

Abstract
CD1d is a nonclassical Ag-presenting molecule that presents glycolipid Ags to NKT cells that are involved in immune defense and tumor rejection. It also plays a role in immunoregulatory functions in the epidermis. The mechanisms controlling the expression of CD1d are not well understood. Therefore, we cloned the CD1d gene promoter and characterized its activities in primary human keratinocytes and other cell lines of epithelial origin. We found that a CCAAT box in the CD1d promoter is required for its expression in keratinocytes. We show here that transcription factor C/EBP-β binds to the CCAAT box in the CD1d promoter in vitro and in vivo. Consistent with these observations, deletion of the gene encoding for C/EBP-β caused a loss of CD1d expression. The in vivo regulation of CD1d has significant implications for the pathologic mechanisms of certain immunologic skin diseases in which NKT cells play a role, such as allergic contact dermatitis and psoriasis. Together, these data show a central role for C/EBP-β in regulating CD1d transcription.
Human keratinocytes (KC)3 can influence immune responses in the skin through the production of cytokines and by presentation of Ags to T cells (1). Besides MHC class I and II that present Ag peptide to CD8 and CD4 T cells, respectively (2, 3), KC also express a MHC class I-like, nonpolymorphic Ag-presenting molecule CD1d that presents glycolipids to NKT cells (4, 5). CD1d is a glycoprotein noncovalently associated with β2-microglobulin and forms a heterodimeric structure similar to the MHC class I molecules (6–9). This molecule is highly conserved across species and is the only member of the group CD1 molecules functionally present in mice and rats (6). In humans, there are two subgroups of CD1 genes. Group 1 members include CD1A, CD1B, CD1C, and CD1E, which share homology in their 5′ untranslated region (6). Group 2 is constituted by CD1D alone. CD1d is expressed centrally in the thymus and by several other peripheral epithelia or stromal cells of the skin, intestine, liver, kidney, pancreas, and uterus (8). CD1d is also expressed by some bone marrow-derived cells such as T cells, B cells, monocytes, and monocyte-derived dendritic cells (10–13). Human CD1d expression can be up-regulated in the intestinal epithelial cells and KC by IFN-γ (4, 14, 15) and in peripheral blood T cells by mitogens (16). Overexpression of CD1d is observed in the epidermal KC of the skin lesions of allergic contact dermatitis (ACD) (17), as well as in psoriatic lesions (4). During allergic reactions to cow’s milk, CD1d is overexpressed by cells located in the duodenal lamina propria (18). Similarly, in primary biliary cirrhosis, epithelial cells of the small bile ducts overexpress CD1d (19). Thus, CD1d is overexpressed in allergic, inflammatory, and autoimmune tissue-based pathologies. The presence of infiltrating NKT cells in these tissues raises the possibility that tissue-homing NKT cells play a major role in these varied inflammatory conditions. CD1d expression varies significantly among T lymphocytes, monocytes, and monocyte-derived dendritic cells of different individuals (16). However, the reason for this individual variations in CD1d expression is not clearly understood, but may be related to genetic background and/or environmental stimuli. Thus, very little is known about the transcriptional control of human CD1d at the tissue level.
A previous report (20) demonstrated that the human gene-encoding CD1d molecule (CD1D) has the TATA-less promoter with multiple transcription initiation sites. These sequences were found to be active in Jurkat and K562 cells (both are immortalized leukemia cell lines). The proximal promoter (located within the region of −106 to +24) was more active in Jurkat cells while the distal promoter (located between −665 to −202) was more active in K562 cells, indicating a cell type-specific activity of the two promoters (20).
In vivo, human epidermal KC have been demonstrated to express CD1d (5), but there is little understanding of the regulation of this Ag-presenting molecule in the skin. To understand the mechanism of CD1d gene expression in epidermal KC, we isolated the human CD1d promoter and investigated its activity in primary human KC, some KC-derived cell lines, as well as other cell lines of epithelial origin. We found a significant stimulatory role for a CCAAT box located in the CD1d proximal promoter, which binds to the C/EBP-β. In this report, we demonstrate that C/EBP-β controls CD1d, which in turn plays a role in presenting glycolipids to invariant NKT (iNKT) cells. These data have implications for the common inflammatory skin diseases ACD and psoriasis.
Materials and Methods
Cell culture and reagents
Normal human epidermal KC cultures were explanted from neonatal foreskins and were generated as described previously (5) and maintained in EpiLife HKGS medium (Invitrogen). Newly established primary KC (two to five passages) cultures were used in all experiments. Cell lines used in this study were maintained in DMEM supplemented with 10% FBS.
Promoter constructs cloning and probes
A 1762-nt sequence from −1738 to +24 of the promoter and 5′ untranslated region of the human CD1D gene was generated by PCR amplification of human genomic DNA that was isolated from whole blood using a DNeasy Blood & Tissue Kit (Qiagen) according to the manufacturer’s protocol. PCR was conducted using 100 ng of the isolated genomic DNA as template and dedicated primers from the CD1d gene (forward, cggCTC GAGctagagtgtggtgcagtagc and reverse. gggggAAGCTTcagcagaaacagcag gca) containing XhoI and HindIII sites incorporated into the forward and reverse primers, respectively. The reaction was run for 35 cycles in the presence of 0.2 mM dNTP, 8% DMSO, 1.5 mM MgCl2, and 1 U of Platinum TaqDNA polymerase (Invitrogen Life Technologies). The amplified fragment (−1738 to +24) was subcloned into the pGL3 basic plasmid vector (Promega) after digestion with XhoI and HindIII (Fig. 1A). Using the above sequence as template, subsequent deletional constructs starting at −985, −665, −354, −106, −65, and −39 with a common 3′ end at +24 were generated in the pGL3 vector. Primers used for generating the deletion mutants are listed in Table I. All promoter fragments were sequenced and verified to the reported CD1d sequence (GenBank accession no. BC027926; http://www.ncbi.nlm.nih.gov/GenBank/).
FIGURE 1.

Significant basal level of CD1d promoter activities observed in normal human KC and other cell lines of epithelial origin. A, Schematic representation of the CD1d promoter fragments studied. The depicted fragments were cloned into the pGL3 expression vector, which contains the luciferase gene. The expression constructs were transiently transfected into primary KC, KC-derived cell lines, and other epithelial cell lines, as well as K562 cells to study CD1d promoter activity. The indicated cell lines were transiently transfected with indicated gene reporter constructs using Lipofectamine reagent and, 48 h after transfection, reporter activity was assayed (luminometry, see Materials and Methods). B, Primary human KC; C, KC cell lines HaCaT (black histogram) and gastrointestinal epithelial cell line CaCo2 (white bar graph); and D, qPCR examining CD1d gene expression in six different sets of primary KC, KC- derived cell lines (HaCat and Ker-1), and non-KC epithelial cell lines (HeLa and CaCo2). Data that are depicted for gene reporter assay represents mean and SD for triplicate samples (B–C). For qPCR (D), the data represent the mean and SD for replicate cDNA for each specimen. Undiff, Undifferentiated.
Table I. Primers used to make CD1d promoter constructs.
| Primer Type | Ends at | Added Restriction Enzyme | Sequence |
|---|---|---|---|
| Forward | −985 | XhoI | cggCTCGAGctgcagcccaggttctgagt |
| Forward | −665 | XhoI | cggCTCGAGcaggaggaaagagaggctag |
| Forward | −354 | XhoI | cggCTCGAGtcctgcttccaaaacataaat |
| Forward | −225 | XhoI | cggCTCGAGtgggaccccgacctctttg |
| Forward | −112 | XhoI | cggCTCGAGaattggctggcacccagcg |
| Forward | −106 | XhoI | cggCTCGAGctggcacccagcggaaaggg |
| Forward | −65 | XhoI | cggCTCGAGagaagagtgcgcaggtcag |
| Forward | −45 | XhoI | cggCTCGAGgggcggcgcgcagcggcgc |
| Forward | −39 | XhoI | cggCTCGAGcgcgcagcggcgctccgcg |
| Reverse | −24 | HindIII | gggggAAGCTTcagcagaaacagcaggca |
Added restriction enzyme sites are written in uppercase letters. Extra sequences of cgg and ggggg were added to XhoI and HindIII sequences, respectively, to facilitate the efficient cutting by these enzymes.
Transient transfection by lipofection/electroporation and luciferase assays
KC and other cells were transfected at ~70% confluence using the Fu-GENE-6 liposomal transfection reagent (Roche Diagnostics). Briefly, cells were transfected with 2 μg of supercoiled plasmid DNA per well in transfection of 6-well plates. An internal control plasmid, pTK-RL, which expresses the Renilla luciferase gene, was also used for correcting variations in the transfection efficiency. After 15 min, 1 ml of growth medium was added and incubated for 48 h. Lysates were harvested and extracts were prepared using freeze-thaw lysis. Lysates were monitored for luciferase activities using a commercially available Dual Luciferase (firefly and Renilla) Reagent Kit (Promega) in a Turner Biosystems 20/20n luminometer. Firefly luciferase activity (which reports the CD1d promoter) was normalized to the Renilla luciferase activity. Transient transfection by electroporation was performed using an Amaxa nucleofector device and a Amaxa Human KC Nucleofector Kit according to the manufacturer’s protocol. Briefly, trypsinized 1 million KC were electroporated with 3 μg of DNA (or small interfering oligonucleotide) and incubated for 48 h before harvesting the cellular lysates. All gene reporter assays were repeated at least three times.
EMSA
EMSA was done using nuclear extracts from KC prepared according to Klinke et al. (21). Briefly, EMSA probes CCAAT and mutCCAAT were generated by annealing the following sequences: CCAAT sense, GCTT GGGAATTGGCTGGCACCCA and CCAAT anti-sense: TGGGTGCC AGCCAATTCCCAAGC; mutCCAAT sense, GCTTGGGATTACGCT GGCACCCA and mutCCAAT anti-sense, TGGGTGCCAGCGTAATCC CAAGC). Two picomoles of the double-stranded probes were radiolabeled with 40 μCi of [γ-32P]ATP (Amersham Biosciences) using T4 polynucleotide kinase (Invitrogen) at 37°C for 1 h. Ten micrograms of nuclear extract was incubated in the presence of EMSA buffer (10 mM Tris-HCl (pH 7.5), 1 mM DTT, 1 mM EDTA, and 5% glycerol) and 1 μg of poly(dI:dC) and labeled probe (50,000 counts) at room temperature for 20 min, and the reaction was resolved on a 4% nondenaturing polyacrylamide gel. The gels were dried and autoradiographed. For competition or supershift, reaction mixtures were preincubated with either cold probe or Ab (C/EBP-β, C/EBP-α, or isotype control; purchased from Santa Cruz Biotechnology) for 30 min before addition of the labeled probe. The EMSA was repeated at least three times.
Chromatin immunoprecipitation (ChIP)
We used a modification of the ChIP protocol as described by Mukhopadhyay et al. (22). Cells (1 × 106 cells/well) were cross-linked with 1% formaldehyde for 10 min, washed with ice-cold PBS, and lysed by incubating for 10 min in radioimmunoprecipitation assay buffer containing protease inhibitors (catalog no. 8340; Sigma-Aldrich). Cell lysates were sonicated on ice to shear DNA to lengths between 400 and 600 bp using a Sonic Dismembrator model 100 (Fisher Scientific). Lysates were sonicated at 50% power for a total of 50 one-second pulses with 20–30 s cooling on ice after every 10 pulses. Sonicated cell supernatant was diluted 10-fold in ChIP Dilution Buffer (ChIP kit, catalog no. 17–295; Upstate Biologicals) with added protease inhibitors and precleared with protein A-agarose/salmon sperm DNA (Upstate Biologicals) for 30 min at 4°C with agitation. Precleared supernatant fraction was divided equally in three tubes for C/EBP-β Ab (catalog no. sc-150; Santa Cruz Biotechnology), normal rabbit IgG control, and no Ab control. Five micrograms of the immunoprecipitating Ab or control IgG was added and incubated for 4 h at 4°C with rotation. After 4 h, 30 μl of protein G magnetic beads (Active Motif part no. 101945) was added to each tube and incubated overnight at 4°C with rotation. Protein G magnetic bead-bound immune complexes were collected in a magnet stand and washed sequentially with 1 ml of the following washing buffers: low-salt immune complex wash buffer, high-salt immune complex wash buffer, LiCl immune complex wash buffer, and Tris-EDTA buffer. After the final wash, the immune complex was eluted using 250 μl of freshly prepared elution buffer (1% SDS, 0.1 M NaHCO3), de-cross-linked by heating at 65°C for 4 h, digested with proteinase K, and DNA recovered by a Qiaquick PCR Purification Kit (catalog no. 28104; Qiagen). DNA was eluted in 30 μl of water and 2 μl of the eluted DNA was used as template for PCR using the following forward primer, 5′-TGGGACCCCGACCTCTTTG-3′ and reverse primer, 5′-CAGCAGAA ACAGCAGGCA-3′ that amplified the proximal promoter region of CD1d from −223 to +24. All ChIP experiments were repeated at least three times.
RNA extraction and real-time RT-PCR analysis
RNA extraction and DNase I digestion was performed using a Qiagen RNeasy kit according to the manufacturer’s protocols. One microgram of total RNA was used for reverse transcription using a First Strand cDNA Synthesis Kit (Amersham Biosciences) and random primers. Two micro-liters of the synthesized cDNA was used in quantitative real-time PCR (qRT-PCR) reactions. A Roche light cycler (Roche Diagnostics) was used in PCR with the following cycling parameters: 95°C for 10 min, followed by 40 cycles of 95°C for 10 s, 60°C for 10 s, and 72°C for 15 s. CD1d primers sequences were as follows: CD1dF, 5′-AGACATGGTATCTCCG AGCAAC-3′ and CD1dR, 5′-CTGAGCAGACCAGGACTGAA-3′. CD1d probes sequences were as following: CD1d_FL, 5′-TGTAGCTCCCACCC CAGTAGAGGAC-3′ and CD1d_LC, ATGTCCTGGCCCTCTAGACT GCTGTG-3′. Reaction conditions included MgCl2 at 4 mM. CD1d transcript values were normalized to 18S rRNA levels measured using CYBR green reaction. Fold induction of CD1d transcript was calculated using the ΔΔCT method as described previously (5). All quantitative PCR (qPCR) experiments were repeated at least three times.
Gene silencing via small interfering RNA (siRNA)
KC were transfected with 3 μg of FlexiTube siRNA oligonucleotide (with target sequence of CGGGCCCTGAGTAATCGCTTA; purchased from Qiagen) using electroporation (Amaxa) as described above. Gene silencing was verified by qRT-PCR using primers form Qiagen (catalog no. 249900) and a QuantiFast SYBR Green PCR kit (Qiagen). A control siRNA oligonucleotide with scrambled sequence was also used in these studies (Qiagen).
Western blot
Western blotting was performed as previously described (5). Equal quantities of total protein from different samples were separated on a 10% Bis-Tris NuPage denaturing gel (Invitrogen Life Technologies). Gels were transferred to nitrocellulose membranes and probed with appropriate first Abs and detected using a Western Breeze Chemiluminescent detection kit (Invitrogen Life Technologies). The bands were detected using a Chemi-Doc densitometer and Quantity-One software (Bio-Rad). Rabbit polyclonal anti-C/EBP-β (clone C-19) Ab was purchased from Santa Cruz Biotech-nology and rabbit polyclonal anti-β-actin Ab from Cell Signaling Technology. β-Actin signals from each sample were used for normalizing the intensity of specific bands of interest. Anti-CD1d antiserum (Abnova) was used in some experiments.
Fluorescence microscopy, immunohistochemistry, and flow cytometry
Five-micrometer frozen sections of the skin biopsy specimens were cut and fixed in ice-cold methanol. The primary CD1d Ab (Nor 3.2) was diluted 1/100 in PBS containing 0.1% BSA and 1% FCS and incubated at room temperature for 1 h, followed by a 1-h incubation at room temperature with the FITC-conjugated goat anti-mouse IgG (1/100 dilution). Fluorescence microscopy was performed using a Nikon Eclipse E600 epifluorescence microscope equipped with a digital camera (RT-spot slide; Diagnostic Instruments).
Cold acetone fixed 5-μm frozen sections of skin from psoriatic lesions or the uninvolved skin were used for immunohistochemistry with anti-C/EBP-β Abs. A rabbit polyclonal Ab against C/EBP-β (clone C-19; Santa Cruz Biotechnology) and Vectastain Elite ABC Kit (Vector Laboratories) were used in these experiments. Images were captured by a Nikon Eclipse E600 epifluorescence microscope fitted with the Spot imaging system (Diagnostic Instruments).
For flow cytometry, at least 1 × 104 gated cells were acquired using a BD Biosciences LSRII flow cytometer. All samples were analyzed using FACSDiva software (BD Biosciences).
Human subjects
Human subjects, who were being evaluated for contact dermatitis, were recruited from our clinical practice. The criteria for enrollment of subjects into this study was the presence of a 1–2+ score (North American Contact Dermatitis Research Group Scoring System) of the patch test allergic reaction at 48- or 96-h clinical readings (23). Psoriatic and control skin biopsy specimens were also collected after informed consent of the subjects under an institutional review board-approved protocol at the University of Maryland Baltimore. The allergens studied are described in a previous manuscript (17). Immunochemical analysis was performed in five different skin biopsies of ACD and psoriasis. Staining patterns observed with CD1d and C/EBP-β Abs were compared with paired normal skin.
NKT cell clones
The human NKT cell clone F6 was generously provided by Dr. T. Yamamura (National Institute of Neuroscience, Tokyo, Japan). The cells were cultured using standard techniques as previously described (24). For the Ag-presenting assays, the NKT cell clone was cultured at 100,000 cells/well, in the presence or absence of α-galactosylceramide at 100 ng/ml, along with the indicated numbers of THP-1 (a monocyte cell line that had been electroporated with control siRNA or C/EPB-β siRNA 48 h before the experiment). After 24 h of coculture of the THP-1 APC with the NKT cell clone, cell-free culture supernatants were collected and analyzed for cytokine content (IFN-γ, IL-4, or TNF-α) using a Luminex bead assay (25).
Mice
The wild-type and the C/EBP-β gene-targeted mice (C57BL/6 background) have been described earlier (26). All experimental protocols using mice were approved by the Institutional Animal Care and Use Committee at the University of Maryland School of Medicine (Baltimore). Total RNA and protein were extracted from liver or skin using previously described methods (5).
Statistical analysis
Quantitative data were analyzed for statistically significant differences using the GraphPad InStat Software Program. When multiple comparisons were examined, a one-way ANOVA was applied to the quantitative data ( p < 0.05 was considered significant).
Results
Significant basal level of CD1d promoter activities in normal human KC and other cell lines of epithelial origin
Previous reports on the transcriptional control of human CD1d have been primarily focused on bone marrow-derived cells (20). Our past studies on CD1d expression in human epidermis indicated that there is polarized expression of this molecule in normal skin, which is maximal in the outermost layer of viable epidermis, in the subcorneal area (5). In cultured KC, CD1d gene expression as well as CD1d Ag (Western blotting) increased with Ca2+-induced terminal differentiation (5), which correlated well with the observed polarized expression of CD1d in normal human skin. To determine whether the human KC support the CD1d gene expression, we first cloned the human CD1d gene promoter spanning from −1738 to +24 that harbors the proximal and distal CD1d promoter (20) upstream of a luciferase reporter gene. Then, various deletions of the promoter region from 5′ end (−985, −665, −354, −106, −65, and −39) were introduced into this fragment (Fig. 1A). We tested these constructs for their transcriptional control activity by transfecting them into primary KC and KC-derived cell lines. Primary human undifferentiated KC exhibited readily detectable basal CD1d promoter activity (Fig. 1B), which was found to be highly reproducible in 10 different sets of primary KC (data not shown). This is in contrast to previous studies of bone marrow- derived cell lines, which required a stimulation with PHA/PMA to detect significant CD1d promoter activity (20). We found a gradual increase in the promoter activities with the deletional constructs in the order −985 < −665 < −354. The −354/+24 sequence-containing construct fragment showed the highest promoter activity, which was significantly decreased in −106/+24 construct. The −65/+24 construct gained some transcriptional activity, but all activity was lost in the −39/+24 construct. We found a similar pattern of promoter activities when we introduced these constructs in the KC-derived HaCat (Fig. 1C) and Ker-1 cell lines (data not shown), except that the magnitude of the reporter activity was higher in HaCat than in primary KC. In primary KC and HaCat, the observed pattern of promoter activity with deletional constructs indicates the presence of potential negative regulatory elements between −985 to −665 and −665 to −354. They also strongly indicate the presence of enhancer element(s) between −354 and −106. Two cell lines of epithelial origin, CaCo2 (a colorectal cancer cell line) (27) (Fig. 1C) and HeLa (a cervical cancer cell line; data not shown) (28) exhibited negligible promoter activities above that of the empty vector. The reporter expression data correlated well with steady-state CD1d mRNA, as studied by quantitative PCR analysis of the CD1d transcripts (Fig. 1D) in HaCat and Ker-1. These cells had a higher CD1d expression than those cells with intermediate basal promoter activities (K562 and primary KC). Thus, basal CD1d gene promoter activity in primary, cultured KC, and in KC-derived cell lines and other epithelial cells correlated well with basal CD1d gene expression (qPCR).
Identification of a CCAAT box as critical regulator of the CD1d proximal promoter
Since enhanced expression of CD1d is related with several skin disorders such as ACD (17) and psoriasis (4), we set out to investigate the putative enhancing element(s) between −354 and −106 of CD1d. A previous study (20) reported the presence of a CCAAT element in this region. This sequence, AATTGG, is located between −112 and −107 of the CD1d promoter (Fig. 2A). However, its function in regulating CD1d expression has not been studied. To test whether it is critical for the promoter activity, we generated two more CD1d promoter constructs. The −112 construct which contains the CCAAT element exhibited promoter activity by ~3-fold over the −106 construct, which lacked the CCAAT element (Fig. 2B). The activity of −112 was almost similar to that observed with the −354 and −225 constructs in the HaCaT cell line, indicating the absence of any inhibitory sequence between −112 and −354. This also shows that in primary KC (NHK in Fig. 2), maximal CD1d promoter activity can be achieved by the CCAAT box and its downstream proximal promoter region. CD1d promoter activities were first defined in K562, a leukemic cell line (20). Therefore, we also examined the expression of these reporters in K562 cells and found that the deletion of the CCAAT element (−106 fragment) resulted in a significant loss of the CD1d promoter activity (Fig. 2C). This confirms that the CCAAT element exhibits significant promoter activity in both KC as well as hematopoietic cells.
FIGURE 2.

A stimulatory inverted CCAAT box is located in the CD1d proximal promoter region. A, −225/+24 region of human CD1d promoter showing inverted CCAAT box between -112 and −107(boxed and bold). B, Deletion of this CCAAT box results in >50% reduction in the CD1d promoter activities both in primary KC (designated as normal human KC (NHK) in figure) and HaCaT (comparing the −112 fragment, which contains the CCAAT box to the −106 fragment, which lacks the CCAAT box). C, Deletion of CCAAT box in K562 (bone marrow-derived cell line) results in loss of promoter activity (comparing the −354 fragment, which contains the CCAAT box, to the −106 fragment, which lacks the CCAAT box).
C/EBP-β binds to the CCAAT box of CD1d promoter
The CCAAT element is found upstream to the initial transcription site in many eukaryotic promoters and is the binding site for C/EBP, a family of the basic leucine zipper transcription factors composed of six members, C/EBP-α, -β, -γ, -δ, -ε, and -ζ (29). C/EBPs are expressed in many cell types, including KC (30), and all of the members of the C/EBP family (except C/EBP-ζ) can induce transcription by interacting with components of the basal transcription apparatus (31). The presence of a putative CCAAT element (Fig. 2, B and C) led us to the hypothesis that a member of the C/EBP transcription factor family might be involved in the CD1d transcriptional regulation. To test this hypothesis, we conducted EMSAs (Fig. 3). When a radiolabeled 23-mer oligonucleotide probe that overlapped the CCAAT box was incubated with HaCaT cell nuclear extract, a complex was observed (band A in Fig. 3A, lane 2) that could be competed out by the presence of 100-fold excess of the cold probe (lane 3). However, a mutant oligonucleotide probe in which three nucleotides within the CCAAT element were substituted (CCAAT to CGTAA) or an unrelated oligonucleotide (SP-1) abolished the development of this complex with the CCAAT box (31) and failed to compete with the radiolabeled CCAAT oligonucleotide at an 100-fold excess of the cold probe (Fig. 3A, lane 4). These observations showed the specificity of binding at the CCAAT box. Furthermore, when Abs were used for supershifting, we observed a supershift with antiC/EBP-β Ab (Fig. 3A, band B in lane 7) but not with either rabbit IgG (isotype control, Fig. 3A, lane 6) or anti-C/EBP-α Ab (Fig. 3A, lane 8), further indicating that the interacting protein was C/EBP-β. We found the similar results with nuclear extract derived from primary KC (Fig. 3B), although the supershifted band (band B, lane 7 in Fig. 3B) was lighter compared with HaCaT cells (Fig. 3A, lane 7). This suggests that the nuclear content of C/EBP-β may be lower in primary KC than in HaCaT, which would be consistent with the lower basal CD1d gene expression observed in primary KC compared with HaCaT (Fig. 2C). These results clearly indicate the in vitro interaction of C/EBP-β transcription factor with the inverted CCAAT region (−112 to −107) located in the CD1d promoter.
FIGURE 3.

C/EBP-β binds with CCAAT box located in CD1d proximal promoter region. EMSA with radiolabeled inverted CCAAT probe in nuclear extracts derived from (A) HaCaT KC (A) and (B) primary human KC identifies C/EBP-β (but not C/EBP-α) as CD1d-binding protein in vitro. In these experiments, the shifted probe (lane 2) was competed with excess unlabeled probe (lane 3), but not with an unlabeled mutated CCAAT probe (CCAAT sequence changed to CGTAA; changed sequences underlined) (lane 4); similarly, an unlabeled SP-1 probe did not interfere with the nuclear extracts shifting of the labeled CCAAT probe (lane 5). Neither isotype control Ab nor anti-C/EBP-α Ab supershifted the labeled CCCAT probe incubated with nuclear extracts (lanes 6 and 8), whereas an anti-C/EBP-β Ab supershifted the labeled CCAAT probe that had been incubated with HaCaT or primary KC (lane 7). C, Comparison of the nucleotide sequence of the wild-type and mutant CD1d CCAAT elements (used in the above EMSA) with that of the consensus C/EBP-β binding site showing significant homology of the CD1d CCAAT element to the C/EBP-β consensus sequence (C/EBP-β cons). D, Agarose gel of ChIP reveals in vivo binding of C/EBP-β in CD1d proximal promoter region in nuclear chromatin derived from HaCaT KC. E, Same ChIP as depicted on agarose gel, in this instance, ChIP was assayed using qPCR (see Materials and Methods). Irrel., Irrelevant.
To determine whether C/EBP-β interaction with the CD1d promoter also occurred in vivo, we conducted the ChIP assay. Chromatin-bound proteins from HaCaT cells were immunoprecipitated with anti-C/EBP-β Ab (or control antibodies), followed by PCR using primers that covered the inverted CCAAT box-containing proximal promoter region. ChIP data clearly revealed a positive band for C/EBP-β Ab (Fig. 3D, lane 3) which was undetectable in the normal rabbit IgG controls or no Ab controls (Fig. 3D, lanes 1 and 2). The ChIP results clearly demonstrate the in vivo interaction of C/EBP-β with the CD1d proximal promoter region in our Ha-CaT cell model. This same ChIP experiment was assayed using qPCR (Fig. 3E). This data clearly demonstrated the in vivo binding of C/EBP-β with CD1d at its proximal promoter region that harbors the inverted CCAAT box.
Increases in the extracellular Ca2+ concentration are known to trigger KC terminal differentiation and have been demonstrated to induce CD1d gene expression (5). We simultaneously assayed C/EBP-β gene expression during this Ca2+-induced terminal differentiation and found that both C/EBP-β and CD1d gene expression increased during terminal differentiation (Fig. 4A). We next asked the question whether such terminally differentiating KC will increase CD1d promoter activity. Compared with undifferentiated KC (cultured in low concentrations of extracellular Ca2+), terminally differentiated KC (cultured in high concentrations of extra-cellular Ca2+) exhibited significantly higher promoter activity (Fig. 4B). KC are known to increase retinoic acid synthesis during terminal differentiation (33). To test whether retinoic acid, when complexed with retinoic acid receptors, would be involved in the transcriptional response to Ca2+-induced terminal differentiation, we tested promoter fragments that contain retinoic acid response elements (RARE) (20). Surprisingly, the −1738 fragment (which contains a RARE) and the −985 fragment (which lacks a RARE) exhibited similar reporter activity in undifferentiated and differentiated KC, suggesting that the Ca2+-induced increase in promoter activity is independent of RARE. A qPCR analysis of ChIP products from undifferentiated and differentiated KC demonstrated that there was significantly more C/EBP-β bound to the CCAAT box in terminally differentiated KC than in the undifferentiated ones (Fig. 4C). This establishes that CD1d transcription is dynamically affected by C/EBP-β binding to the inverted CCAAT box region, at least for Ca2+-induced KC terminal differentiation, an event linked to enhanced CD1d gene expression in KC (5). Thus, Ca2+-induced perturbations in CD1d gene expression in human KC correlated well with C/EBP-β gene expression, increased promoter activity, and increased binding of C/EBP-β to the inverted CCAAT box region as measured by ChIP-qPCR.
FIGURE 4.
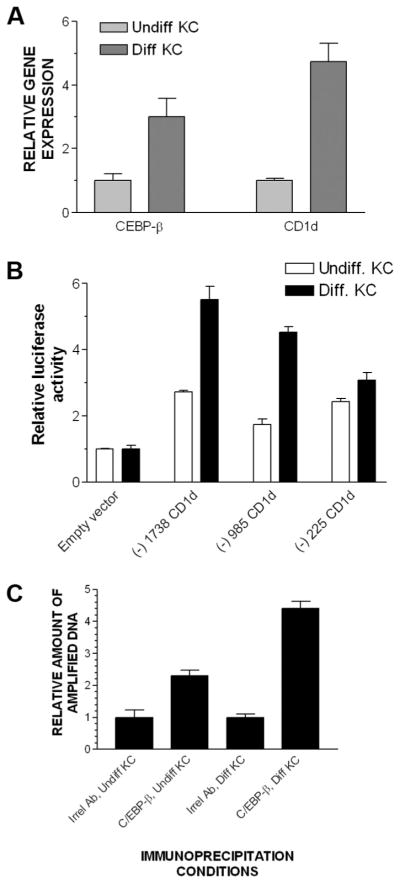
KC terminal differentiation increases C/EBP-β gene expression, CD1 promoter fragment reporter activity, as well as C/EBP-β binding to the CD1d CCAAT box region in KC. Primary human KC were cultured in medium containing low extracellular Ca2+ concentrations (0.05 mM) to keep them undifferentiated; 24 h after inducing differentiation by increasing the extracellular Ca2+ concentration to 1.5 mM, RNA was extracted, cDNA synthesized, and both CD1d and C/EBP-β gene expression were assayed using qPCR (A). The same experiment was repeated after transiently transfecting KC with indicated gene reporter constructs using Lipofectamine reagent (B) or ChIP assay in undifferentiated or differentiated KC (C). Irrel., Irrelevant.
C/EBP-β regulates CD1d transcription in cultured, primary KC, and bone marrow-derived cells
To further confirm that C/EBP-β binding with CD1d regulatory elements is linked to transcriptional control capacity in KC, we used two approaches. First, we took the gene knockdown approach. We knocked down C/EBP-β in KC using RNAi which resulted in more than a 40% reduction in C/EBP-β gene expression. Loss of C/EBP-β expression also resulted in a 50% decrease in CD1d gene expression (Fig. 5A). We confirmed that knockdown of C/EBP-β led to a decline in C/EBP-β and CD1d proteins in Western blot analyses (Fig. 5B). In the second approach, we examined the effects of C/EBP-β overexpression on CD1d gene expression in KC. Real-time PCR analysis (Fig. 5C) showed a 6-fold increase in CD1d mRNA in C/EBP-β-transfected KC compared with empty vector-transfected control cells. Western blot analyses confirmed that the CD1d levels increased at the protein level in the presence of gene transfer (overexpression) of C/EBP-β (Fig. 5D). To determine whether the observed regulatory effect of C/EBP-β on CD1d gene expression in KC was applicable to bone marrow-derived cells, we studied the knockdown of C/EBP-β in THP-1, a CD1d+ monocyte-derived cell line. Ninety-six hours after electroporating THP-1 with siRNA for C/EBP-β or control siRNA, flow cytometry was studied (Fig. 6A). The mean fluorescence intensity of THP-1 treated with control siRNA (mean fluorescence intensity, 1867 linear units) was greater than that of THP-1 that had C/EBP-β knocked down (mean fluorescence intensity, 1378 linear units). Examination of cell suspensions for membrane CD1d staining revealed that control siRNA-treated THP-1 exhibited brighter membrane staining than that of C/EBP-β siRNA-treated THP-1 (Fig. 6B). To determine whether THP-1 with low CD1d (C/EBP-β siRNA treated) exhibited impaired ability to trigger cytokine production in iNKT cells, we studied the CD1d-dependent presentation of stimulatory glycolipids to iNKT cells (using the iNKT cell clone F6). Those THP-1 treated with C/EBP-β siRNA exhibited an impaired ability to trigger iNKT cells to secrete the cytokines IFN-γ, IL-4, and TNF-α compared with THP-1 treated with control siRNA (Fig. 6, C–E).
FIGURE 5.

C/EBP-β overexpression or silencing in primary KC control CD1d gene expression. For qPCR analysis, assays were performed 48 h after electroporation. For Western blot analysis, assays were performed 72 h after electroporation. A, Primary undifferentiated KC were electroporated with medium alone or medium containing an irrelevant (Irrel) or C/EBP-β siRNA. C/EBP-β and CD1d gene expression were assayed using qPCR. B, Lysate was prepared from another set of electroporated KC and CD1d, C/EBP-β, and β-actin were Western blotted. C, Another set of undifferentiated KC were electroporated with medium alone, empty CMV vector, or the CMV vector driving the expression of C/EBP-β cDNA and CD1d gene expression was assayed by qPCR. D, After the overexpression of C/EBP-β in primary KC, a lysate was prepared for Western blotting.
FIGURE 6.

Silencing C/EBP-β in a CD1d+ monocytic cell line down-regulates cell surface CD1d and impairs presentation of stimulatory glycolipids to a NKT cell clone. The CD1d+ monocytic cell line THP-1 was electroporated with control siRNA or C/EBP-β siRNA, stained for (A) flow cytometry (gray histogram is isotype control, dashed line represents the CD1d staining of THP-1 treated with C/EBP-β siRNA; solid line represents the CD1d staining for THP-1 treated with control (Con) siRNA). B, Fluorescence photomicrograph of stained cells (×40 original magnification), left panel is the THP-1 treated with control siRNA, right panel is the THP-1 treated with C/EBP-β siRNA, (green staining is CD1d; blue nuclear staining is 4′,6-diamindo-2-phenylindole counterstain; ×40 original magnification). C–E, Luminex assay to detect cytokine secretion (IFN-γ, IL-4, and TNF-α) by the NKT cell clone F6 stimulated by the indicated THP-1 and α-galactosylceramide (*, p < 0.05 and **, p < 0.001; ANOVA).
Biological relevance of C/EBP-β to CD1d expression in vivo
To validate our observations further, we studied C/EBP-β expression in human ACD, a pathologic condition in which we have demonstrated that CD1d is overexpressed by epidermal KC (17). In normal skin, nuclear expression of C/EBP-β was localized to the subcorneal area (Fig. 7A), which is precisely the location that CD1d is maximally expressed in normal human skin (5). In ACD, a strong nuclear localization of C/EBP-β was noted in most layers of the epidermis (Fig. 7B). A similar staining pattern was observed in five independent skin biopsy specimens of ACD (data not shown). Lastly, we confirmed that both C/EBP-β and CD1d gene expression was increased in ACD compared with normal controls, paired skin in a total of five cases of ACD. Both C/EBP-β and CD1d gene expression were ~5-fold higher in ACD compared with the paired normal human skin from the same patient (Fig. 7C). All of these data established a direct role for C/EBP-β in the transcriptional regulation of CD1d in human KC.
FIGURE 7.

C/EBP-β controls CD1d expression in vivo in humans. A, Normal human skin exhibits prominent C/EBP-β localization to stratum granulosum area of epidermis (×100 original magnification). B, In ACD (a skin biopsy specimen derived from a 48-h positive patch test reaction), the skin exhibits nuclear localization of C/EBP-β more prominently throughout most of the epidermis (×200 original magnification). Reaction product is dark purple; counterstain is with methyl green. Irrelevant primary Ab did not result in any staining of epidermal nuclei (data not shown). C, qPCR analysis of C/EBP-β and CD1d gene expression in paired normal skin and ACD skin (n = 5 cases).
Discussion
CD1d expression is regulated in bone marrow-derived cells by transcriptional control (13–16), as well as by posttranscriptional mechanisms such as the association of the CD1d polypeptide with lipid transporter proteins that are rate limiting in the trafficking of this MHC-like molecule between the endosomal compartment and the cell surface (34–39). In this report, we showed the transcriptional control mechanisms of CD1d in primary human KC as well as KC-derived cell lines. Although CD1d promoter activities have been studied in some immortalized bone marrow-derived cell lines, to our knowledge, this is the first report that investigated CD1d promoter activities in primary cultured KC and other cells of epithelial origin. We found that primary KC reproducibly exhibit significant basal CD1d promoter activity (Fig. 1B) equivalent to that of bone marrow-derived cell lines that have been previously studied (Fig. 2). Some cultured cell lines such as HaCaT and Ker-1 drive even higher CD1d promoter activities (Fig. 1C and data not shown). Some cell lines such as HeLa and CaCo2 exhibit negligible promoter activity (Fig. 1C). The promoter activities correlated well with steady-state CD1d gene expression as measured by qRT-PCR (Fig. 1D) and cell surface expression (data not shown). This study of transcriptional regulation of CD1d is important because of the overexpression of this Ag-presenting molecule by KC in some human immune-mediated inflammatory skin diseases such as ACD (17) and psoriasis (6). ACD is an acute inflammatory skin disorder triggered by skin exposures to environmental xenobiotic compounds that are able to penetrate into the skin and elicit a T lymphocyte-dependent immune response. CD1d-restricted iNKT cells have been demonstrated to be necessary for the afferent phase of ACD in a mouse model (40 – 42). Recently, we demonstrated that NKT cells are present in all cases of the elicitation phase of ACD (i.e., positive patch test reactions (17). In the elicitation phase of ACD, NKT cells have been found both in the epidermis and dermis. Those epidermal-homing NKT cells have been noted to be in direct physical apposition to KC, which may be presenting microbial and/or self-glycolipids to NKT cells. It is therefore proposed that increased CD1d expression by KC plays an important role in modulating NKT cells during ACD.
In this study, we observed a basal level of CD1d promoter activity in KC and cell lines of epithelial origin. However, we were more interested in any mechanism that would enhance the expression of CD1d in these cells. We identified a CCAAT element to be active as an enhancer that contributed to the CD1d transcription. Although this CCAAT element was noted in the study published by Chen and Jackson (20), these authors did not attempt to characterize the role of the CCAAT box in their study, because the CCAAT element deletion studies were not attempted. We also identified this region as the binding site of the transcription factor C/EBP-β, both in vitro and in vivo (Fig. 3). A comparison of the nucleotide sequence of the CD1d CCAAT element to that of the consensus sequence of C/EBP-β revealed significant homology (Fig. 3C), supporting the molecular basis for the role of this transcription factor in driving basal and stimulated CD1d gene expression.
To clarify the role of C/EBP-β as a transcriptional regulator of CD1d, we conducted overexpression of C/EBP-β as well as knocking down by siRNA in KC. Both of these approaches showed a direct correlation between the C/EBP-β level present in the cell and CD1d expression. We also found increased binding of C/EBP-β to the CCAAT box in the proximal promoter region leads to increased CD1d message. These findings were further validated using tissues derived from C/EBP-β knockout mice, where we found a significant decrease in CD1d (both at RNA and protein levels) in the knockout mice compared with the wild-type mice (data not shown).
Our previous studies on KC CD1d expression have demonstrated that when KC are induced to undergo terminal differentiation, CD1d gene expression increases and the cytoplasmic pool of CD1d found in the undifferentiated KC migrates to the cell membrane in terminally differentiated KC (5). We thus used this model to demonstrate that CD1d promoter activity is further increased above basal conditions when KC are induced to increase CD1d gene expression by triggering terminal differentiation (Fig. 4B). C/EBP-β has been found to play a role in KC terminal differentiation (30) and we thus demonstrated that both C/EBP-β and CD1d gene expression increased during terminal differentiation. Furthermore, we also demonstrated that there is increased C/EBP-β binding to the CCAAT box in KC during terminal differentiation using a ChIP assay. Another molecular event associated with terminal differentiation in KC is increased synthesis of retinoic acid (33). Because retinoic acid has been demonstrated to play a role in increasing CD1d gene expression in dendritic cells induced by peroxisome proliferator-activated receptor-γ agonists (43), we investigated the RARE (present in CD1d promoter fragment −1738) played a role in the KC response to terminal differentiation signals. Surprisingly, our promoter fragment characterization data failed to detect a role in the KC CD1d transcriptional control during terminal differentiation (Fig. 4B). There were no significant differences between the promoter activities of the −1785/+24 construct (which contains a RARE) and the −985/+24 construct (which lacks RARE) during KC terminal differentiation. Our ChIP and qPCR data suggest that C/EBP-β is more important than RARE in regulating CD1d expression during terminal differentiation of KC (Fig. 4) and during inflammation (Fig. 7).
Our finding that C/EBP-β is involved in the enhanced transcription of CD1d is indirectly supported by a recent finding that shows increased nuclear expression of C/EBP-β in psoriasis (42). We also have confirmed the correlation between increased CD1d and increased C/EBP-β in human psoriatic lesions (45). Additionally, our finding of increased CD1d and C/EBP-β expression (Fig. 7, A–C) in another inflammatory skin condition, ACD, extends the observation that C/EBP-β plays a role in the control of CD1d in a variety of inflammatory conditions in the skin beyond psoriasis.
The different members of the C/EBP family can form homodimers or heterodimers with one another or with another transcription factor (that may or may not have a leucine zipper domain) (29). The dimerization is needed for the binding of C/EBP protein with its target DNA. Our EMSA data suggest that C/EBP-α does not partner with C/EBP-β in CD1d transcription in KC. Since C/EBP-α is the major partner of C/EBP-β in heterodimerizing (29), it is possible that C/EBP-β forms homodimer to facilitate its binding to CD1d, although we cannot rule out heterodimerization with other members of the C/EBP family of transcription factors. Therefore, it is of interest to determine in future study whether any other C/EBP family transcription factors are involved in C/EBP-β-dependent regulation of CD1d transcription.
In conclusion, this is the first demonstration that C/EBP-β controls CD1d expression by directly interacting with the CCAAT box of the CD1d promoter. Our findings that C/EBP-β augments CD1d expression in human KC identifies C/EBP-β as a key element in regulating expression of CD1d, which is known to play a role in regulating iNKT cell responses. Thus, this transcription factor appears to play a prominent role in controlling innate immunity, which is important in the pathogenesis of inflammatory skin diseases such as ACD and psoriasis.
Footnotes
This work was supported by Grant R0-1-AR46108-05 (to A.A.G.), CA78282 and CA105005 (to D.V.K.), and the Intramural Research Program of the Center for Cancer Research, National Cancer Institute (to P.F.J.).
Abbreviations used in this paper: KC, keratinocyte; ChIP, chromatin immunoprecipitation; qPCR, quantitative PCR; siRNA, small interfering RNA; ACD, allergic contact dermatitis; RARE, retinoid acid response element; iNKT, invariant NKT; qRT-PCR, quantitative real-time RT-PCR.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Gaspari AA. The role of KCs in the pathophysiology of contact dermatitis. Immunol Allergy Clin North Am. 1997;17:377–405. [Google Scholar]
- 2.Gaspari AA, Katz SI. Induction and functional characterization of class II MHC (Ia) antigens on murine KCs. J Immunol. 1988;140:2956–2963. [PubMed] [Google Scholar]
- 3.Gaspari AA, Jenkins MK, Katz SI. Class II MHC-bearing KCs induce antigen-specific unresponsiveness in hapten-specific TH1 clones. J Immunol. 1988;141:2216–2220. [PubMed] [Google Scholar]
- 4.Bonish B, Jullien D, Dutronc Y, Huang BB, Modlin R, Spada FM, Porcelli SA, Nickoloff BJ. Overexpression of CD1d by KCs in psoriasis and CD1d-dependent IFN-γ production by NKT cells. J Immunol. 2000;165:4076– 4085. doi: 10.4049/jimmunol.165.7.4076. [DOI] [PubMed] [Google Scholar]
- 5.Fishelevich R, Malanina A, Luzina I, Atamas S, Smyth MJ, Porcelli SA, Gaspari AA. Ceramide-dependent regulation of human epidermal KC CD1d expression during terminal differentiation. J Immunol. 2006;176:2590–2599. doi: 10.4049/jimmunol.176.4.2590. [DOI] [PubMed] [Google Scholar]
- 6.Porcelli SA, Modlin RL. CD1 and the expanding universe of T cell antigens. J Immunol. 1995;155:3709–3710. [PubMed] [Google Scholar]
- 7.Exley M, Garcia J, Balk SP, Porcelli SA. Requirements for CD1d recognition by human invariant Va24+CD4−CD8− T cells. J Exp Med. 1997;186:109–120. doi: 10.1084/jem.186.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Canchis PW, Bhan AK, Landau SB, Yang L, Balk SP, Blumberg RS. Tissue distribution of the non-polymorphic major histo-compatibility complex class I-like molecule CD1d. Immunology. 1993;80:561–565. [PMC free article] [PubMed] [Google Scholar]
- 9.Paduraru C, Spridon L, Yan W, Bricard G, Valencia X, Porcelli SA, Illarionov PA, Besra GS, Petrescu SM, Petrescu AJ, Cresswell PJ. An N-linked glycan modulates the interaction between the CD1d heavy chain and β2-microglobulin. J Biol Chem. 2006;281:40369– 40378. doi: 10.1074/jbc.M608518200. [DOI] [PubMed] [Google Scholar]
- 10.Brossay L, Julien D, Cardell S, Sydora BC, Burdin N, Modlin RL, Kronenberg M. Mouse CD1 is mainly expressed on hemopoietic derived cells. J Immunol. 1997;159:1216–1224. [PubMed] [Google Scholar]
- 11.Spada FM, Borriello F, Sugita M, Watts GFM, Koezuka Y, Porcelli SA. Low expression level but potent antigen presenting function of CD1d on monocyte lineage cells. Eur J Immunol. 2000;30:3468–3477. doi: 10.1002/1521-4141(2000012)30:12<3468::AID-IMMU3468>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 12.Skold M, Xiong X, Illarionov PA, Besra GS, Behar SM. Interplay of cytokines and microbial signals in the regulation of CD1d expression and NKT cell activation. J Immunol. 2005;175:3584–3593. doi: 10.4049/jimmunol.175.6.3584. [DOI] [PubMed] [Google Scholar]
- 13.Dougan SK, Kaser S, Blumberg RS. CD1 expression on antigen-presenting cells. Curr Topics Microbiol Immunol. 2007;314:113–141. doi: 10.1007/978-3-540-69511-0_5. [DOI] [PubMed] [Google Scholar]
- 14.Blumberg RS, Terhorst C, Bleicher P, McDermott FV, Allan CH, Landau SB, Trier JS, Balk SP. Expression of a non-polymorphic MHC class I-like molecule, CD1D, by human intestinal epithelial cells. J Immunol. 1991;147:2518–2524. [PubMed] [Google Scholar]
- 15.Colgan SP, V, Morales M, Madara JL, Polischuk JE, Balk SP, Blumberg RS. IFN-γ modulates CD1d surface expression intestinal epithelia. Am J Physiol. 1996;271:C276–C283. doi: 10.1152/ajpcell.1996.271.1.C276. [DOI] [PubMed] [Google Scholar]
- 16.Exley M, Garcia J, Wilson SB, Spada F, Gerdes D, Tahir SMA, Patton KT, Blumberg RS, Porcelli SA, Chott A, Balk SP. CD1d structure and regulation on human thymocytes, peripheral blood T-cells, B-cells and monocytes. Immunology. 2000;100:37–47. doi: 10.1046/j.1365-2567.2000.00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gober M, Fishelevich R, Zhao YM, Unutmaz D, Gaspari AA. Human natural killer T cells infiltrate into the skin at elicitation sites of allergic contact dermatitis. J Invest Dermatol. 2008;128:1460–1469. doi: 10.1038/sj.jid.5701199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyer EH, DeKruyff RH, Umetsu DT. iNKT cells in allergic disease. Curr Top Microbiol Immunol. 2007;314:269–291. doi: 10.1007/978-3-540-69511-0_11. [DOI] [PubMed] [Google Scholar]
- 19.Tsuneyama K, Yasoshima M, Harada K, Hiramatsu K, Gershwin ME, Nakanuma Y. Increased CD1d expression on small bile duct epithelium and epitheliod granuloma in livers in primary biliary cirrhosis. Hepatology. 1998;28:620– 623. doi: 10.1002/hep.510280303. [DOI] [PubMed] [Google Scholar]
- 20.Chen QY, Jackson N. Human CD1d gene has TATA boxless dual promoters: an SP-1-binding element determines the function of the proximal promoter. J Immunol. 2004;172:5512–5521. doi: 10.4049/jimmunol.172.9.5512. [DOI] [PubMed] [Google Scholar]
- 21.Klinke DJ, II, Ustyugova IV, Brundage KM, Barnett JB. Modulating temporal control of NF-κB activation: implications for therapeutic and assay selection. Biophys J. 2008;94:4249– 4259. doi: 10.1529/biophysj.107.120451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mukhopadhyay A, Deplancke B, Walhout AJ, Tissenbaum HA. Chromatin immunoprecipitation (ChIP) coupled to detection by quantitative real-time PCR to study transcription factor binding to DNA in Caenorhabditis elegans. Nat Prot. 2008;3:698–709. doi: 10.1038/nprot.2008.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Belsito DV. Patch testing with a standard allergen (“screening”) tray: rewards and risks. Dermatol Ther. 2004;17:231–239. doi: 10.1111/j.1396-0296.2004.04033.x. [DOI] [PubMed] [Google Scholar]
- 24.Sakuishi K, Oki S, Araki M, Porcelli SA, Takashi MS, Yamamura T. Invariant NKT cells biased for IL-5 production act as crucial regulators of inflammation. J Immunol. 2007;179:3452–3462. doi: 10.4049/jimmunol.179.6.3452. [DOI] [PubMed] [Google Scholar]
- 25.Carson RT, Vignali DAA. Simultaneous quantitation of 15 cytokines using a multiplexed flow cytometric assay. J Immunol Methods. 1999;227:41–52. doi: 10.1016/s0022-1759(99)00069-1. [DOI] [PubMed] [Google Scholar]
- 26.Sterneck E, Tessarollo L, Johnson PF. An essential role of C/EBP-β in female reproduction. Genes Dev. 1997;11:2153–2162. doi: 10.1101/gad.11.17.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fogh J, Fogh JM, Orfeo T. One hundred and twenty-seven cultured human tumor cell lines producing tumors in nude mice. J Natl Cancer Inst. 1977;22:221–226. doi: 10.1093/jnci/59.1.221. [DOI] [PubMed] [Google Scholar]
- 28.George P, Journey LJ, Goldstein MN. Effect of vincristine on the fine structure of HeLa cells during mitosis. J Natl Cancer Inst. 1965;35:55–75. [PubMed] [Google Scholar]
- 29.Li H, Gade P, Xiao W, Kalvakolanu D. The interferon-signaling network and transcription factor C/EBP-β. Cell Mol Immunol. 2007;4:407–418. [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu S, Oh HS, Shim M, Steneck E, Johnson PF, Smart RC. C/EBP-β modulates the early events of KC differentiation involving growth arrest and keratin 1 and keratin 10 expression. Mol Cell Biol. 1999;19:7181–7190. doi: 10.1128/mcb.19.10.7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsukagoshi N. Regulation of gene expression in Aspergilli. Nippon Ishinkin Gakkai Zasshi. 1998;39:85–90. doi: 10.3314/jjmm.39.85. [DOI] [PubMed] [Google Scholar]
- 32.Roy SK, Wachira SJ, Weihua X, Hu J, Kalvakolanu DV. CCAAT/enhancer-binding protein-β regulates interferon-induced transcription through a novel element. J Biol Chem. 2000;275:12626–12632. doi: 10.1074/jbc.275.17.12626. [DOI] [PubMed] [Google Scholar]
- 33.Fisher GJ, Voorhees JJ. Molecular mechanisms of retinoic acid actions in skin. FASEB J. 1996;10:1002–1013. doi: 10.1096/fasebj.10.9.8801161. [DOI] [PubMed] [Google Scholar]
- 34.Zhou D, Cantu C, Sagiv Y, Schrantz N, Kulkarni AB, Qi X, Mahuran DJ, Morales CR, Grabowski GA, Benhlaha K, et al. Editing of CD1d-bound lipid antigens by endospomal lipid transfer proteins. Science. 2004;303:523–527. doi: 10.1126/science.1092009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sagiv Y, Hudspeth K, Mattner J, Schrantz N, Stern RK, Zhou D, Savage PB, Teyton L, Bendelac A. Impaired glycosphingolipid trafficking and NKT cell development in mice lacking Niemann-Pick type C1 protein. J Immunol. 2006;177:26–30. doi: 10.4049/jimmunol.177.1.26. [DOI] [PubMed] [Google Scholar]
- 36.Schrantz N, Sagiv Y, Liu Y, Savage PB, Bendelac A, Teyton L. The Niemann-Pick type C2 protein loads isoglobtrihexosylceramide onto CD1d molecules and contributes to the thymic selection of NKT cells. J Exp Med. 2007;204:841–852. doi: 10.1084/jem.20061562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brozovic S, Nagaishi T, Yoshida M, Betz S, Salas A, Chen D, Kaser A, Glickman J, Kuo T, Little A, et al. CD1d function is regulated by microsomal triglyceride transfer protein. Nat Med. 2004;10:535–539. doi: 10.1038/nm1043. [DOI] [PubMed] [Google Scholar]
- 38.Dougan SK, Salas A, Rava P, Agyemang A, Kaser A, Morrison J, Khurana A, Kronenberg M, Johnson C, Exley M, et al. Microsomal triglyceride transfer protein lipidation and control of CD1d on antigen-presenting cells. J Exp Med. 2005;202:529–539. doi: 10.1084/jem.20050183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sagiv Y, Bai L, Wei DG, Agami R, Savage PB, Teyton L, Bendelac A. A distal effect of microsomal triglyceride transfer protein deficiency on the lysosomal recycling of CD1d. J Exp Med. 2007;204:921–928. doi: 10.1084/jem.20061568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Campos RA, Szczepanik M, Itakura A, Akahira-Azuma M, Sidobre S, Kronenberg M, Askenase PW. Cutaneous immunization rapidly activates liver invariant Va14 NKT cells stimulating B-1 B cells to initiate T cell recruitment for elicitation of contact hypersensitivity. J Exp Med. 2003;198:1785–1796. doi: 10.1084/jem.20021562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Askenase PW, Szczepanik M, Itakura A. Extravascular T cell recruitment requires initiation begun by Vα14+ NKT cells and B-1 B cells. Trends Immunol. 2004;25:441–449. doi: 10.1016/j.it.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 42.Nieuwenhuis EES, Eillessen S, Scheper RJ, Exley MA, Taniguchi M, Balk SK, Strominger JL, Dranoff G, Blumenberg RS, Wilson SB. CD1d and CD1d-restricted iNKT cells play a pivotal role in contact hypersensitivity. Exp Dermatol. 2005;14:250–258. doi: 10.1111/j.0906-6705.2005.00289.x. [DOI] [PubMed] [Google Scholar]
- 43.Szatmari I, Pap A, Ruhl R, Ma JX, Illarionov PA, Besra GS, Rajnavolgyi E, Dezso D, Nagy L. PPAR-γ controls CD1d by turning on retinoic acid synthesis in developing human DC. J Exp Med. 2006;203:2351–2362. doi: 10.1084/jem.20060141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Komine M, Karakawa M, Takekoshi T, Sakurai N, Minatani Y, Mitsui H, Tada Y, Saeki H, Asahina A, Tamaki K. Early inflammatory changes in the “perilesional skin” of psoriatic plaques: is there interaction between dendritic cells and KCs? J Invest Dermatol. 2007;127:1915–1922. doi: 10.1038/sj.jid.5700799. [DOI] [PubMed] [Google Scholar]
- 45.Zhao YM, Fishelevich R, Petrali JP, Zheng L, Anatolieva MA, Deng A, Eckert RL, Gaspari AA. Activation of keratinocyte protein kinase c-zeta in psoriasis plaques. J Invest Dermatol. 2008;128:2190–2197. doi: 10.1038/jid.2008.81. [DOI] [PMC free article] [PubMed] [Google Scholar]