disease. 2012;46:137–146. doi: 10.1016/j.nbd.2011.12.055. [DOI] [PubMed] [Google Scholar]
- Jung C, Higgins CMJ, Xu Z. Mitochondrial electron transport chain complex dysfunction in a transgenic mouse model for amyotrophic lateral sclerosis. Journal of Neurochemistry. 2002;83:535–545. doi: 10.1046/j.1471-4159.2002.01112.x. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Magranè J, Starkov AA, Manfredi G. The mitochondrial calcium regulator cyclophilin D is an essential component of oestrogen-mediated neuroprotection in amyotrophic lateral sclerosis. Brain. 2012;135:2865–2874. doi: 10.1093/brain/aws208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkinezos IG, Bacman SR, Hernandez D, Oca-Cossio J, Arias LJ, Perez-Pinzon MA, Bradley WG, Moraes CT. Cytochrome c Association with the Inner Mitochondrial Membrane Is Impaired in the CNS of G93A-SOD1 Mice. The Journal of Neuroscience. 2005;25:164–172. doi: 10.1523/JNEUROSCI.3829-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klivenyi P, Ferrante RJ, Matthews RT, Bogdanov MB, Klein AM, Andreassen OA, Mueller G, Wermer M, Kaddurah-Daouk R, Beal MF. Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nat Med. 1999;5:347–350. doi: 10.1038/6568. [DOI] [PubMed] [Google Scholar]
- Kong J, Xu Z. Massive Mitochondrial Degeneration in Motor Neurons Triggers the Onset of Amyotrophic Lateral Sclerosis in Mice Expressing a Mutant SOD1. The Journal of Neuroscience. 1998;18:3241–3250. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowaltowski AJ, Costa ADT, Vercesi AE. Activation of the potato plant uncoupling mitochondrial protein inhibits reactive oxygen species generation by the respiratory chain. FEBS Letters. 1998;425:213–216. doi: 10.1016/s0014-5793(98)00231-2. [DOI] [PubMed] [Google Scholar]
- Loizzo S, Pieri M, Ferri A, Carrì MT, Zona C, Fortuna A, Vella S. Dynamic NAD(P)H post-synaptic autofluorescence signals for the assessment of mitochondrial function in a neurodegenerative disease: Monitoring the primary motor cortex of G93A mice, an amyotrophic lateral sclerosis model. Mitochondrion. 2010;10:108–114. doi: 10.1016/j.mito.2009.11.001. [DOI] [PubMed] [Google Scholar]
- ML, FFZ, JJT, CJS, YF, JHD, JSB, GH The neuroprotection of hydrogen sulfide against MPTP-induced dopaminergic neuron degeneration involves uncoupling protein 2 rather than ATP-sensitive potassium channels. Antioxidants & Redox Signaling. 2012;17:10. doi: 10.1089/ars.2011.4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredi G, Yang L, Gajewski CD, Mattiazzi M. Measurements of ATP in mammalian cells. Methods. 2002;26:317–326. doi: 10.1016/S1046-2023(02)00037-3. [DOI] [PubMed] [Google Scholar]
- Martin L. Mitochondrial pathobiology in ALS. J Bioenerg Biomembr. 2011;43:569–579. doi: 10.1007/s10863-011-9395-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiazzi M, D’Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G. Mutated Human SOD1 Causes Dysfunction of Oxidative Phosphorylation in Mitochondria of Transgenic Mice. Journal of Biological Chemistry. 2002;277:29626–29633. doi: 10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- Nakase T, Yoshida Y, Nagata K. Amplified expression of uncoupling proteins in human brain ischemic lesions. Neuropathology. 2007;27:442–447. doi: 10.1111/j.1440-1789.2007.00815.x. [DOI] [PubMed] [Google Scholar]
- Nègre-Salvayre A, Hirtz C, Carrera G, Cazenave R, Troly M, Salvayre R, Pénicaud L, Casteilla L. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. The FASEB Journal. 1997;11:809–815. [PubMed] [Google Scholar]
- Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiological reviews. 2000;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- Panov A, Kubalik N, Zinchenko N, Hemendinger R, Dikalov S, Bonkovsky HL. Respiration and ROS production in brain and spinal cord mitochondria of transgenic rats with mutant G93a Cu/Zn-superoxide dismutase gene. Neurobiology of Disease. 2011;44:53–62. doi: 10.1016/j.nbd.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Reinholz MM, Merkle CM, Poduslo JF. Therapeutic Benefits of Putrescine-Modified Catalase in a Transgenic Mouse Model of Familial Amyotrophic Lateral Sclerosis. Experimental Neurology. 1999;159:204–216. doi: 10.1006/exnr.1999.7142. [DOI] [PubMed] [Google Scholar]
- Shaw IC, Fitzmaurice PS, Mitchell JD, Lynch PG. Studies on Cellular Free Radical Protection Mechanisms in the Anterior Horn from Patients with Amyotrophic Lateral Sclerosis. Neurodegeneration. 1995;4:391–396. doi: 10.1006/neur.1995.0047. [DOI] [PubMed] [Google Scholar]
- Shibata N. Transgenic mouse model for familial amyotrophic lateral sclerosis with superoxide dismutase-1 mutation. Neuropathology. 2001;21:82–92. doi: 10.1046/j.1440-1789.2001.00361.x. [DOI] [PubMed] [Google Scholar]
- Shibata N, Hirano A, Hedley-Whyte T, Dal Canto M, Nagai R, Uchida K, Horiuchi S, Kawaguchi M, Yamamoto T, Kobayashi M. Selective formation of certain advanced glycation end products in spinal cord astrocytes of humans and mice with superoxide dismutase-1 mutation. Acta Neuropathol. 2002;104:171–178. doi: 10.1007/s00401-002-0537-5. [DOI] [PubMed] [Google Scholar]
- Starkov AA. Protein-mediated energy-dissipating pathways in mitochondria. Chemico-biological interactions. 2006;163:133–144. doi: 10.1016/j.cbi.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Starkov AA. Measurement of Mitochondrial ROS Production. T Protein Misfolding and Cellular Stress in Disease and Aging. 2010:245–255. doi: 10.1007/978-1-60761-756-3_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenker M, Malli R, Fertschai I, Levak-Frank S, Graier WF. Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat Cell Biol. 2007;9:445–452. doi: 10.1038/ncb1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner BJ, Talbot K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog Neurobiol. 2008;85:94–134. doi: 10.1016/j.pneurobio.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Yim HS, Kang JH, Chock PB, Stadtman ER, Yim MB. A Familial Amyotrophic Lateral Sclerosis-associated A4V Cu, Zn-Superoxide Dismutase Mutant Has a Lower Km for Hydrogen Peroxide: Correlation Between Clinical Severity and the Km Value. Journal of Biological Chemistry. 1997;272:8861–8863. doi: 10.1074/jbc.272.14.8861. [DOI] [PubMed] [Google Scholar]
- Yim MB, Kang JH, Yim HS, Kwak HS, Chock PB, Stadtman ER. A gain-of-function of an amyotrophic lateral sclerosis-associated Cu, Zn-superoxide dismutase mutant: An enhancement of free radical formation due to a decrease in Km for hydrogen peroxide. Proceedings of the National Academy of Sciences. 1996;93:5709–5714. doi: 10.1073/pnas.93.12.5709. [DOI] [PMC free article] [PubMed] [Google Scholar]